

Abstract
The humoral innate immune system is composed of three major branches, complement, coagulation, and natural antibodies. To persist in the host, pathogens, such as bacteria, viruses, and cancers must evade parts of the innate humoral immune system. Disruptions in the humoral innate immune system also play a role in the development of autoimmune diseases. This review will examine how gram positive bacteria, viruses, cancer, and the autoimmune conditions Systemic Lupus Erythematosus and Anti-phospholipid syndrome, interact with these immune system components. Through examining evasion techniques it becomes clear that interplay between these three systems exists. By exploring the interplay and the evasion/disruption of the humoral innate immune system, we can develop a better understanding of pathogenic infections, cancer, and autoimmune disease development.
Keywords: complement, coagulation, natural antibodies, B1 B cells, pathogens, cancer, SLE
1.0 The role of complement evasion in disease
The complement system serves as a first line of defense, by directly lysing abnormal cells and pathogens, and recruiting other immune cells such as macrophages and neutrophils. Complement activation occurs by three distinct, but connected pathways: classical, lectin, and alternative. C1q and Mannose Binding Lectin (MBL) initiate the classical and lectin pathways, respectively. Both pathways cleave C4 and C2 to form the C3 convertase C2aC4b. The alternative initiation pathway follows constitutive C3 hydrolysis to C3b which, after interaction with Factors B, D and P form the stable C3 convertase, C3bBb [1, 2]. Both C3 convertases cleave C3 into C3a and C3b. C3b binds the C3 convertases to form the C5 convertase to cleave C5. C3a and C5a are anaphylatoxins which stimulate inflammation. C5b initiates the formation of the membrane attack complex (MAC or C5b-9) which leads to cytolysis and cell or pathogen death [3] (Fig. 1). Regulatory factors including CD55, CD46 and complement receptor 1 interact with co-factor, Factor I to control the convertases [4]. In addition, Factor H (FH) with the Factor I (FI) degrades the alternative C3 convertase.
Figure 1. Schematic representation of the complement cascade.

Upon recognition of a foreign or damage cell surface, one or more complement pathway is activated. The classical and lectin pathways differ in their initiation. The alternative pathway is distinct from the other two pathways but all three pathways converge toward the formation of a C5 convertase, which is necessary for the MAC formation. The
 indicate a cleavage. Ag, antigen; MAC, Membrane Attack Complex.
indicate a cleavage. Ag, antigen; MAC, Membrane Attack Complex.
1.1 Complement and Gram positive bacteria
Bacteria use three main methods of evading complement that include binding host inhibitors to the pathogen surface, using bacterial enzymes to cleave active complement components, and degrading surface bound proteins to prevent further activation of the complement cascade.
The ability to bind host inhibitors is prevalent among many different bacterial species, though this paper will focus only on gram positive bacteria. Species such as Haemophilus influenzae, Bordetella pertussis, and the gram positive Streptococcus pneumoniae use a conserved “superevasion” site to bind FH which results in degradation of C3b bound to the bacterial surface [5]. Both the streptococcal family and Staphylococcus aureus also use multiple, and sometimes redundant, proteins to bind FH to facilitate the formation of FH:C3b complexes which inhibit complement activation and increase bacterial survival [6–10]. S. aureus proteins not only recruit FH to the bacterial surface but also recruit FI which together inactivates C3b to form iC3b [8]. S. pneumoniae uses multiple proteins to bind and sequester C1q, as well as modulate complement FH, C4bp, and the C3 convertase [9, 11]. Members of the Microbial Surface Components Recognizing Adhesive Matrix Molecules protein family commonly expressed on S. aureus, Streptococcus equi, and Streptococcus mutans also bind C1q [12]. Finally, bacterial spores utilize similar survival strategies as Bacillus anthracis spores recruit FH, Factor H Related Protein 1, C1 inhibitor, and C4bp to their cell surfaces [13]. Thus, both spore forming and non-spore forming, gram positive bacteria evade destruction by recruiting natural host complement inhibitors or sequestering pathway initiators to prevent complement activation.
While recruiting complement pathway inhibitors to the bacterial surface is a common method of immune evasion, bacteria also use their own proteins to cleave active members of the complement pathway, preventing the cascade from proceeding. The most common target of gram positive bacterial enzymes is C3b. For example, S. aureus uses at least three proteins to degrade C3b and yet another protein to inhibit the formation of the C3 convertase [8, 14–16]. S. aureus also degrades C3 by activating plasminogen, a member of the coagulation pathway [8, 14, 17]. Other bacteria also bind fibronectin (streptococcal family) or interact with plasminogen (B. anthracis) to decrease the amount of C3b deposited on the bacteria [7, 13].
Other targets for complement evasion include direct inhibition of the common complement pathway and formation of convertases that lead to this step. Proteins expressed on the S. aureus bacterial surface prevent formation of the MAC and bacterial lysis [7, 9]. In addition, at least four S. aureus proteins bind to C3b and prevent the formation of further convertases [8, 14, 16], while an additional S. aureus protein prevents formation of the C3 convertase halting the complement cascade [18].
Finally, gram positive bacteria prevent complement activation by degrading bound Ab, initiator of the classical pathway. S. aureus accomplishes this by using over a half dozen different proteins which work together to degrade the surface bound Ab [19]. This exemplifies an interconnection between the complement pathways and natural antibodies (NAb), discussed below. Gram positive bacteria evolved multiple methods of complement evasion which play a crucial role in bacterial survival during infection.
1.2 Viruses
Viruses have evolved a wide range of novel mechanisms to evade the complement response to host infection. Mechanisms include preventing molecules such as C1q from binding to the antibody-antigen complex on the surface of infected cells, and pathogens. Additionally, viruses can express proteins that mimic complement regulators, thereby allowing the pathogen to enter the cell, and protecting the infected cell from lysis. Still, other viruses incorporate complement regulatory proteins, which are capable of upregulating or downregulating the complement response to the invasion [20, 21].
Specific viruses appear to interfere with the formation of the MAC, crucial to ridding the body of virus infected cells. One example is West Nile virus (WNV), a member of the flavivirus family, which encodes a Nonstructural protein 1 believed to bind to the regulatory protein FH. This binding leads to the inhibition of C3b deposition and to the formation of the MAC [22]. Similarly, Hepatitis C, also of the flavivirus family, prevents the formation of the MAC by interfering with C3, C4 and C9 complement components [23].
Viruses may also express cellular complement regulators on their envelope, which use molecular mimicry to interfere with complement regulation. Human immunodeficiency virus activates the complement system through the classical and lectin pathways, however, the virus appears to incorporate complement regulatory molecules on the virion envelope, preventing complement mediated neutralization [24–26]. One study shows that Nipah virus, of the paramyxoviridae family, can evade the complement pathways using FI to inactivate C3b through cleavage. This evasion mechanism may contribute to the high mortality rate and systemic infection associated with this virus [27]. All of these examples demonstrate mechanisms, which a variety of viruses have evolved, to evade the complement mediated response to invasion by viral pathogens.
1.3 Systemic Lupus Erythematosus (SLE) and anti-phospholipid syndrome (APLS)
Complement plays a major role in the pathogenesis of multiple autoimmune diseases. The mechanisms that cause the development of autoimmune disease are complex and include the dysregulation or depletion of complement cascade components. SLE is a syndrome consisting of a group of systemic, self-reactive, chronic inflammatory responses that may affect the skin (butterfly rash), joints, kidneys (glomerulonephritis), and other organ systems. APLS is an autoimmune disease in which thrombosis occurs in both veins and arteries due to anti-phospholipid antibodies (aPLs) and often results in spontaneous fetal abortion. As SLE patients may develop APLS, the interconnection appears to include complement playing a significant role in manifestation of both these autoimmune diseases [28, 29].
Deficiencies of the complement proteins C1q, C4 and C3 correlate with development of SLE disease [30, 31]. In 88% of an SLE cohort, a homozygous deficiency in C1q correlated to the development of SLE [32] and lower C1q levels occur during glomerulonephritis flares in SLE patients [33]. C4 deficiency is also correlated to SLE but the risk is higher for patients with C1q deficiency [34, 35]. As a central complement component, a lack of C3 appears to prevent APLS and development of SLE [36]. Similar effects are found with C5 deficiency in APLS and these effects are mirrored in mice treated with anti-C5 Ab [37]. Compared to healthy people, primary APLS patients appear to have unregulated and/or excessive complement activation as demonstrated by low C3 and C4 serum levels and increased levels of anaphylatoxins C3a and C4a [38]. In summary, deficiencies in distinct complement components do not result in a universal phenotype of SLE or APLS disease, though it appears that the further upstream in the complement cascade the deficient protein is located, the higher the risk of disease development.
SLE and APLS both develop significant auto-reactive Ab to initiate complement activation. Specifically, anti-C1q autoantibodies form aggregates with the collagen-like portion of C1q and correlate with SLE development [39]. Low complement levels and another autoreactive Ab, anti-double stranded DNA are also phenotypic of lupus nephritis [40]. The increase in autoreactivity towards both single and double stranded DNA, histones, and chromatin in SLE correlate with defects in the complement receptor 2 (CD21) [41, 42]. Taken together, deficiencies in complement pathways, as well as complement receptors, frequently lead to autoimmune disease development.
1.4 Cancer
Cancer is a disease characterized by uncontrolled cellular growth and possible dissemination of cells from the primary site to distant tissues. Abnormal cells are targets of the complement system, however, some cancer cells take advantage of activation while others inhibit this system. In a phase II clinical trial, pancreatic cancer was shown to inhibit the MBL pathway and treatment with ω-3–rich fatty acids and gemcitabine restored MBL activity, improving patient survival [43]. In contrast, lung cancer cells evade complement-mediated cell lysis by overexpressing the alternative pathway inhibitor FH [44]. Additional studies indicate that cancer cells utilize the classical pathway to their advantage. Ligation of the cell surface receptor for C1q, also known as C1q binding protein, stimulates tumor proliferation and increases the metastatic potential of lung and breast cancer [45–47]. Similarly, C4b, which is cleaved following C1 activation, is overexpressed in cancers such as melanoma [48], pancreatic [49], and breast cancers [50].
Tumor cells also circumvent MAC formation preventing cell lysis by overexpressing CD59, a MAC inhibitor [51–54]. Gastric cancer cells overexpress C9 suggesting it plays a role independent of MAC formation in cancer [55]. These findings could explain the failure of some monoclonal antibody (mAb) therapies requiring complement mediated cell lysis. Ab activation of complement-induced inflammation may be beneficial for multiple cancer types and inhibition of MAC formation could reduce the therapeutic activity of the mAb. If the Ab targets mucins away from the cell membrane, the MAC will not form [56], causing inflammation without cytotoxicity. Consequently, targeting the complement dysregulation [43, 55] in association with mAb may be a promising chemotherapy [57].
Activation of any complement pathway produces the analyphylatoxins, C3a and C5a [58]. Overproduction of C5a and/or C3a stimulates tumor progression in lymphoma [59], ovarian [60], lung [61, 62], and breast cancer [63]. Metastasis also involves anaphylatoxins as C3a is required for cellular cohesion during cell migration, [64] and C5a contributes to immune regulation [65, 66]. In summary, cancer cells may down regulate the lectin and alternative pathways to escape the immune response but may utilize the classical pathway, to induce inflammation for growth and metastasis. Cancer cell expression of complement inhibitors prevent MAC formation and cell lysis.
2.0 The role of coagulation evasion in disease
The coagulation pathways function to maintain vascular integrity, protecting the vascular system from injury and invading pathogens. Composed of proteolytic cascades, cleavage of inactive plasma enzymes releases an active factor, which acts on the next enzyme in the cascade leading to clot formation. There are two coagulation pathways, intrinsic and extrinsic, which merge into the common pathway, resulting in the formation of a fibrin clot (Fig. 2) [67]. Damage to endothelial cell surfaces activates the intrinsic pathway through Factor XII. This activation allows binding of kallikrein (KLK), and high molecular weight kininogen (HK) to the cell surface which is crucial for the propagation of cleavage events producing additional Factor XIIa as a product. Factor XIIa then cleaves Factor XI which prompts the cleavage of subsequent Factors IX, VIII and X in the intrinsic cascade. Trauma may induce exposure of Tissue Factor (TF) and initiate the extrinsic pathway with subsequent cleavage of Factor VII into VIIa, or direct activation of TF resulting in the cleavage of Factor IX and Factor X. Activation of Factor X initiates the common pathway and when complexed with Factor Va activates thrombin. Thrombin, the primary regulator of coagulation, cleaves fibrinogen in the formation of a fibrin clot. Pathogens persist in the host by interfering with the function of the coagulation cascade. Dysregulation within these pathways occasionally leads to autoimmune disease and is implicated in some cancers.
Figure 2. Diagram of the coagulation cascades.
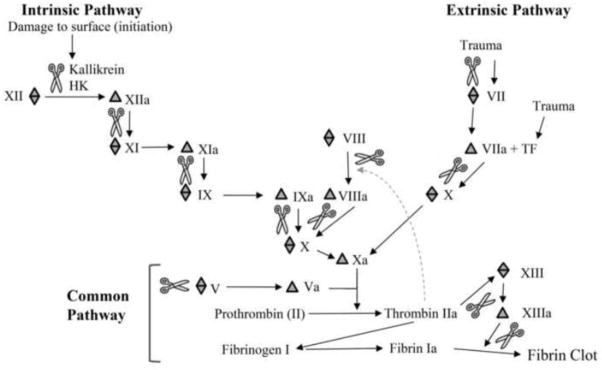
There are two coagulation cascades, intrinsic and extrinsic, which converge into the common pathway. These cascades are advanced through proteolytic cleavage of coagulation factors.
 represents factor prior to cleavage,
represents factor prior to cleavage,
 represents the cleavage product and
represents a proteolytic cleavage. HK, High molecular weight kininogen.
represents the cleavage product and
represents a proteolytic cleavage. HK, High molecular weight kininogen.
2.1 Gram positive bacteria
During bacterial infections, coagulation is often used to contain bacteria, making them easier for the host to destroy [68]. Some bacterial species disrupt the coagulation pathway by breaking up fibrin clots, while others cause a fatal overactivation of the coagulation pathways leading to disseminated clots [68]. In the case of B. anthracis, the peptidoglycan layer activates platelets and causes coagulation dysfunction [69, 70]. Clostridium perfringens can occasionally induce intravascular coagulation by secreting an extracellular alpha toxin that forms complexes composed of activated platelets, fibrin, and leukocytes [71].
S. pyogenes infections significantly impact host coagulation as shown by decreased fibrinogen levels in the plasma, increased levels of fibrin degradation products, and up to a 50% reduction in platelet count [71, 72]. Killed bacteria, washed to remove any non-S. pyogenes proteins, increase the production of TF from cells, as shown in vitro [71]. The M protein complexes with up to four fibrinogen molecules and platelets already activated by anti-S. pyogenes Ab [72, 73]. These activated platelets then interact with neutrophils and monocytes to increase the release of TF, increasing clotting and releasing heparin-binding protein. [72–74]. The bacteria also produce the enzyme streptokinase, which cleaves fibrinogen [75]. Additional redundancy includes, S. pyogenes upregulates the host’s coagulation system by sequestering protein S [71].
Sequestering coagulation pathway components is an additional mechanism by which S. pyogenes inhibits clotting. The M proteins recruit HK to the bacterial surface exposing it to a secreted protein that cleaves HK releasing bradykinin, leading to vascular permeability [75, 76]. Additional S. pneumoniae surface proteins adhere to host plasminogen, bringing it into contact with the urokinase-type plasminogen activator, leading to the activation of plasmin [74, 77].
S. aureus has a variety of other mechanisms for altering the coagulation pathway. Two secreted homeostasis factors activate prothrombin without enzymatic cleavage and prevent clot formation [15, 73, 78, 79]. As another important survival mechanism, S. aureus forms a pseudo-capsule of fibrin and fibrinogen [73, 79]. Extracellular Binding Protein complexes with fibrinogen and surface bound C3b to create a shield. This shield hides immune factors, such as Ab and C3b, that are bound to the bacterial surface preventing phagocytosis [15]. Other S. aureus surface proteins associate directly with multiple members of the coagulation pathway. For example, coagulase binds to thrombin, fibrinogen, and fibronectin, while Von Willebrand Factor Binding Protein recognizes thrombin and factor XII [79]. Multiple additional S. aureus proteins are substrates of members of the coagulation cascade including factors VII, IX, X, Xa, XIIa and prothrombin [79, 80]. Studies with S. aureus suggest that clot formation prevention is required for bacterial survival, as coagulase mutants cannot survive in the skin of infected mice, and the host clears bacteria immobilized in a clot [73, 79]. Redundancy in S. aureus coagulation evasion is critical and the loss of key bacterial proteins can lead to bacterial clearing. By avoiding immobilization within a clot, gram positive bacteria escape detection and destruction. Given this important role, it is easy to see why a variety of evasion mechanisms have evolved.
2.2 Viruses
Viruses which cause hemorrhagic fever (HF), are in principle distinct, because these viruses specifically interfere with the coagulation pathways, among other essential defense mechanisms. Ebola virus, a filovirus responsible for over 10,000 death in 2014 alone, causes hemorrhagic disease in infected patients. Over-expression of TF leads to excessive activation of the TF activated coagulation pathway and fibrin accumulation in tissue associated with viral Ag, as well as a loss of endothelial barrier function, which are also characteristic of Ebola HF [81]. Dengue HF, caused by Dengue virus, a flavivirus, activates fibrinolysis through the direct degradation of fibrinogen, initiating a secondary activation of pro-coagulants such as TF. Dengue virus expresses molecules, which mimic coagulation factors, contribute to the inhibition of thrombin activity, and enhance fibrinolysis [82].
Enveloped viruses acquire envelopes from host cells, thus expressing and utilizing host cell and cell membrane properties. Herpes Simplex Virus 1 uses host components to activate thrombin and enhance infection and encodes glycoprotein C which contributes to the enhancement of factor X activation [83]. Adenovirus type 5 also uses a host coagulation factor to overcome a complement-mediated blockade and infect the liver. Additionally, Adenovirus type 5 associates with factor X which protects this virus from complement-mediated and Ab neutralization [84].
Viruses interfere with the regulation of the coagulation pathways in multiple ways. Some viruses cause the overexpression of coagulation activation molecules. Others utilize host molecules to interfere with important regulatory components, while some associate with coagulation factors which decrease the immune response.
2.3 Systemic Lupus Erythematosus and anti-phospholipid syndrome
SLE and APLS may be characterized by hypercoagulability. SLE and APLS patients have increased TF expression in monocytes and aPL Ab found in these patients increase TF expression [85, 86]. Endothelial cell activation by aPL leads to production of TF and upregulation of adhesion molecules [87]. In APLS and SLE this leads to pathogenesis and hypercoagulability. In addition, compared to healthy people, SLE patient plasma contains significantly reduced levels of TF pathway inhibitor [88].
A reduction of annexin A5 anticoagulant activity is correlated with SLE pathogenesis in children [89]. Annexin A5 binds anionic phospholipids and blocks coagulation enzymatic reactions [90]. In addition, aPL Ab can interfere with annexin A5 binding to phospholipids and promote thrombosis events in APLS patients. This is influenced by an increased level of anti-domain 1 Ab to beta-2-glycoprotein I, which are found in children and adolescents with SLE [89] and significantly increase the risk of thrombosis [91].
Activation of the intrinsic pathway in coagulation occurs through activation of factor XII specifically by preformed fibrin clots [92]. High levels of factor XIIa-antithrombin (AT) complex are found in APLS and SLE patients with previous venous and arterial thrombosis [92, 93] and SLE patients have a higher risk of developing thrombosis compared to APLS [94]. Thus, multiple mechanisms alter the coagulation cascade in SLE and APLS patients.
2.4 Cancer
The association between thromboembolic events and cancer was reported over a century ago by Armand Trousseau who called them phlegmasia alba dolens (Latin for painful white inflammation). He later gave his name to the vessel inflammations caused by blood clots [95]. The incidence of thromboembolic events in cancer patients is extremely variable between studies (3–40%) but is always higher in cancer patients [96, 97]. Three main risk factors have been reported: patient-related (such as age, race, or comorbid conditions), site of primary tumor and treatment (surgery and chemotherapy increasing the risk of developing a thromboembolic event) [96–98].
Human kallikreins (KLK), a group of 15 serine proteases, initiate the intrinsic pathway and play a role in carcinogenesis; however, not all KLK play the same role in carcinogenesis or the same role between cancer types. Upregulated KLK13 is associated with a good prognosis in non-small cell lung cancer [99], whereas KLK5 is downregulated in some breast cancer subtypes leading to a misregulation of miRNAs and expression of genes involved in metastasis [100]. KLK6 activates the MAPK pathway through the receptor PAR2 in non-small cell lung cancer and increases tumor survival [101]. In addition, increased expression of both KLK6 and KLK10 are potential prognostic marker in gastric cancer and correlate with poor survival. Indeed, there is a positive correlation between high expression and nodal involvement as well as shorter survival (KLK6) or advanced-stage disease (KLK10) [102]. KLK7 regulation varies in different cancer types: it is overexpressed in colon [103] and cervical cancer [104] whereas it is down regulated in prostate cancer [105]. Factor XII, which is activated by the KLKs, seems to increase the metastatic potential of ovarian cancer by transforming monocytes/macrophages into tumor-associated macrophages [106].
In the extrinsic pathway, the androgen receptor in breast cancer [107], or hypoxia-inducible factors in ovarian cancer [108], may upregulate factor VII. The role of TF is complex with full length TF cooperating with PAR2 and FVIIa in stimulating the angiogenic response, and the alternatively spliced isoform of TF in cancer being independent of PAR2. The alternatively spliced variant of TF activates the PI3K, MAPK pathway. This leads to increased migration and angiogenesis, though the newly formed vessels are leaky [109]. The overexpression of TF is associated with high incidence of thrombotic events and poor survival, making it a useful diagnostic tool [110].
In the common pathway, thrombin is associated with cancer proliferation, invasion and angiogenesis [111, 112]. Thrombin stimulates inflammation at the tumor site in breast cancer patients [113] and plays a role in tumorigenesis of colitis-associated colon cancer [114]. Fibrinogen levels may provide a prognostic parameter, since a high level of fibrinogen is associated with advanced stage and poor survival in multiple types of cancer including cervical [115], ovarian [116, 117], endometrial, [118] or vulvar cancers [119]. A strong correlation also exists between coagulation and cancer progression and as such, anticoagulants including low molecular weight heparin, constitute a treatment of choice against cancer. This treatment is even more prevalent when patients present with co-morbidity factors [96, 97, 105, 120]. Other treatments include drugs targeting KLK [121], or novel oral anticoagulants which directly target Factor X and thrombin [122].
In conclusion, cancer cells activate almost every step of the coagulation cascade leading to clot formation. In turn, the clot may serve as a scaffold for cancer cells to metastasize. This explains the high incidence of thromboembolic events in cancer patients compared to controls. When the clots detach from the endothelial cells small capillaries may be blocked.
3.0 The role of natural antibodies in disease
Natural antibodies (NAb) are polyreactive Ab, composed of mostly IgM, but also IgG and IgA [123–125]. The Ab originate from the germ line and can be found in newborns and germ-free animals [123–125]. Ab are produced by recombination of multiple gene segments, including the variable region alleles. This rearrangement provides for Ab diversity and specificity. However, NAb utilize only a limited number of variable region alleles as many alleles are inaccessible during Ab rearrangement [126]. NAb are structurally highly flexible, which enables low affinity binding to a broad range of microbial and self Ag [124, 125, 127] (Fig. 3). NAb interact with serum lectins and the complement cascade similar to Ab from the adaptive response [124, 127, 128]. As such, these Ab protect the host with a basal level of immunity to pathogens but as NAb recognize self antigens, an excessive response may result in pathology.
Figure 3. B1 B cells produce self-reactive NAb.
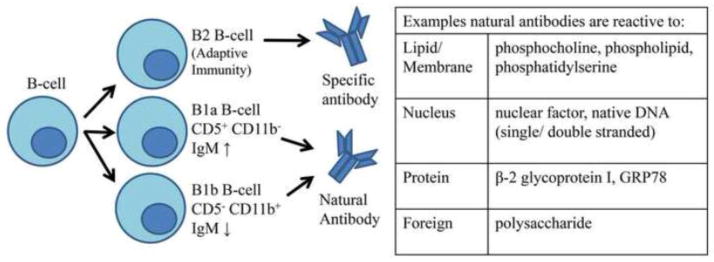
B-cells include two distinct populations, B1 and B2 B-cells. B2 cells participate in the adaptive immune response while B1 cells participate in the humoral innate immune response. Based on surface marker expression, B1 B-cells are further divided into two subpopulations B1a and B1b. The NAb produced by the B1 B-cell subpopulations are reactive to “self” proteins, membrane, and nucleus components as well as foreign molecules.
B1 B-cells, a subpopulation of B-cells, are the main producers of NAb (Fig. 3). Unlike B2 B-cells, B1 B-cells do not require Ag presentation by T-cells to produce Ab [129]. T-cell independent spontaneous Ab responses, such as lipopolysaccharides, can develop more quickly than T-cell dependent Ab, helping prevent infection in the earliest stages and play a critical role in protection of the vital organs [129, 130]. B1 B-cells are divided into two groups, B1a B-cells (CD5+, CD11b−) which secrete high levels of IgM and have low T-cell interaction [131] and B1b B-cells (CD5−, CD11b+) which have lower levels of IgM and do not express CD5, but higher levels of CD86 and are more likely to interact with T-cells [131].
3.1 Gram positive bacteria
Among other functions, NAb protect the host from Ag that do not develop monoreactive Ab, such as capsular polysaccharides [128]. Pathogens will encounter these Ab almost immediately as they are constantly present in normal serum, though they degrade more quickly than monoreactive Ab [123, 127, 132]. Many pathogens attempt to circumvent the host immune system by using antigenic variation, but NAb’s ability to bind to a number of Ag helps the host fight infections that they have never seen before or with antigenic variation [130].
Some of the specific targets of NAb are found on S. pneumoniae and B. anthracis [126]. A specific idiotype, T15, recognizes phosphocholine on the surfaces of some bacteria, including S. pneumoniae [100, 124, 133]. NAb against the S. pyogenes capsule have been found in rabbits and in humans, NAb target cell wall polysaccharides [134, 135]. Mice injected with the polysaccharides found on S. pneumoniae developed low affinity, high avidity, polyreactive germline derived Ab, suggesting that NAb are an important line of defense against the bacteria [136]. These Ab also selectively used the VH1 and IGHJ6 genes, which encodes for a tyrosine rich CDR3, indicative of polyreactive Ab [133, 136]. Mice who fail to develop these Ab are more susceptible to S. pneumoniae infections, but NAb alone are not sufficient to clear the infection. [126, 132].
While B1a B-cells are long lived (longevity and replacement rates for B1b-B-cells are still unknown), their rate of replacement is very slow and their loss may be irreversible. One S. aureus protein, Staphylococcal aureus Protein A, is a B-cell superantigen causing cross linking of B-cell receptors, leading to cell death and a hole in the Ab repertoire. This depletion can still be detected over one year after exposure to the protein [137]. Protein L of Peptostreptococcus magnus also functions similarly as a B-cell superantigen leading to cell death and producing an irreparable immune system gap [138].
3.2 Viruses
B1 B-cells are unique due to a poor response to receptor-mediated activation but a rapid and strong response to cytokines and pathogen-encoded signals. This enhances the ability of B1 B-cells to attenuate early viral replication and greatly decreases the chance of autoimmunity [139]. Studies show that influenza virus infection may be neutralized by natural IgM mediated activation of the complement system. Through the binding of C1q to a complement-fixing immunoglobulin such as IgM, the complement cascade is activated resulting in not only the neutralization of influenza, but the prevention of viral pneumonia [140]. Based on this mechanism, NAb mediated activation of the complement system is crucial in attenuation of the early influenza virus infection.
Although the role of IgM in WNV infection remains poorly understood, a mouse model study of WNV infection demonstrated that induction of natural IgM plays a role in early protection, but does not appear to neutralize or attenuate the infection [141]. The authors of this study hypothesized that natural IgM helps link the innate and adaptive immune response during infection. In contrast, vesicular stomatitis virus (VSV) infection was attenuated by NAb when μMT (B-cell deficient) mice are passively immunized with NAb following infection. NAb appear to influence the initial distribution of VSV, preventing viral dissemination to vital organs, and directing dissemination to secondary lymphatic organs accelerating and enhancing the immune response [142].
These contrasting results suggest that although NAb are important in the early attenuation of viral infections, and the protection from invading pathogens, NAb are not consistently sufficient to neutralize infection. This variability in effectivity is likely the result of differences in viral evolution and adaptation to host immune responses.
3.3 Systemic Lupus Erythematosus and anti-phospholipid syndrome
The highly variable clinical manifestations make SLE a challenge to accurately diagnose but, a major component includes auto-reactive Ab, composed primarily of IgG isotypes. The absence of IgM in lupus prone mice significantly increased IgG autoantibody levels [143]. Recent studies demonstrated that auto-reactive, NAb may decrease the risk of SLE development [144–146]. Initial studies showed that patients with higher IgM, compared to IgG, had a lower risk of developing lupus nephritis [144]. Importantly, subsequent studies showed that anti-apoptotic cell, natural IgM Ab decreased the inflammatory response and blocked in vitro Toll-like receptor activation of dendritic cells [146, 147]. Together these studies suggest that NAb may down regulate the inflammatory response in lupus and that the IgM to IgG ratio may be crucial in autoimmune disease development.
Specific autoreactive IgM includes anti-lipid Ab, anti-nuclear Ab, and Ab that recognize proteins bound to lipids. In SLE, anti-phosphatidylcholine IgM levels decrease while anti-phosphatidylserine IgM levels increase possibly due to increased apoptosis levels leading to increased phosphatidylserine on the cell membrane [145]. Studies also showed that Ab which recognize oxidative epitopes on apoptotic cells, and both single and double stranded DNA are prevalent in patients with SLE [144]. These patients specifically develop anti-nuclear factor and anti-native DNA IgM [148]. Compared to healthy controls, APLS patients have elevated levels of IgG to the serum protein, β2-GPI, which binds phosphatidylserine. In addition, APLS patients produce low affinity IgM and IgA anti-β2-GPI Ab [149], suggesting that these Ab are NAb. Elevated levels of IgG aPL may increase thrombotic events and abortion in APLS and SLE patients [149, 150].
The excess Ab produced in SLE lead to formation of immune complexes that lower the threshold for platelet activation [151]. SLE patients with aPLs Ab show increased complement activation on platelets compared to healthy individuals [30, 152, 153]. The platelet bound aPL initiate the classical pathway resulting in complement deposition and platelet activation selectively via p38 MAPK [154–156]. A new approach to treat and prevent thrombotic events in APLS patients utilizes inhibitor of p38 MAPK [157].
B1 B-cells may directly contribute to SLE as lupus patients appear to have an increased number of B1b B-cells [131]. SLE patients, but not their healthy counterparts, express increased levels of CXCL13 which recruits B1 B-cells to the kidney and results in increased renal damage [158]. However, the latter study did not determine the specific B1 B-cell subtype in the kidney. Similarly, a mouse model for lupus expressed an increased B1 B-cell population compared to wild type mice [159]. In addition, Sialic acid-binding Ig-like lectin (Siglec)-G protein, an inhibitory receptor that downregulates the B1 B-cell response [160], contributes to increased autoantibody production and kidney damage in a mouse model of lupus [161].
3.4 Cancer
Many studies report that NAb in cancer patients may result in apoptosis of cancer cells [162–165]. In particular, the NAb SAM-6 exhibits an anti-cancer effect on myeloma by targeting the protein GRP 78. This heat-shock protein localizes to the lumen of the reticulum under physiological conditions, but translocates to the surface of myeloma cells [166]. SAM-6 NAb also induce lipoptosis, an excess lipid accumulation in cancer cells but not in normal cells, that leads to cell death [167, 168] and reviewed in [169]. However, the role of IgM NAb in tumorigenesis is not always clear. For example, the natural NAb isoagglutinins cross-react with the tumor cell Ag T and Tn but it is unclear if these Ab are beneficial or detrimental. Also, the level of isoagglutinins is reduced in pancreatic cancer but there is no known correlation with outcome [170].
Independent of NAb, B1a-B-cells play an important role in cancer development. The tumor microenvironment contains lymphocytes which express a decreased level of CD5. Because CD5 inhibits T-cell receptor and B-cell receptor signaling, it may be hypothesized that decreasing CD5 may be a strategy to maintain the T-cell anti-tumor effect [171]. However, some cancer cells seem to escape this downregulation of CD5, as B1a B-cells are present in the melanoma cells microenvironment and enhance metastasis [172, 173]. B1a B-cells, like any other cell type, may also accumulate mutations, transform, and become the primary tumor. Many chronic lymphocytic leukemia derive from NAb producing cells in which natural IgM stimulates proliferation [174–176]. Binding of IgM activates the MAPK cascade, which in turn enhance the oncogene MYC expression [177].
NAb are utilized in the treatment of tumors. For example, intra-tumor injection of the anti-gal NAb increased survival of some renal or pancreatic cancer patients [178]. This required a complement-dependent cytotoxicity, which illustrated the important interconnections that exist between the different systems involved in innate immunity. Contrary to what was expected, NAb activated not only the classical complement pathway, but also the lectin pathway through an interaction with H-ficolin [179].
In conclusion, B1 B-cells and NAb play a role as gatekeeper in cancer progression. Efficacy is limited since we observe cancer formation; however, an overexpression of NAb through direct injection demonstrates promising results in patients [178, 180]. Unfortunately, this strong interrelation between the different systems can also be deleterious as IgM elimination of cancer cells depends in part on complement activation which is dysregulated. Additionally, NAb activate coagulation, which may be beneficial for metastasis. Finally, mutation of B1 B-cells biases the entire system by producing IgM which provides a positive feedback to the B1 B-cells, especially in CD38-positive cells [181].
4.0 Interactions, Redundancies, and Conclusions
While the complement system, coagulation cascades, and NAb are three distinct entities, these cascades are highly interconnected in multiple clinical conditions (Fig. 4). Evasion of these immune responses is important for bacterial and viral survival. This is illustrated by the failure of the coagulase mutants in S. aureus to survive in mouse models [73, 79]. Cancer cells are less sensitive to the immune response than some bacteria and viruses, as these cells derive from the host itself. Cancer cells continually accumulate mutations which allow escape from the host’s regulatory signals. Finally, either over-stimulation or suppression of the immune system is detrimental to the host and may result in disease pathogenesis. In autoimmune diseases, symptoms of dysregulation are comparable in severity to those caused by pathogens or cancer.
Figure 4. The interplay between complement, coagulation, and natural antibodies.

The three pathways for activation of the complement pathway (solid lines) are the classical, lectin, and alternative pathways. The classical pathways interacts with Factor XII from the coagulation pathways (dotted lines) also leading to C3 activation. Natural antibodies (dashed lines) also interact with the classical pathway to activate C3. Plasminogen, from the coagulation pathway, also activates C3 directly. C3 activation leads to two outcomes, both a continuation of activation in the complement cascade to C5, but also leading to the release of tissue factor, which activates prothrombin, leading to the activation of thrombin, both members of the coagulation pathway. Thrombin is also able to lead to both C3 and C5 activation, causing formation of the MAC.
All three pathways of complement activation lead to the formation of C3 convertase. NAbs and Factor XII interact with the classical pathway also leading to proteolytic cleavage of C3. Another coagulation cascade member, plasminogen, also leads to activation of C3 and this further activates C5 and leads to TF release. This release leads to the cleavage of prothrombin into thrombin, and initiates clot formation further downstream. Thrombin also interacts with the complement system activating both C3 and C5 resulting in formation of the MAC.
The interaction between complement and coagulation systems is further exemplified in the generation of the anaphylatoxin, C5a, by KLK and thrombin [112, 182]. These complex relations are evident in patients with autoimmune diseases. SLE patients develop venous thrombosis and present with an increased deposition of C1q, C3, and C4 on platelets. It is still unclear why there is no increased deposition of complement factors in SLE patient with arterial thrombosis [30, 155].
NAb and coagulation also interconnect as demonstrated in two specific studies: a case of disseminated intravascular coagulation caused by IgM-mediated hemolysis in an 11-month old baby, and a xenograft rejection leading to DIC in baboons [183, 184]. NAb initiate complement activation not only through the classical pathway but also through the lectin pathway. In an animal model of myocardial infarction, for example, damage to the heart requires both natural IgM and the lectin pathway [185].
Increased understanding of the humoral innate immune system components and their interplay can lead to the development of novel or improved therapeutics for these specific clinical condition. In a recent African green monkey model study of avian influenza, treatment with anti-C5a mAb improved survival and decreased acute lung injuries in monkeys infected with H7N9, normally associated with high morbidity and mortality rates in humans [186]. As the first line of immune defense, advances in the understanding of the role of humoral innate immunity in the pathogenesis of pathogenic infection, cancer, and autoimmune disease provides a new approach to treatment.
Highlights.
Pathogens evade the immune response with multiple, frequently redundant proteins.
Cancer may evade or utilize the innate response for growth and metastasis.
A dysregulated humoral innate response may result in autoimmunity.
Complement, coagulation and natural antibodies interact to prevent disease.
Acknowledgments
This work was supported by grants from National Institutes of Health (R01 AI061691 to S.D.F.); National Bio and Agro-Defense Facility Fund (J.N.H.) and Kansas State University. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Institute of Health or National Science Foundation.
Abbreviations
- Ab
antibody/antibodies
- MBL
Mannose Binding Lectin/s
- Ag
Antigen/s
- FH
Factor H
- FI
Factor I
- MAC
Membrane Attack Complex
- mAb
monoclonal antibody/ies
- TF
Tissue Factor
- HF
Hemorrhagic Fever
- APLS
Antiphospholipid syndrome
- aPL
antiphospholipid
- SLE
Systemic Lupus Erythematosus
- WNV
West Nile Virus
- NAb
natural antibody
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mastellos D, Andronis C, Persidis A, Lambris JD. Novel biological networks modulated by complement. Clin Immunol. 2005;115:225–235. doi: 10.1016/j.clim.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Meri T, Amdahl H, Lehtinen MJ, Hyvärinen S, McDowell JV, Bhattacharjee A, Meri S, Marconi R, Goldman A, Jokiranta TS. Microbes bind complement inhibitor factor H via a common site. PLOS pathogens. 2013;9:e1003308. doi: 10.1371/journal.ppat.1003308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pian Y, Gan S, Wang S, Guo J, Wang P, Zheng Y, Cai X, Jiang Y, Yuan Y. Fhb, a novel factor H-binding surface protein, contributes to the antiphagocytic ability and virulence of Streptococcus suis. Infection and immunity. 2012;80:2402–2413. doi: 10.1128/IAI.06294-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi M, Terao Y, Kawabata S. Pleiotropic virulence factor - Streptococcus pyogenes fibronectin-binding proteins. Cellular microbiology. 2013;15:503–511. doi: 10.1111/cmi.12083. [DOI] [PubMed] [Google Scholar]
- 8.Zecconi A, Scali F. Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases. Immunology letters. 2013;150:12–22. doi: 10.1016/j.imlet.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal V, Hammerschmidt S, Malm S, Bergmann S, Riesbeck K, Blom A. Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. The journal of immunology. 2012;189:3575–3584. doi: 10.4049/jimmunol.1102934. [DOI] [PubMed] [Google Scholar]
- 10.Pérez-Caballero D, García-Laorden I, Cortés G, Wessels MR, de Córdoba SR, Albertí S. Interaction between Complement Regulators and Streptococcus pyogenes: Binding of C4b-Binding Protein and Factor H/Factor H-Like Protein 1 to M18 Strains Involves Two Different Cell Surface Molecules. The journal of immunology. 2004;173:6899–6904. doi: 10.4049/jimmunol.173.11.6899. [DOI] [PubMed] [Google Scholar]
- 11.Agarwal V, Sroka M, Fulde M, Bergmann S, Riesbeck K, Blom A. Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. Journal of biological chemistry. 2014;289:15833–15844. doi: 10.1074/jbc.M113.530212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang M, Ko Y-P, Liang X, Ross C, Liu Q, Murray B, Höök M. Collagen-binding microbial surface components recognizing adhesive matrix molecule (MSCRAMM) of Gram-positive bacteria inhibit complement activation via the classical pathway. Journal of biological chemistry. 2013;288:20520–20531. doi: 10.1074/jbc.M113.454462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung MC, Tonry J, Narayanan A, Manes N, Mackie R, Gutting B, Mukherjee D, Popova T, Kashanchi F, Bailey C, Popov S. Bacillus anthracis interacts with plasmin(ogen) to evade C3b-dependent innate immunity. PLoS ONE. 2011;6:e18119. doi: 10.1371/journal.pone.0018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amdahl H, Jongerius I, Meri T, Pasanen T, Hyvärinen S, Haapasalo K, van Strijp J, Rooijakkers S, Jokiranta TS. Staphylococcal Ecb protein and host complement regulator factor H enhance functions of each other in bacterial immune evasion. The journal of immunology. 2013;191:1775–1784. doi: 10.4049/jimmunol.1300638. [DOI] [PubMed] [Google Scholar]
- 15.Ko Y-P, Kuipers A, Freitag C, Jongerius I, Medina E, van Rooijen W, Spaan A, van Kessel KPM, Höök M, Rooijakkers SHM. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLOS pathogens. 2013;9:e1003816. doi: 10.1371/journal.ppat.1003816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Kessel KPM, Bestebroer J, van Strijp JAG. Neutrophil-Mediated Phagocytosis of Staphylococcus aureus. Frontiers in Immunology. 2014;5:467. doi: 10.3389/fimmu.2014.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorham R, Rodriguez W, Morikis D. Molecular analysis of the interaction between staphylococcal virulence factor Sbi-IV and complement C3d. Biophysical journal. 2014;106:1164–1173. doi: 10.1016/j.bpj.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woehl J, Stapels DAC, Garcia B, Ramyar K, Keightley A, Ruyken M, Syriga M, Sfyroera G, Weber A, Zolkiewski M, Ricklin D, Lambris J, Rooijakkers SHM, Geisbrecht B. The extracellular adherence protein from Staphylococcus aureus inhibits the classical and lectin pathways of complement by blocking formation of the C3 proconvertase. The journal of immunology. 2014;193:6161–6171. doi: 10.4049/jimmunol.1401600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jusko M, Potempa J, Kantyka T, Bielecka E, Miller H, Kalinska M, Dubin G, Garred P, Shaw L, Blom A. Staphylococcal proteases aid in evasion of the human complement system. Journal of innate immunity. 2014;6:31–46. doi: 10.1159/000351458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Favoreel HW, Van de Walle GR, Nauwynck HJ, Pensaert MB. Virus complement evasion strategies. J Gen Virol. 2003;84:1–15. doi: 10.1099/vir.0.18709-0. [DOI] [PubMed] [Google Scholar]
- 21.Ojha H, Panwar HS, Gorham RD, Jr, Morikis D, Sahu A. Viral regulators of complement activation: structure, function and evolution. Mol Immunol. 2014;61:89–99. doi: 10.1016/j.molimm.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Chung KM, Nybakken GE, Thompson BS, Engle MJ, Marri A, Fremont DH, Diamond MS. Antibodies against West Nile Virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J Virol. 2006;80:1340–1351. doi: 10.1128/JVI.80.3.1340-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim H, Meyer K, Di Bisceglie AM, Ray R. Inhibition of c3 convertase activity by hepatitis C virus as an additional lesion in the regulation of complement components. PLoS ONE. 2014;9:e101422. doi: 10.1371/journal.pone.0101422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ballegaard V, Haugaard AK, Garred P, Nielsen SD, Munthe-Fog L. The lectin pathway of complement: advantage or disadvantage in HIV pathogenesis? Clin Immunol. 2014;154:13–25. doi: 10.1016/j.clim.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Saifuddin M, Hedayati T, Atkinson JP, Holguin MH, Parker CJ, Spear GT. Human immunodeficiency virus type 1 incorporates both glycosyl phosphatidylinositol-anchored CD55 and CD59 and integral membrane CD46 at levels that protect from complement-mediated destruction. J Gen Virol. 1997;78(Pt 8):1907–1911. doi: 10.1099/0022-1317-78-8-1907. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz J, Zimmer JP, Kluxen B, Aries S, Bogel M, Gigli I, Schmitz H. Antibody-dependent complement-mediated cytotoxicity in sera from patients with HIV-1 infection is controlled by CD55 and CD59. J Clin Invest. 1995;96:1520–1526. doi: 10.1172/JCI118190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson JB, Borisevich V, Rockx B, Parks GD. A novel factor I activity in Nipah virus inhibits human complement pathways through cleavage of C3b. J Virol. 2015;89:989–998. doi: 10.1128/JVI.02427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barilla-Labarca ML, Toder K, Furie R. Targeting the complement system in systemic lupus erythematosus and other diseases. Clin Immunol. 2013;148:313–321. doi: 10.1016/j.clim.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 29.Cervera RC. Antiphospholipid Syndrome in Systemic Autoimmune Diseases. In: Ricard Cervera JCR, Munther K, editors. Handbook of Systemic Autoimmune Diseases. Elsevier: 2009. p. iii. [Google Scholar]
- 30.Lood C, Eriksson S, Gullstrand B, Jonsen A, Sturfelt G, Truedsson L, Bengtsson AA. Increased C1q, C4 and C3 deposition on platelets in patients with systemic lupus erythematosus--a possible link to venous thrombosis? Lupus. 2012;21:1423–1432. doi: 10.1177/0961203312457210. [DOI] [PubMed] [Google Scholar]
- 31.Marquart HV, Schejbel L, Sjoholm A, Martensson U, Nielsen S, Koch A, Svejgaard A, Garred P. C1q deficiency in an Inuit family: identification of a new class of C1q disease-causing mutations. Clin Immunol. 2007;124:33–40. doi: 10.1016/j.clim.2007.03.547. [DOI] [PubMed] [Google Scholar]
- 32.Schejbel L, Skattum L, Hagelberg S, Ahlin A, Schiller B, Berg S, Genel F, Truedsson L, Garred P. Molecular basis of hereditary C1q deficiency--revisited: identification of several novel disease-causing mutations. Genes Immun. 2011;12:626–634. doi: 10.1038/gene.2011.39. [DOI] [PubMed] [Google Scholar]
- 33.Sturfelt G, Johnson U, Sjoholm AG. Sequential studies of complement activation in systemic lupus erythematosus. Scand J Rheumatol. 1985;14:184–196. doi: 10.3109/03009748509165503. [DOI] [PubMed] [Google Scholar]
- 34.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 35.Aringer M, Gunther C, Lee-Kirsch MA. Innate immune processes in lupus erythematosus. Clin Immunol. 2013;147:216–222. doi: 10.1016/j.clim.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 36.Pierangeli SS, Espinola RG, Liu X, Harris EN. Thrombogenic effects of antiphospholipid antibodies are mediated by intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1, and P-selectin. Circ Res. 2001;88:245–250. doi: 10.1161/01.res.88.2.245. [DOI] [PubMed] [Google Scholar]
- 37.Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, Hollmann TJ, Casali P, Caroll MC, Wetsel RA, Lambris JD, Holers VM, Salmon JE. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112:1644–1654. doi: 10.1172/JCI18817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, Yasuda S, Koike T. Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis. 2009;68:1030–1035. doi: 10.1136/ard.2008.090670. [DOI] [PubMed] [Google Scholar]
- 39.Sjowall C, Olin AI, Skogh T, Wettero J, Morgelin M, Nived O, Sturfelt G, Bengtsson AA. C-reactive protein, immunoglobulin G and complement co-localize in renal immune deposits of proliferative lupus nephritis. Autoimmunity. 2013;46:205–214. doi: 10.3109/08916934.2013.764992. [DOI] [PubMed] [Google Scholar]
- 40.Orbai AM, Truedsson L, Sturfelt G, Nived O, Fang H, Alarcon GS, Gordon C, Merrill J, Fortin PR, Bruce IN, Isenberg DA, Wallace DJ, Ramsey-Goldman R, Bae SC, Hanly JG, Sanchez-Guerrero J, Clarke AE, Aranow CB, Manzi S, Urowitz MB, Gladman DD, Kalunian KC, Costner MI, Werth VP, Zoma A, Bernatsky S, Ruiz-Irastorza G, Khamashta MA, Jacobsen S, Buyon JP, Maddison P, Dooley MA, Van Vollenhoven RF, Ginzler E, Stoll T, Peschken C, Jorizzo JL, Callen JP, Lim SS, Fessler BJ, Inanc M, Kamen DL, Rahman A, Steinsson K, Franks AG, Jr, Sigler L, Hameed S, Pham N, Brey R, Weisman MH, McGwin G, Jr, Magder LS, Petri M. Anti-C1q antibodies in systemic lupus erythematosus. Lupus. 2015;24:42–49. doi: 10.1177/0961203314547791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boackle SA, Holers VM, Chen X, Szakonyi G, Karp DR, Wakeland EK, Morel L. Cr2, a candidate gene in the murine Sle1c lupus susceptibility locus, encodes a dysfunctional protein. Immunity. 2001;15:775–785. doi: 10.1016/s1074-7613(01)00228-x. [DOI] [PubMed] [Google Scholar]
- 42.Wu X, Jiang N, Deppong C, Singh J, Dolecki G, Mao D, Morel L, Molina HD. A role for the Cr2 gene in modifying autoantibody production in systemic lupus erythematosus. J Immunol. 2002;169:1587–1592. doi: 10.4049/jimmunol.169.3.1587. [DOI] [PubMed] [Google Scholar]
- 43.Arshad A, Chung W, Isherwood J, Steward W, Metcalfe M, Dennison A. Restoration of mannose-binding lectin complement activity is associated with improved outcome in patients with advanced pancreatic cancer treated with gemcitabine and intravenous omega-3 fish oil. JPEN J Parenter Enteral Nutr. 2014;38:214–219. doi: 10.1177/0148607113476304. [DOI] [PubMed] [Google Scholar]
- 44.Ajona D, Castano Z, Garayoa M, Zudaire E, Pajares MJ, Martinez A, Cuttitta F, Montuenga LM, Pio R. Expression of complement factor H by lung cancer cells: effects on the activation of the alternative pathway of complement. Cancer Res. 2004;64:6310–6318. doi: 10.1158/0008-5472.CAN-03-2328. [DOI] [PubMed] [Google Scholar]
- 45.Kim KB, Yi JS, Nguyen N, Lee JH, Kwon YC, Ahn BY, Cho H, Kim YK, Yoo HJ, Lee JS, Ko YG. Cell-surface receptor for complement component C1q (gC1qR) is a key regulator for lamellipodia formation and cancer metastasis. J Biol Chem. 2011;286:23093–23101. doi: 10.1074/jbc.M111.233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jane Scully O, Yu Y, Salim A, Aye Thike A, Wai-Cheong Yip G, Hun Baeg G, Tan PH, Matsumoto K, Huat Bay B. Complement component 1, q subcomponent binding protein is a marker for proliferation in breast cancer. Exp Biol Med (Maywood) 2015 doi: 10.1177/1535370214565075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Zhang F, Guo L, Wang Y, Zhang P, Wang R, Zhang N, Chen R. Interactome analysis reveals that C1QBP (complement component 1, q subcomponent binding protein) is associated with cancer cell chemotaxis and metastasis. Mol Cell Proteomics. 2013;12:3199–3209. doi: 10.1074/mcp.M113.029413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang MR, Wang YX, Guo S, Han SY, Wang D. CSMD1 exhibits antitumor activity in A375 melanoma cells through activation of the Smad pathway. Apoptosis. 2012;17:927–937. doi: 10.1007/s10495-012-0727-0. [DOI] [PubMed] [Google Scholar]
- 49.Chen J, Wu W, Zhen C, Zhou H, Yang R, Chen L, Hu L. Expression and clinical significance of complement C3, complement C4b1 and apolipoprotein E in pancreatic cancer. Oncol Lett. 2013;6:43–48. doi: 10.3892/ol.2013.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Escudero-Esparza A, Kalchishkova N, Kurbasic E, Jiang WG, Blom AM. The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J. 2013;27:5083–5093. doi: 10.1096/fj.13-230706. [DOI] [PubMed] [Google Scholar]
- 51.Cui W, Zhao Y, Shan C, Kong G, Hu N, Zhang Y, Zhang S, Zhang W, Zhang Y, Zhang X, Ye L. HBXIP upregulates CD46, CD55 and CD59 through ERK1/2/NF-kappaB signaling to protect breast cancer cells from complement attack. FEBS Lett. 2012;586:766–771. doi: 10.1016/j.febslet.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 52.Jarvis GA, Li J, Hakulinen J, Brady KA, Nordling S, Dahiya R, Meri S. Expression and function of the complement membrane attack complex inhibitor protectin (CD59) in human prostate cancer. Int J Cancer. 1997;71:1049–1055. doi: 10.1002/(sici)1097-0215(19970611)71:6<1049::aid-ijc22>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 53.Kesselring R, Thiel A, Pries R, Fichtner-Feigl S, Brunner S, Seidel P, Bruchhage KL, Wollenberg B. The complement receptors CD46, CD55 and CD59 are regulated by the tumour microenvironment of head and neck cancer to facilitate escape of complement attack. Eur J Cancer. 2014;50:2152–2161. doi: 10.1016/j.ejca.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Watson NF, Durrant LG, Madjd Z, Ellis IO, Scholefield JH, Spendlove I. Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer Immunol Immunother. 2006;55:973–980. doi: 10.1007/s00262-005-0055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chong PK, Lee H, Loh MC, Choong LY, Lin Q, So JB, Lim KH, Soo RA, Yong WP, Chan SP, Smoot DT, Ashktorab H, Yeoh KG, Lim YP. Upregulation of plasma C9 protein in gastric cancer patients. Proteomics. 2010;10:3210–3221. doi: 10.1002/pmic.201000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ragupathi G, Liu NX, Musselli C, Powell S, Lloyd K, Livingston PO. Antibodies against tumor cell glycolipids and proteins, but not mucins, mediate complement-dependent cytotoxicity. J Immunol. 2005;174:5706–5712. doi: 10.4049/jimmunol.174.9.5706. [DOI] [PubMed] [Google Scholar]
- 57.Guo B, Ma ZW, Li H, Xu GL, Zheng P, Zhu B, Wu YZ, Zou Q. Mapping of binding epitopes of a human decay-accelerating factor monoclonal antibody capable of enhancing rituximab-mediated complement-dependent cytotoxicity. Clin Immunol. 2008;128:155–163. doi: 10.1016/j.clim.2008.03.507. [DOI] [PubMed] [Google Scholar]
- 58.Zhou W. The new face of anaphylatoxins in immune regulation. Immunobiology. 2012;217:225–234. doi: 10.1016/j.imbio.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 59.Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y, Hu X, Aggarwal D, Zhang HG, Yan J. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. 2012;189:2985–2994. doi: 10.4049/jimmunol.1200846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu YQ, DeAngelis RA, Lambris JD, Coukos G, Scholler N. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. 2012;14:994–1004. doi: 10.1593/neo.121262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao X. Regulatory T cells and immune tolerance to tumors. Immunol Res. 2010;46:79–93. doi: 10.1007/s12026-009-8124-7. [DOI] [PubMed] [Google Scholar]
- 62.Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, Rouzaut A, Pajares MJ, Montuenga LM, Pio R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189:4674–4683. doi: 10.4049/jimmunol.1201654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu Y, Hu XB. C5a stimulates the proliferation of breast cancer cells via Akt-dependent RGC-32 gene activation. Oncol Rep. 2014;32:2817–2823. doi: 10.3892/or.2014.3489. [DOI] [PubMed] [Google Scholar]
- 64.Carmona-Fontaine C, Theveneau E, Tzekou A, Tada M, Woods M, Page KM, Parsons M, Lambris JD, Mayor R. Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev Cell. 2011;21:1026–1037. doi: 10.1016/j.devcel.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piao C, Cai L, Qiu S, Jia L, Song W, Du J. Complement 5a Enhances Hepatic Metastases of Colon Cancer via Monocyte Chemoattractant Protein-1-Mediated Inflammatory Cell Infiltration. J Biol Chem. 2015 doi: 10.1074/jbc.M114.612622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, Manne S, Fairlie DP, Gorczyca W, Almanza O, Karbowniczek M, Markiewski MM. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014;74:3454–3465. doi: 10.1158/0008-5472.CAN-14-0157. [DOI] [PubMed] [Google Scholar]
- 67.Johari V, Loke C. Brief overview of the coagulation cascade. Dis Mon. 2012;58:421–423. doi: 10.1016/j.disamonth.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 68.Lupu F, Keshari RS, Lambris JD, Mark Coggeshall K. Crosstalk between the coagulation and complement systems in sepsis, Thrombosis Research. 2014;133(Supplement 1):S28–S31. doi: 10.1016/j.thromres.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Remy KE, Qiu P, Li Y, Cui X, Eichacker PQ. B. anthracis associated cardiovascular dysfunction and shock: the potential contribution of both non-toxin and toxin components. BMC Medicine. 2013;11:217–217. doi: 10.1186/1741-7015-11-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun D, Popescu N, Raisley B, Keshari R, Dale G, Lupu F, Coggeshall KM. Bacillus anthracis peptidoglycan activates human platelets through FcγRII and complement. Blood. 2013;122:571–579. doi: 10.1182/blood-2013-02-486613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bryant A. Biology and pathogenesis of thrombosis and procoagulant activity in invasive infections caused by group A streptococci and Clostridium perfringens. Clinical microbiology reviews. 2003;16:451–462. doi: 10.1128/CMR.16.3.451-462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reglinski M, Sriskandan S. The contribution of group A streptococcal virulence determinants to the pathogenesis of sepsis. Virulence. 2014;5:127–136. doi: 10.4161/viru.26400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loof T, Goldmann O, Naudin C, Mörgelin M, Neumann Y, Pils M, Foster S, Medina E, Herwald H. Staphylococcus aureus-induced clotting of plasma is an immune evasion mechanism for persistence within the fibrin network. Microbiology. 2015;161:621–627. doi: 10.1099/mic.0.000019. [DOI] [PubMed] [Google Scholar]
- 74.Okumura CYM, Nizet V. Subterfuge and Sabotage: Evasion of Host Innate Defenses by Invasive Gram-Positive Bacterial Pathogens. Annual Review of Microbiology. 2014;68:439–458. doi: 10.1146/annurev-micro-092412-155711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Loof TG, Deicke C, Medina E. The role of coagulation/fibrinolysis during Streptococcus pyogenes infection. Frontiers in Cellular and Infection Microbiology. 2014;4:128. doi: 10.3389/fcimb.2014.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Linder A, Johansson L, Thulin P, Hertzen E, Morgelin M, Christensson B, Bjorck L, Norrby-Teglund A, Akesson P. Erysipelas Caused by Group A Streptococcus Activates the Contact System and Induces the Release of Heparin-Binding Protein. J Invest Dermatol. 2010;130:1365–1372. doi: 10.1038/jid.2009.437. [DOI] [PubMed] [Google Scholar]
- 77.Mohan S, Hertweck C, Dudda A, Hammerschmidt S, Skerka C, Hallström T, Zipfel P. Tuf of Streptococcus pneumoniae is a surface displayed human complement regulator binding protein. Molecular immunology. 2014;62:249–264. doi: 10.1016/j.molimm.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 78.Dacombe PJ, Evans J, Gosling OB, Heal J. Stage 3 pyomyositis of the gluteus minimus; Staphylococcus aureus sepsis, autoanticoagulation, proximal femoral osteomyelitis and the role of surgical intervention. BMJ Case Reports. 2013;2013 doi: 10.1136/bcr-2013-201357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomer L, Schneewind O, Missiakas D. Multiple ligands of von Willebrand factor-binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. Journal of biological chemistry. 2013;288:28283–28292. doi: 10.1074/jbc.M113.493122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Itoh S, Yokoyama R, Kamoshida G, Fujiwara T, Okada H, Takii T, Tsuji T, Fujii S, Hashizume H, Onozaki K. Staphylococcal superantigen-like protein 10 (SSL10) inhibits blood coagulation by binding to prothrombin and factor Xa via their γ-carboxyglutamic acid (Gla) domain. Journal of biological chemistry. 2013;288:21569–21580. doi: 10.1074/jbc.M113.451419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis. 2003;188:1618–1629. doi: 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- 82.Chuang YC, Lin YS, Liu HS, Yeh TM. Molecular mimicry between dengue virus and coagulation factors induces antibodies to inhibit thrombin activity and enhance fibrinolysis. J Virol. 2014;88:13759–13768. doi: 10.1128/JVI.02166-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pryzdial EL, Sutherland MR, Ruf W. The procoagulant envelope virus surface: contribution to enhanced infection. Thromb Res. 2014;133(Suppl 1):S15–17. doi: 10.1016/j.thromres.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nemerow GR. Coagulation factor defends adenovirus from immune attack. Nat Med. 2013;19:406–407. doi: 10.1038/nm.3149. [DOI] [PubMed] [Google Scholar]
- 85.Nojima J, Masuda Y, Iwatani Y, Suehisa E, Futsukaichi Y, Kuratsune H, Watanabe Y, Takano T, Hidaka Y, Kanakura Y. Tissue factor expression on monocytes induced by anti-phospholipid antibodies as a strong risk factor for thromboembolic complications in SLE patients. Biochem Biophys Res Commun. 2008;365:195–200. doi: 10.1016/j.bbrc.2007.10.173. [DOI] [PubMed] [Google Scholar]
- 86.Teruel R, Perez-Sanchez C, Corral J, Herranz MT, Perez-Andreu V, Saiz E, Garcia-Barbera N, Martinez-Martinez I, Roldan V, Vicente V, Lopez-Pedrera C, Martinez C. Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J Thromb Haemost. 2011;9:1985–1992. doi: 10.1111/j.1538-7836.2011.04451.x. [DOI] [PubMed] [Google Scholar]
- 87.Kornberg A, Blank M, Kaufman S, Shoenfeld Y. Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J Immunol. 1994;153:1328–1332. [PubMed] [Google Scholar]
- 88.Adams MJ, Palatinus AA, Harvey AM, Khalafallah AA. Impaired control of the tissue factor pathway of blood coagulation in systemic lupus erythematosus. Lupus. 2011;20:1474–1483. doi: 10.1177/0961203311418267. [DOI] [PubMed] [Google Scholar]
- 89.Wahezi DM, Ilowite NT, Wu XX, Pelkmans L, Laat B, Schanberg LE, Rand JH. I. Atherosclerosis Prevention in Pediatric Lupus Erythematosus, Annexin A5 anticoagulant activity in children with systemic lupus erythematosus and the association with antibodies to domain I of beta2-glycoprotein I. Lupus. 2013;22:702–711. doi: 10.1177/0961203313490241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andree HA, Stuart MC, Hermens WT, Reutelingsperger CP, Hemker HC, Frederik PM, Willems GM. Clustering of lipid-bound annexin V may explain its anticoagulant effect. J Biol Chem. 1992;267:17907–17912. [PubMed] [Google Scholar]
- 91.de Laat B, Pengo V, Pabinger I, Musial J, Voskuyl AE, Bultink IE, Ruffatti A, Rozman B, Kveder T, de Moerloose P, Boehlen F, Rand J, Ulcova-Gallova Z, Mertens K, de Groot PG. The association between circulating antibodies against domain I of beta2-glycoprotein I and thrombosis: an international multicenter study. J Thromb Haemost. 2009;7:1767–1773. doi: 10.1111/j.1538-7836.2009.03588.x. [DOI] [PubMed] [Google Scholar]
- 92.Back J, Lood C, Bengtsson AA, Ekdahl KN, Nilsson B. Contact activation products are new potential biomarkers to evaluate the risk of thrombotic events in systemic lupus erythematosus. Arthritis Res Ther. 2013;15:R206. doi: 10.1186/ar4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Amengual O, Atsumi T, Koike T. Antiprothombin antibodies and the diagnosis of antiphospholipid syndrome. Clin Immunol. 2004;112:144–149. doi: 10.1016/j.clim.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 94.Brey RL, Petri MA. Neuropsychiatric systemic lupus erythematosus: miles to go before we sleep. Neurology. 2003;61:9–10. doi: 10.1212/wnl.61.1.9. [DOI] [PubMed] [Google Scholar]
- 95.Samuels MA, King ME, Balis U. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 31-2002. A 61-year-old man with headache and multiple infarcts. N Engl J Med. 2002;347:1187–1194. doi: 10.1056/NEJMcpc020117. [DOI] [PubMed] [Google Scholar]
- 96.Falanga A, Marchetti M, Vignoli A. Coagulation and cancer: biological and clinical aspects. J Thromb Haemost. 2013;11:223–233. doi: 10.1111/jth.12075. [DOI] [PubMed] [Google Scholar]
- 97.Khorana AA. Cancer and coagulation. Am J Hematol. 2012;87(Suppl 1):S82–87. doi: 10.1002/ajh.23143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lysov Z, Swystun LL, Kuruvilla S, Arnold A, Liaw PC. Lung cancer chemotherapy agents increase procoagulant activity via protein disulfide isomerase-dependent tissue factor decryption. Blood Coagul Fibrinolysis. 2015;26:36–45. doi: 10.1097/MBC.0000000000000145. [DOI] [PubMed] [Google Scholar]
- 99.Gueugnon F, Barascu A, Mavridis K, Petit-Courty A, Marchand-Adam S, Gissot V, Scorilas A, Guyetant S, Courty Y. Kallikrein-related peptidase 13: an independent indicator of favorable prognosis for patients with nonsmall cell lung cancer. Tumour Biol. 2015 doi: 10.1007/s13277-015-3148-1. [DOI] [PubMed] [Google Scholar]
- 100.Sidiropoulos KG, White NM, Bui A, Ding Q, Boulos P, Pampalakis G, Khella H, Samuel JN, Sotiropoulou G, Yousef GM. Kallikrein-related peptidase 5 induces miRNA-mediated anti-oncogenic pathways in breast cancer. Oncoscience. 2014;1:709–724. doi: 10.18632/oncoscience.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Michel N, Heuze-Vourc’h N, Lavergne E, Parent C, Jourdan ML, Vallet A, Iochmann S, Musso O, Reverdiau P, Courty Y. Growth and survival of lung cancer cells: regulation by kallikrein-related peptidase 6 via activation of proteinase-activated receptor 2 and the epidermal growth factor receptor. Biol Chem. 2014;395:1015–1025. doi: 10.1515/hsz-2014-0124. [DOI] [PubMed] [Google Scholar]
- 102.Kolin DL, Sy K, Rotondo F, Bassily MN, Kovacs K, Brezden-Masley C, Streutker CJ, Yousef GM. Prognostic significance of human tissue kallikrein-related peptidases 6 and 10 in gastric cancer. Biol Chem. 2014;395:1087–1093. doi: 10.1515/hsz-2014-0143. [DOI] [PubMed] [Google Scholar]
- 103.Walker F, Nicole P, Jallane A, Soosaipillai A, Mosbach V, Oikonomopoulou K, Diamandis EP, Magdolen V, Darmoul D. Kallikrein-related peptidase 7 (KLK7) is a proliferative factor that is aberrantly expressed in human colon cancer. Biol Chem. 2014;395:1075–1086. doi: 10.1515/hsz-2014-0142. [DOI] [PubMed] [Google Scholar]
- 104.Li W, Zhao Y, Ren L, Wu X. Serum human kallikrein 7 represents a new marker for cervical cancer. Med Oncol. 2014;31:208. doi: 10.1007/s12032-014-0208-0. [DOI] [PubMed] [Google Scholar]
- 105.Zhang CY, Zhu Y, Rui WB, Dai J, Shen ZJ. Expression of kallikrein-related peptidase 7 is decreased in prostate cancer. Asian J Androl. 2015;17:106–110. doi: 10.4103/1008-682X.137613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang R, Zhang T, Ma Z, Wang Y, Cheng Z, Xu H, Li W, Wang X. The interaction of coagulation factor XII and monocyte/macrophages mediating peritoneal metastasis of epithelial ovarian cancer. Gynecol Oncol. 2010;117:460–466. doi: 10.1016/j.ygyno.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 107.Naderi A. Coagulation factor VII is regulated by androgen receptor in breast cancer. Exp Cell Res. 2015;331:239–250. doi: 10.1016/j.yexcr.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Koizume S, Ito S, Miyagi E, Hirahara F, Nakamura Y, Sakuma Y, Osaka H, Takano Y, Ruf W, Miyagi Y. HIF2alpha-Sp1 interaction mediates a deacetylation-dependent FVII-gene activation under hypoxic conditions in ovarian cancer cells. Nucleic Acids Res. 2012;40:5389–5401. doi: 10.1093/nar/gks201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kocaturk B, Versteeg HH. Tissue factor isoforms in cancer and coagulation: may the best isoform win. Thromb Res. 2012;129(Suppl 1):S69–75. doi: 10.1016/S0049-3848(12)70020-8. [DOI] [PubMed] [Google Scholar]
- 110.Sato R, Obonai T, Tsumura R, Tsumoto K, Koga Y, Yasunaga M, Matsumura Y. Preparation and characterization of anti-tissue factor single-chain variable fragment antibody for cancer diagnosis. Cancer Sci. 2014;105:1631–1637. doi: 10.1111/cas.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhong YC, Zhang T, Di W, Li WP. Thrombin promotes epithelial ovarian cancer cell invasion by inducing epithelial-mesenchymal transition. J Gynecol Oncol. 2013;24:265–272. doi: 10.3802/jgo.2013.24.3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Danckwardt S, Hentze MW, Kulozik AE. Pathologies at the nexus of blood coagulation and inflammation: thrombin in hemostasis, cancer, and beyond. J Mol Med (Berl) 2013;91:1257–1271. doi: 10.1007/s00109-013-1074-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ohshiro K, Bui-Nguyen TM, Divijendra Natha RS, Schwartz AM, Levine P, Kumar R. Thrombin stimulation of inflammatory breast cancer cells leads to aggressiveness via the EGFR-PAR1-Pak1 pathway. Int J Biol Markers. 2012;27:e305–313. doi: 10.5301/JBM.2012.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Turpin B, Miller W, Rosenfeldt L, Kombrinck K, Flick MJ, Steinbrecher KA, Harmel-Laws E, Mullins ES, Shaw M, Witte DP, Revenko A, Monia B, Palumbo JS. Thrombin drives tumorigenesis in colitis-associated colon cancer. Cancer Res. 2014;74:3020–3030. doi: 10.1158/0008-5472.CAN-13-3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Polterauer S, Grimm C, Seebacher V, Concin N, Marth C, Tomovski C, Husslein H, Leipold H, Hefler-Frischmuth K, Tempfer C, Reinthaller A, Hefler L. Plasma fibrinogen levels and prognosis in patients with ovarian cancer: a multicenter study. Oncologist. 2009;14:979–985. doi: 10.1634/theoncologist.2009-0079. [DOI] [PubMed] [Google Scholar]
- 116.Hefler-Frischmuth K, Lafleur J, Hefler L, Polterauer S, Seebacher V, Reinthaller A, Grimm C. Plasma fibrinogen levels in patients with benign and malignant ovarian tumors. Gynecol Oncol. 2015;136:567–570. doi: 10.1016/j.ygyno.2014.12.041. [DOI] [PubMed] [Google Scholar]
- 117.Polterauer S, Seebacher V, Hefler-Frischmuth K, Grimm C, Heinze G, Tempfer C, Reinthaller A, Hefler L. Fibrinogen plasma levels are an independent prognostic parameter in patients with cervical cancer. Am J Obstet Gynecol. 2009;200:647 e641–647. doi: 10.1016/j.ajog.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 118.Seebacher V, Polterauer S, Grimm C, Husslein H, Leipold H, Hefler-Frischmuth K, Tempfer C, Reinthaller A, Hefler L. The prognostic value of plasma fibrinogen levels in patients with endometrial cancer: a multi-centre trial. Br J Cancer. 2010;102:952–956. doi: 10.1038/sj.bjc.6605547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seebacher V, Polterauer S, Grimm C, Tempfer C, Hefler-Frischmuth K, Reinthaller A, Hefler L. The impact of plasma fibrinogen levels on patients with vulvar cancer. Eur J Obstet Gynecol Reprod Biol. 2012;161:88–91. doi: 10.1016/j.ejogrb.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 120.Kim JY, Al-Hilal TA, Chung SW, Kim SY, Ryu GH, Son WC, Byun Y. Antiangiogenic and anticancer effect of an orally active low molecular weight heparin conjugates and its application to lung cancer chemoprevention. J Control Release. 2015;199:122–131. doi: 10.1016/j.jconrel.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 121.Mavridis K, Avgeris M, Scorilas A. Targeting kallikrein-related peptidases in prostate cancer. Expert Opin Ther Targets. 2014;18:365–383. doi: 10.1517/14728222.2014.880693. [DOI] [PubMed] [Google Scholar]
- 122.Franchini M, Bonfanti C, Lippi G. Cancer-associated thrombosis: investigating the role of new oral anticoagulants. Thromb Res. 2015 doi: 10.1016/j.thromres.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 123.Zhou ZH, Tzioufas AG, Notkins AL. Properties and function of polyreactive antibodies and polyreactive antigen-binding B cells. Journal of Autoimmunity. 2007;29:219–228. doi: 10.1016/j.jaut.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Panda S, Ding JL. Natural Antibodies Bridge Innate and Adaptive Immunity. The journal of immunology. 2015;194:13–20. doi: 10.4049/jimmunol.1400844. [DOI] [PubMed] [Google Scholar]
- 125.Racine R, Winslow G. IgM in microbial infections: taken for granted? Immunology letters. 2009;125:79–85. doi: 10.1016/j.imlet.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shriner AK, Liu H, Sun G, Guimond M, Alugupalli KR. IL-7–Dependent B Lymphocytes Are Essential for the Anti-polysaccharide Response and Protective Immunity to Streptococcus pneumoniae. The journal of immunology. 2010;185:525–531. doi: 10.4049/jimmunol.0902841. [DOI] [PubMed] [Google Scholar]
- 127.Zhou ZH, Zhang Y, Hu YF, Wahl LM, Cisar JO, Notkins AL. The broad antibacterial activity of the natural antibody repertoire is due to polyreactive antibodies. Cell Host & Microbe. 2007;1:51–61. doi: 10.1016/j.chom.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cunningham AF, Flores-Langarica A, Bobat S, Dominguez Medina CC, Cook CN, Ross EA, Lopez-Macias C, Henderson IR. B1b cells recognize protective antigens after natural infection and vaccination. Front Immunol. 2014;5:535. doi: 10.3389/fimmu.2014.00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tumang JR, Frances R, Yeo SG, Rothstein TL. Spontaneously Ig-secreting B-1 cells violate the accepted paradigm for expression of differentiation-associated transcription factors. J Immunol. 2005;174:3173–3177. doi: 10.4049/jimmunol.174.6.3173. [DOI] [PubMed] [Google Scholar]
- 130.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b Lymphocytes Confer T Cell-Independent Long-Lasting Immunity. Immunity. 2004;21:379–390. doi: 10.1016/j.immuni.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 131.Griffin DO, Rothstein TL. A small CD11b(+) human B1 cell subpopulation stimulates T cells and is expanded in lupus. J Exp Med. 2011;208:2591–2598. doi: 10.1084/jem.20110978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b Cells Exhibit Distinct Developmental Requirements and Have Unique Functional Roles in Innate and Adaptive Immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 133.Mold C, Rodic-Polic B, Du Clos TW. Protection from Streptococcus pneumoniae Infection by C-Reactive Protein and Natural Antibody Requires Complement But Not Fcγ Receptors. The journal of immunology. 2002;168:6375–6381. doi: 10.4049/jimmunol.168.12.6375. [DOI] [PubMed] [Google Scholar]
- 134.Skurnik D, Kropec A, Roux D, Theilacker C, Huebner J, Pier GB. Natural Antibodies in Normal Human Serum Inhibit Staphylococcus aureus Capsular Polysaccharide Vaccine Efficacy. Clinical infectious diseases. 2012;55:1188–1197. doi: 10.1093/cid/cis624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stamp L, Hobbs JR. A natural “antibody” in rabbit serum producing a capular reaction with Staphylococcus pyogenes and related to immunity. British Journal of Experimental Pathology. 1967;48:1–10. [PMC free article] [PubMed] [Google Scholar]
- 136.Baxendale HE, Johnson M, Stephens RCM, Yuste J, Klein N, Brown JS, Goldblatt D. Natural human antibodies to pneumococcus have distinctive molecular characteristics and protect against pneumococcal disease. Clinical and Experimental Immunology. 2008;151:51–60. doi: 10.1111/j.1365-2249.2007.03535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Silverman GJ, Goodyear CS. Confounding B-cell defences: lessons from a staphylococcal superantigen. Nat Rev Immunol. 2006;6:465–475. doi: 10.1038/nri1853. [DOI] [PubMed] [Google Scholar]
- 138.Goodyear CS, Silverman GJ. B cell superantigens: a microbe’s answer to innate-like B cells and natural antibodies. Springer Semin Immunopathol. 2005;26:463–484. doi: 10.1007/s00281-004-0190-2. [DOI] [PubMed] [Google Scholar]
- 139.Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin Immunopathol. 2005;26:347–362. doi: 10.1007/s00281-004-0182-2. [DOI] [PubMed] [Google Scholar]
- 140.Jayasekera JP, Moseman EA, Carroll MC. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J Virol. 2007;81:3487–3494. doi: 10.1128/JVI.02128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, Zinkernagel RM. Control of early viral and bacterial distribution and disease by natural antibodies. Science. 1999;286:2156–2159. doi: 10.1126/science.286.5447.2156. [DOI] [PubMed] [Google Scholar]
- 143.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci U S A. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gronwall C, Vas J, Silverman GJ. Protective Roles of Natural IgM Antibodies. Front Immunol. 2012;3:66. doi: 10.3389/fimmu.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Su J, Hua X, Concha H, Svenungsson E, Cederholm A, Frostegard J. Natural antibodies against phosphorylcholine as potential protective factors in SLE. Rheumatology (Oxford) 2008;47:1144–1150. doi: 10.1093/rheumatology/ken120. [DOI] [PubMed] [Google Scholar]
- 146.Vas J, Gronwall C, Marshak-Rothstein A, Silverman GJ. Natural antibody to apoptotic cell membranes inhibits the proinflammatory properties of lupus autoantibody immune complexes. Arthritis Rheum. 2012;64:3388–3398. doi: 10.1002/art.34537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen Y, Khanna S, Goodyear CS, Park YB, Raz E, Thiel S, Gronwall C, Vas J, Boyle DL, Corr M, Kono DH, Silverman GJ. Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J Immunol. 2009;183:1346–1359. doi: 10.4049/jimmunol.0900948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Harisdangkul V, McDougal JS, Knapp M, Christian CL. Naturally occurring low molecular weight IgM in patients with rheumatoid arthritis, systemic lupus erythematosus and macroglobulinemia. I. Purification and immunologic studies. J Immunol. 1975;115:216–222. [PubMed] [Google Scholar]
- 149.Guerin J, Casey E, Feighery C, Jackson J. Anti-Beta 2-glycoprotein I antibody isotype and IgG subclass in antiphospholipid syndrome patients. Autoimmunity. 1999;31:109–116. doi: 10.3109/08916939908994054. [DOI] [PubMed] [Google Scholar]
- 150.Lieby P, Soley A, Knapp AM, Cerutti M, Freyssinet JM, Pasquali JL, Martin T. Memory B cells producing somatically mutated antiphospholipid antibodies are present in healthy individuals. Blood. 2003;102:2459–2465. doi: 10.1182/blood-2003-01-0180. [DOI] [PubMed] [Google Scholar]
- 151.Ekdahl KN, Bengtsson AA, Andersson J, Elgue G, Ronnblom L, Sturfelt G, Nilsson B. Thrombotic disease in systemic lupus erythematosus is associated with a maintained systemic platelet activation. Br J Haematol. 2004;125:74–78. doi: 10.1111/j.1365-2141.2004.04858.x. [DOI] [PubMed] [Google Scholar]
- 152.Navratil JS, Manzi S, Kao AH, Krishnaswami S, Liu CC, Ruffing MJ, Shaw PS, Nilson AC, Dryden ER, Johnson JJ, Ahearn JM. Platelet C4d is highly specific for systemic lupus erythematosus. Arthritis Rheum. 2006;54:670–674. doi: 10.1002/art.21627. [DOI] [PubMed] [Google Scholar]
- 153.Peerschke EI, Yin W, Alpert DR, Roubey RA, Salmon JE, Ghebrehiwet B. Serum complement activation on heterologous platelets is associated with arterial thrombosis in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus. 2009;18:530–538. doi: 10.1177/0961203308099974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hamad OA, Back J, Nilsson PH, Nilsson B, Ekdahl KN. Platelets, complement, and contact activation: partners in inflammation and thrombosis. Adv Exp Med Biol. 2012;946:185–205. doi: 10.1007/978-1-4614-0106-3_11. [DOI] [PubMed] [Google Scholar]
- 155.Lood C, Tyden H, Gullstrand B, Sturfelt G, Jonsen A, Truedsson L, Bengtsson AA. Platelet activation and anti-phospholipid antibodies collaborate in the activation of the complement system on platelets in systemic lupus erythematosus. PLoS ONE. 2014;9:e99386. doi: 10.1371/journal.pone.0099386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Vega-Ostertag M, Harris EN, Pierangeli SS. Intracellular events in platelet activation induced by antiphospholipid antibodies in the presence of low doses of thrombin. Arthritis Rheum. 2004;50:2911–2919. doi: 10.1002/art.20434. [DOI] [PubMed] [Google Scholar]
- 157.Vega-Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum. 2005;52:1545–1554. doi: 10.1002/art.21009. [DOI] [PubMed] [Google Scholar]
- 158.Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, Pfeilschifter J, Young MF, Schaefer RM, Schaefer L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest. 2010;120:4251–4272. doi: 10.1172/JCI42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med. 1983;157:202–218. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jellusova J, Duber S, Guckel E, Binder CJ, Weiss S, Voll R, Nitschke L. Siglec-G regulates B1 cell survival and selection. J Immunol. 2010;185:3277–3284. doi: 10.4049/jimmunol.1001792. [DOI] [PubMed] [Google Scholar]
- 161.Bokers S, Urbat A, Daniel C, Amann K, Smith KG, Espeli M, Nitschke L. Siglec-G deficiency leads to more severe collagen-induced arthritis and earlier onset of lupus-like symptoms in MRL/lpr mice. J Immunol. 2014;192:2994–3002. doi: 10.4049/jimmunol.1303367. [DOI] [PubMed] [Google Scholar]
- 162.Brandlein S, Lorenz J, Ruoff N, Hensel F, Beyer I, Muller J, Neukam K, Illert B, Eck M, Muller-Hermelink HK, Vollmers HP. Human monoclonal IgM antibodies with apoptotic activity isolated from cancer patients. Hum Antibodies. 2002;11:107–119. [PubMed] [Google Scholar]
- 163.Brandlein S, Pohle T, Ruoff N, Wozniak E, Muller-Hermelink HK, Vollmers HP. Natural IgM antibodies and immunosurveillance mechanisms against epithelial cancer cells in humans. Cancer Res. 2003;63:7995–8005. [PubMed] [Google Scholar]
- 164.Brandlein S, Vollmers HP. Natural IgM antibodies, the ignored weapons in tumour immunity. Histol Histopathol. 2004;19:897–905. doi: 10.14670/HH-19.897. [DOI] [PubMed] [Google Scholar]
- 165.Vollmers HP, Brandlein S. Nature’s best weapons to fight cancer. Revival of human monoclonal IgM antibodies. Hum Antibodies. 2002;11:131–142. [PubMed] [Google Scholar]
- 166.Rasche L, Duell J, Morgner C, Chatterjee M, Hensel F, Rosenwald A, Einsele H, Topp MS, Brandlein S. The natural human IgM antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLoS ONE. 2013;8:e63414. doi: 10.1371/journal.pone.0063414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Brandlein S, Rauschert N, Rasche L, Dreykluft A, Hensel F, Conzelmann E, Muller-Hermelink HK, Vollmers HP. The human IgM antibody SAM-6 induces tumor-specific apoptosis with oxidized low-density lipoprotein. Mol Cancer Ther. 2007;6:326–333. doi: 10.1158/1535-7163.MCT-06-0399. [DOI] [PubMed] [Google Scholar]
- 168.Pohle T, Brandlein S, Ruoff N, Muller-Hermelink HK, Vollmers HP. Lipoptosis: tumor-specific cell death by antibody-induced intracellular lipid accumulation. Cancer Res. 2004;64:3900–3906. doi: 10.1158/0008-5472.CAN-03-3149. [DOI] [PubMed] [Google Scholar]
- 169.Shishido SN, Varahan S, Yuan K, Li X, Fleming SD. Humoral innate immune response and disease. Clin Immunol. 2012;144:142–158. doi: 10.1016/j.clim.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hofmann BT, Stehr A, Dohrmann T, Gungor C, Herich L, Hiller J, Harder S, Ewald F, Gebauer F, Tachezy M, Precht C, Izbicki JR, Bockhorn M, Wagener C, Wolters-Eisfeld G. ABO blood group IgM isoagglutinins interact with tumor-associated O-glycan structures in pancreatic cancer. Clin Cancer Res. 2014;20:6117–6126. doi: 10.1158/1078-0432.CCR-14-0716. [DOI] [PubMed] [Google Scholar]
- 171.Dorothee G, Vergnon I, El Hage F, Le Maux Chansac B, Ferrand V, Lecluse Y, Opolon P, Chouaib S, Bismuth G, Mami-Chouaib F. In situ sensory adaptation of tumor-infiltrating T lymphocytes to peptide-MHC levels elicits strong antitumor reactivity. J Immunol. 2005;174:6888–6897. doi: 10.4049/jimmunol.174.11.6888. [DOI] [PubMed] [Google Scholar]
- 172.Perez EC, Machado J, Jr, Aliperti F, Freymuller E, Mariano M, Lopes JD. B-1 lymphocytes increase metastatic behavior of melanoma cells through the extracellular signal-regulated kinase pathway. Cancer Sci. 2008;99:920–928. doi: 10.1111/j.1349-7006.2008.00776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Staquicini FI, Tandle A, Libutti SK, Sun J, Zigler M, Bar-Eli M, Aliperti F, Perez EC, Gershenwald JE, Mariano M, Pasqualini R, Arap W, Lopes JD. A subset of host B lymphocytes controls melanoma metastasis through a melanoma cell adhesion molecule/MUC18-dependent interaction: evidence from mice and humans. Cancer Res. 2008;68:8419–8428. doi: 10.1158/0008-5472.CAN-08-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chu CC, Catera R, Zhang L, Didier S, Agagnina BM, Damle RN, Kaufman MS, Kolitz JE, Allen SL, Rai KR, Chiorazzi N. Many chronic lymphocytic leukemia antibodies recognize apoptotic cells with exposed nonmuscle myosin heavy chain IIA: implications for patient outcome and cell of origin. Blood. 2010;115:3907–3915. doi: 10.1182/blood-2009-09-244251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ugarte-Berzal E, Bailon E, Amigo-Jimenez I, Albar JP, Garcia-Marco JA, Garcia-Pardo A. A novel CD44-binding peptide from the pro-matrix metalloproteinase-9 hemopexin domain impairs adhesion and migration of chronic lymphocytic leukemia (CLL) cells. J Biol Chem. 2014;289:15340–15349. doi: 10.1074/jbc.M114.559187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tavolaro S, Colombo T, Chiaretti S, Peragine N, Fulci V, Ricciardi MR, Messina M, Bonina S, Brugnoletti F, Marinelli M, Di Maio V, Mauro FR, Del Giudice I, Macino G, Foa R, Guarini A. Increased chronic lymphocytic leukemia proliferation upon IgM stimulation is sustained by the upregulation of miR-132 and miR-212. Genes Chromosomes Cancer. 2015;54:222–234. doi: 10.1002/gcc.22236. [DOI] [PubMed] [Google Scholar]
- 177.Krysov S, Potter KN, Mockridge CI, Coelho V, Wheatley I, Packham G, Stevenson FK. Surface IgM of CLL cells displays unusual glycans indicative of engagement of antigen in vivo. Blood. 2010;115:4198–4205. doi: 10.1182/blood-2009-12-254847. [DOI] [PubMed] [Google Scholar]
- 178.Whalen GF, Sullivan M, Piperdi B, Wasseff W, Galili U. Cancer immunotherapy by intratumoral injection of alpha-gal glycolipids. Anticancer Res. 2012;32:3861–3868. [PubMed] [Google Scholar]
- 179.Lei X, Liu C, Azadzoi K, Li C, Lu F, Xiang A, Sun J, Guo Y, Zhao Q, Yan Z, Yang J. A novel IgM-H-ficolin complement pathway to attack allogenic cancer cells in vitro. Sci Rep. 2015;5:7824. doi: 10.1038/srep07824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Galili U, Wigglesworth K, Abdel-Motal UM. Intratumoral injection of alpha-gal glycolipids induces xenograft-like destruction and conversion of lesions into endogenous vaccines. J Immunol. 2007;178:4676–4687. doi: 10.4049/jimmunol.178.7.4676. [DOI] [PubMed] [Google Scholar]
- 181.Cutrona G, Colombo M, Matis S, Fabbi M, Spriano M, Callea V, Vigna E, Gentile M, Zupo S, Chiorazzi N, Morabito F, Ferrarini M. Clonal heterogeneity in chronic lymphocytic leukemia cells: superior response to surface IgM cross-linking in CD38, ZAP-70-positive cells. Haematologica. 2008;93:413–422. doi: 10.3324/haematol.11646. [DOI] [PubMed] [Google Scholar]
- 182.Governa M, Fenoglio I, Amati M, Valentino M, Bolognini L, Coloccini S, Volpe AR, Carmignani M, Fubini B. Cleavage of the fifth component of human complement and release of a split product with C5a-like activity by crystalline silica through free radical generation and kallikrein activation. Toxicol Appl Pharmacol. 2002;179:129–136. doi: 10.1006/taap.2002.9351. [DOI] [PubMed] [Google Scholar]
- 183.Bleakly NT, Fontaine MJ, Pate LL, Sutherland SM, Jeng M. Disseminated intravascular coagulation due to IgM-mediated autoimmune hemolytic anemia. Pediatr Blood Cancer. 2011;57:329–331. doi: 10.1002/pbc.23024. [DOI] [PubMed] [Google Scholar]
- 184.Buhler L, Yamada K, Kitamura H, Alwayn IP, Basker M, Appel JZ, 3rd, Colvin RB, White-Scharf ME, Sachs DH, Robson SC, Awwad M, Cooper DK. Pig kidney transplantation in baboons: anti-Gal(alpha)1-3Gal IgM alone is associated with acute humoral xenograft rejection and disseminated intravascular coagulation. Transplantation. 2001;72:1743–1752. doi: 10.1097/00007890-200112150-00007. [DOI] [PubMed] [Google Scholar]
- 185.Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol Heart Circ Physiol. 2009;297:H1853–1859. doi: 10.1152/ajpheart.00049.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sun S, Zhao G, Liu C, Fan W, Zhou X, Zeng L, Guo Y, Kou Z, Yu H, Li J, Wang R, Li Y, Schneider C, Habel M, Riedemann NC, Du L, Jiang S, Guo R, Zhou Y. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis. 2015;60:586–595. doi: 10.1093/cid/ciu887. [DOI] [PMC free article] [PubMed] [Google Scholar]