

Summary
Meiosis activating sterols (MAS) are substrates of SC4MOL and NSDHL in the cholesterol pathway and are important for normal organismal development. Oncogenic transformation by EGFR or RAS increases the demand for cholesterol, suggesting a possibility for metabolic interference. To test this idea in vivo, we ablated Nsdhl in adult keratinocytes expressing KRASG12D. Strikingly, Nsdhl inactivation antagonized the growth of skin tumors, while having little effect on normal skin. Loss of Nsdhl induced the expression of ATP-binding cassette (ABC) transporters ABCA1 and ABCG1, reduced the expression of low-density lipoprotein receptor (LDLR), decreased intracellular cholesterol and was dependent on the liver X receptor (LXR) α. Importantly, EGFR signaling opposed LXRα effects on cholesterol homeostasis, while an EGFR inhibitor synergized with LXRα agonists in killing cancer cells. Inhibition of SC4MOL or NSDHL, or activation of LXRα by sterol metabolites can be an effective strategy against carcinomas with activated EGFR-KRAS signaling.
Introduction
Oncogene-transformed cancer cells require elevated levels of cholesterol to support their rapid growth. In addition to enhanced de novo cholesterol synthesis (Pitroda et al., 2009; Silvente-Poirot and Poirot, 2014), the process of cholesterol uptake is also tightly regulated by the signaling activity of the epidermal growth factor receptor (EGFR), and increased in cancer (Guo et al., 2011). The cholesterol metabolic pathway proceeds through a series of elongation reactions from mevalonate to an isoprenoid, squalene. Subsequent cyclization of the squalene carbon chains produces the first sterol precursor, lanosterol, which is further metabolized through a series of oxygen-dependent reactions to produce the final product, cholesterol. The role of the isoprenoid precursors in protein prenylation (Clendening et al., 2010a), or of cholesterol in formation of lipid rafts that support oncogenic signaling platforms (Munro, 2003) is well-established. In contrast, relatively little is known about the biological activities of the intermediate sterol metabolites.
Central in the sterol synthesis pathway producing metabolites essential to convert squalene to cholesterol, SC4MOL and NSDHL (Nes, 2011) catalyze two sequential steps of oxidative decarboxylation of the C4 methyl groups from meiosis activating sterols (MAS) (Acimovic and Rozman, 2013). Mutations in SC4MOL or NSDHL lead to significant accumulation of T-MAS and FF-MAS (He et al., 2011; Liu et al., 1999). Under physiological conditions, these MAS are abundant in male and female gonads and regulate meiosis (Byskov et al., 1995) through a mechanism that involves release of EGFR ligands and activated receptor signaling (Park et al., 2004). In previous work, we had pursued suggestions from an RNAi-based sensitization screen to establish that depletion of SC4MOL and NSDHL, but not enzymes operating upstream or downstream in the sterol synthesis pathway, sensitized cancer cells to the EGFR inhibitors cetuximab and erlotinib (Sukhanova et al., 2013). These EGFR inhibitor-sensitizing effects of SC4MOL and NSDHL were mediated by the MAS sterol metabolites, and were observed both in vitro and in xenograft analysis in vivo (Sukhanova et al., 2013), suggesting further study of SC4MOL, NSDHL, and MAS might be informative in cancers dependent on EGFR pathway signaling.
The cellular targets and the effector mechanisms for MAS have for a long time remained unclear. However, one in vitro study using luciferase reporter assays in CV-1 cells suggested an important target of MAS may be the sterol-binding transcription factor, liver X nuclear receptor (LXR) (Janowski et al., 1996), which plays a critical role in regulating de novo synthesis and uptake of cholesterol (Zelcer et al., 2009). LXR, as a heterodimer with retinoid X receptor (RXR), is a transcription factor activated by specific oxysterol metabolites to induce the expression of multiple targets including ABCG1 and ABCA1, cholesterol efflux pumps associated with the endoplasmic reticulum (ER) vesicles or the cell surface membrane, respectively (Venkateswaran et al., 2000). Activation of LXR also reduces cholesterol uptake by promoting the ubiquitin-dependent degradation of low-density lipoprotein receptor (LDLR) and intriguingly, is cytotoxic to cancer cells with hyperactive EGFR signaling (Guo et al., 2011) and is antagonizing angiogenesis and metastatic spread (Pencheva et al., 2014).
Nsdhl was first identified as the gene associated with the X-linked dominant, male lethal mouse mutations bare patches (Bpa) and striated (Str). Heterozygous Bpa/+ female mice are dwarfed compared to wild type female littermates (Liu et al., 1999) and develop hyperkeratotic skin eruptions on postnatal day 5–7 that resolve leaving a striped coat that follows lines of X-inactivation (Liu et al., 1999). Affected male embryos for all murine Nsdhl alleles die by midgestation (Caldas et al., 2005). Heterozygous germline mutations in the human NSDHL gene cause congenital hemidysplasia and skin ichthyosiform nevi with limb deformation (CHILD syndrome) (Hummel et al., 2003). Together, these severe phenotypes raised concerns about the therapeutic potential of targeting MAS, but left open the question of whether effects would be observed targeting MAS in the adult setting.
To further evaluate whether inhibition of NSDHL is a potential therapeutic route in cancer, and to assess the toxicity, selectivity, and mechanism of action of such an approach, we have used here a recently developed, conditional Nsdhl knockout mouse model (Cunningham et al., 2015) in the setting of tumor development driven by EGFR and its effector KRAS. The results of this work have delineated a critical MAS-sensing mechanism mediated by LXR by which tumor cell growth is dependent on endogenous sterol metabolism, and support the further exploration of these enzymes and sterol metabolites as clinical targets.
Results
NSDHL deficiency is tolerated in adult animals
Given the known serious consequences of loss of Nsdhl during embryonic development (Caldas et al., 2005), we first investigated whether more limited loss of the gene could be tolerated in post-natal or adult animals. For this purpose, we used recently generated mice with a “floxed” exon 5 conditional knockout allele of Nsdhlflx5 (Cunningham et al., 2015) crossed to transgenic animals carrying Cre recombinase gene under the control of the keratin 14 promoter (K14Cre), which inactivates Nsdhl (denoted as NsdhlΔ5) selectively in skin keratinocytes, beginning at day E14 (Wang et al., 1997) (Fig. S1). Although Nsdhl is X-linked, this approach yielded viable progenies at term with approximately a 1:1 male to female ratio, suggesting no embryonic lethality in hemizygous K14Cre;NsdhlΔ5/Y males. Male and female mice appeared fully developed at birth, and pups were able to crawl and feed; however, all K14Cre;NsdhlΔ5/Y males with total NSDHL inactivation in the skin died within 24 hours after birth with features of dehydration (Fig. 1A). Heterozygous X chromosome-mosaic for Nsdhl loss, K14Cre;NsdhlΔ5/+ female mice were viable but had impaired hair development at 1 week of age (Fig. 1B), similar to the original Bpa phenotype (Liu et al., 1999). Evaluation of the NSDHL-null areas of 4-week old K14Cre;NsdhlΔ5/+ females showed reduced skin thickness and fewer Ki67-positive proliferating basal layer keratinocytes (Fig. 1C, S2A–C).
Figure 1.
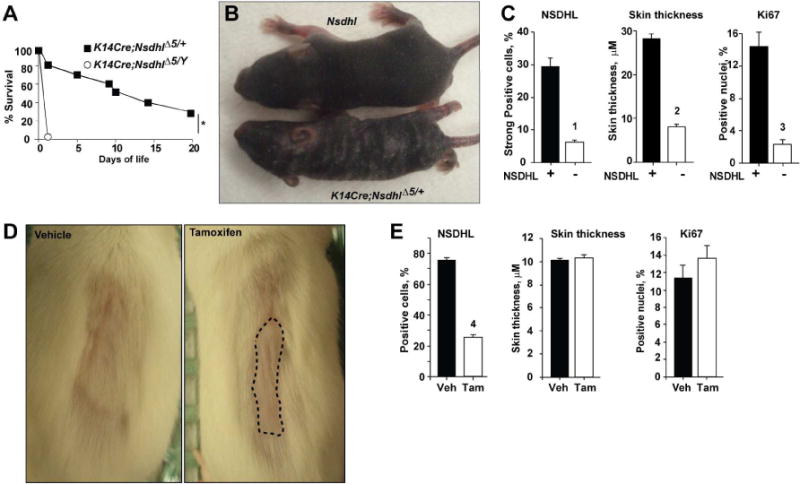
Blockade of keratinocyte proliferation by conditional inactivation of NSDHL. (A) Survival of K14Cre;NsdhlΔ5/+ females and K14Cre;NsdhlΔ5/Y males. Shown is pooled data from 3 independent crosses; *, p=10−4. (B) Conditional inactivation of Nsdhl produces hyperkeratotic, scaly patches and a striped coat in 1 week old heterozygous K14Cre;NsdhlΔ5/+ female pups. (C) NSDHL-deficient areas of the dorsal skin in 4 week-old K14Cre;NsdhlΔ5/+ females show reduced thickness and proliferation of the basal layer keratinocytes (Ki67-positive nuclei). See also Figure S3. Digital imaging data are represented as mean±SEM. (D) Delayed hair regrowth (dashed line) following conditional Nsdhl ablation in the skin of adult K5CreERT;Nsdhlflx5/Y mice following oral administration of vehicle (left, corn oil) or tamoxifen (right, 40 mg/kg) for 5 consecutive days at age of 12 weeks. One week after treatment, the coat from the dorsal skin was depilated and hair regrowth was imaged 1 week after depilation. (E) No change in interfollicular epidermis Ki67-positive cells or epidermis thickness in tamoxifen-treated adult K5CreERT;Nsdhlflx5/Y mice. See also Figure S4. Digital imaging data are represented as mean±SEM, p-values: (1) p=3*10−6; (2) p=3*10−16; (3) p=2*10−5; (4) p= 2*10−16, based on Wilcoxon test.
As an alternative approach, we generated mice in which expression of the floxed allele of Nsdhlflx5 was excised in the keratinocytes of adult animals (Fig. S1A) by tamoxifen-activated CreERT under the control of the murine keratin 5 (K5) promoter (5 days at 12 weeks of age immediately followed by hair removal (Kim et al., 2009b)). In this model, inactivation of Nsdhl gene in the skin of Nsdhlflx5/Y adult males and Nsdhlflx5/flx5 females, delayed the hair regrowth normally expected after 2 weeks after depilation, and was the only apparent phenotypic change (Fig. 1D), with no apparent skin toxicity. Histological analysis of skin from 14 week-old mice with conditional CreERT-inactivation of NSDHL following tamoxifen treatment did not affect the number of Ki67 positive cells or the thickness of interfollicular epidermis in contrast to the K14-Cre;Nsdhlflx5 mice (Fig. 1E, S3A). The more subtle phenotype associated with loss of NSDHL in adults versus newborn animals suggested that therapeutic targeting of NSDHL and its effectors may be a viable strategy.
NSDHL is critical for de novo growth of KRAS-transformed tumors
We next tested the effects of conditional Nsdhl ablation in the skin on tumor development induced by oncogenic KRASG12D (Jackson et al., 2001; Tuveson et al., 2004). We created mice carrying three modified genes: the K5CreERTand LSL-KrasG12D transgenes and the conditional Nsdhlflx5 allele (Methods and Fig. S1). In these animals, the LSL-KrasG12D and Nsdhlflx5 alleles are rearranged after the mice are treated with tamoxifen at 4 weeks of age, which induces the nuclear translocation of CreERT protein (Kim et al., 2009a). Cre-mediated rearrangement of the LSL-KrasG12D allele produced Lox-KrasG12D (Fig. S1E, F), while rearrangement of the Nsdhlflx5 allele resulted in loss of NSDHL expression in the skin and the tumors (Fig. S1G).
Prior to the overt appearance of tumors, the Cre-activated KrasG12D transgene caused hypertrophy of the sebaceous glands in the dermal layer of the skin and increased thickness of the interfollicular epidermis (Fig. 2A, B). These effects were counteracted by concomitant inactivation of NSDHL in Nsdhlflx5/Y mice compared to the control age-matched Nsdhl+/+ and were mosaic in Nsdhlflx5/+ animals (Fig. 2A, B). Importantly, loss of NSDHL significantly influenced KrasG12D-dependent tumor growth. Skin tumors appeared at similar rate in Nsdhlflx5/Y and Nsdhlflx5/+ animals (respectively, in 93% and 97% of animals following tamoxifen treatment). However, the NSDHL-null tumors formed on the Nsdhlflx5/Y background remained small (Fig. 2C–E). In contrast, all KrasG12D-induced Nsdhl+/+ tumors (Fig. 2D–F) and some (13 of 100) of tumors in heterozygous mice (Nsdhlflx5/+, Fig. 2C–E) grew rapidly. The NSDHL-null skin tumors in Nsdhlflx5/Y mice appeared as desiccated papilloma with narrow stem in contrast to broad-based and fleshy Nsdhl+/+ and some of Nsdhlflx5/+ tumors (Fig. 2E). Parallel western analysis of NSDHL expression in the tumors (Fig. 2F, G) indicated essentially complete ablation of NSDHL in the tumors of Nsdhlflx5/Y mice, complete retention in Nsdhl+/+ tumors, and variable expression in Nsdhlflx5/+ tumors (Fig. 2G). NSDHL has been reported to influence immune cell maturation, raising the possibility of a non-cell autonomous action in promoting through tumor growth through influencing microenvironment (Santori et al., 2015; Villablanca et al., 2010). However, we did not detect significant differences in infiltration by inflammatory cells (Fig. S3B) including CD3 T cells (Fig. S3C) between NSDHL-sufficient vs. NSDHL-deficient skin tumors, suggesting the growth impediment was unlikely immune-mediated.
Figure 2.
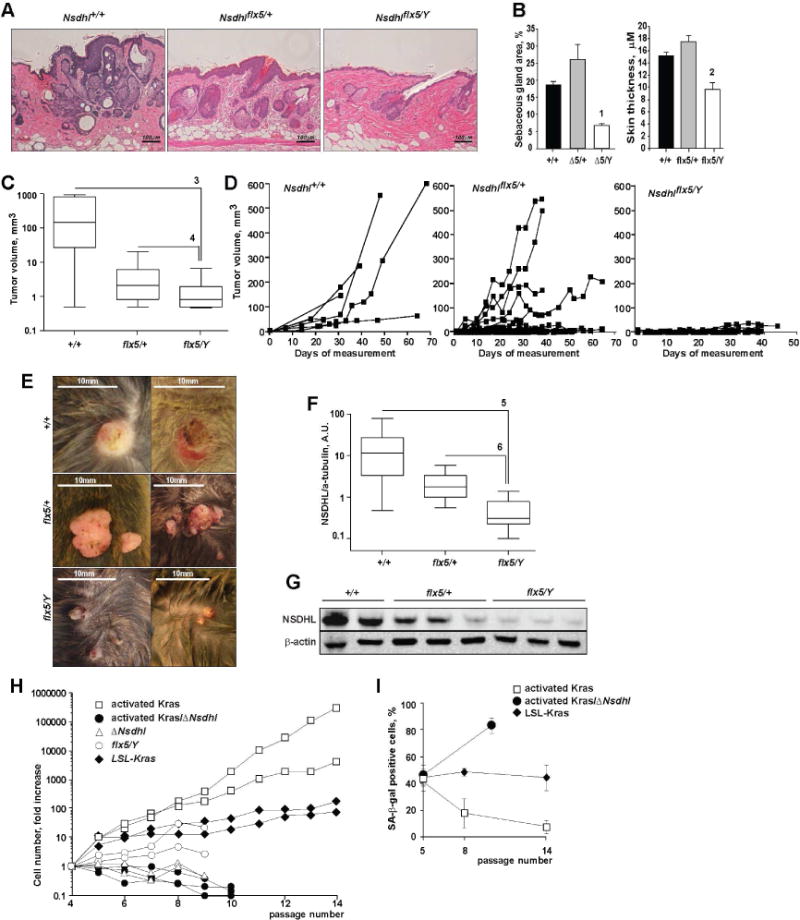
NSDHL deficiency antagonizes the growth of KRASG12D-induced skin tumors. (A) H&E stained sections of the skin 4 weeks after tamoxifen treatment induces K5CreERT;LSL-KrasG12D transgenic animals on indicated Nsdhl backgrounds; 20× magnification. (B) Conditional ablation of Nsdhl antagonized KRASG12D-induced hyperplasia of skin sebaceous glands and interfollicular epidermis. Shown, digital imaging data as mean±SEM percent of dermis area and skin thickness from randomly selected skin sections of 4 animals per group. (C) Tumor volumes and (D) individual tumor growth curves in tamoxifen-induced K5CreERT;LSL-KrasG12D littermates on indicated Nsdhl backgrounds. Pooled data from n=3 independent littermates. (E) Photographs of skin tumors in the littermates. (F) Loss of NSDHL expression was confirmed by Western blot (G) of tumor tissue lysates. Pooled data from n=3 independent littermates. (H) Loss of NSDHL abolishes the proliferative effect of activated KrasG12D in pre-senescent mouse embryonic fibroblasts. (I) Growth arrest of NSDHL-deficient fibroblasts was associated with accelerated senescence as assessed by SA-β-galactosidase positivity. See also Figure S5. Data are represented as mean±SEM, p-values: (1) p=0.03; (2) p=6*10−10; (3) p=6*10−4; (4) p=10−5; (5) p=5*10−4; (6) p=9*10−5, Wilcoxon test.
To confirm the cell-intrinsic nature of Nsdhl loss on KrasG12D-dependent tumor growth, we derived fibroblasts (MEF) from embryos carrying the LSL-KrasG12D transgene with or without a “floxed” allele of Nsdhlflx5 (Jackson et al., 2001) (Fig. S1). At passage 1, these MEF cells were treated with Cre-adenovirus to activate KrasG12D transgene expression, or with a control green fluorescent protein (GFP)-expressing adenovirus (Fig. 2H, I). The cells were propagated in vitro in 10% FBS (fetal bovine serum)-supplemented media and counted at each passage. Cells carrying the Cre-activated oncogenic KrasG12D transgene (Tuveson et al., 2004) showed exponential proliferation compared to the control-treated LSL-KrasG12D cells (Fig. 2H). Notably, Cre-mediated inactivation of Nsdhl (ΔNsdhl cells) in 3 independent MEF lines completely abrogated the proliferative effect of oncogenic KrasG12D. The ΔNsdhl or activated Kras/ΔNsdhl cells could not be passaged beyond P10 and underwent rapid senescence as assessed by SA-β-galactosidase positivity while the KRAS-activated cells escaped senescence for at least 14 passages (Fig. 2I, S4).
Loss of NSDHL activates LXR in vivo
To determine whether NSDHL deficiency activates LXR signaling in vivo, we investigated the expression of two critical LXR target proteins, ABCA1 and LDLR (Bensinger et al., 2008; Guo et al., 2011; Zelcer et al., 2009). Skin biopsies and skin tissue lysates obtained from the newborn K14Cre;NsdhΔ5/Y male pups show striking induction of ABCA1 protein and mRNA in comparison to NSDHL-positive skins (Nsdhlflx5/flx5 in Fig. 3A, B). LDLR has been shown to be degraded following activation of LXR (Zelcer et al., 2009). In keeping with the increased LXR activity, NsdhlΔ5/Y keratinocytes showed reduced expression of LDLR (Fig. 3C, D). Induction of the cholesterol efflux pump ABCA1 and inhibition of LDLR activity are associated with cholesterol depletion from the intracellular membranes (Radhakrishnan et al., 2008), and lead to compensatory cleavage-based activation and nuclear translocation of sterol regulatory element-binding protein 2 – SREBP2 (nSREBP2), which positively regulates genes involved in cholesterol synthesis (Hua et al., 1993). NSDHL-deficient keratinocytes show accelerated proteolytic conversion of SREBP2 to an active nuclear form in comparison to Nsdhlflx5/flx5 controls (Fig. 3A). Serial analyses of skin sections from the mosaic K14Cre;NsdhlΔ5/+ females (4 week-old) revealed an expected patchy staining for NSDHL (Fig. 3E, S2). The NSDHL-negative areas were consistently thinner, had fewer Ki67-expressing keratinocytes and showed markedly reduced LDLR and increased ABCA1 staining. Together, these data support LXR activation in NSDHL deficiency in vivo.
Figure 3.
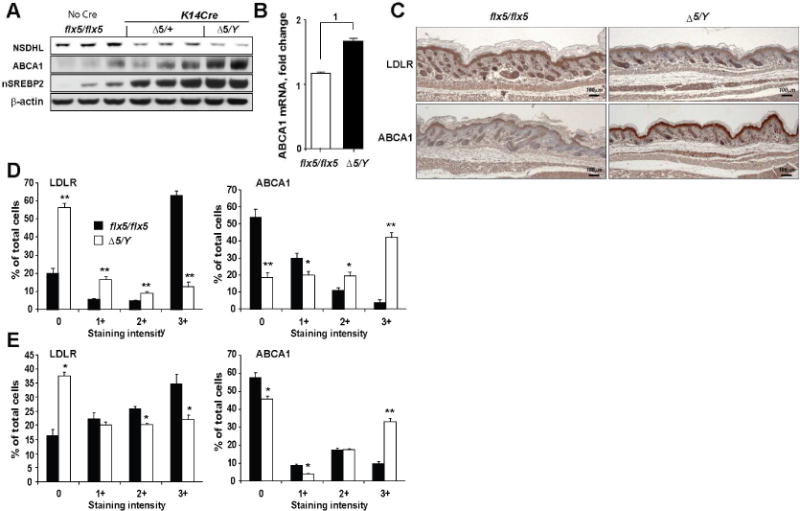
Conditional inactivation of Nsdhl in skin keratinocytes activates LXR. (A) NSDHL, ABCA1 and nuclear SREBP2 protein levels on Western blots in skin tissue lysates of newborn mice. (B) Increased expression of ABCA1 mRNA in the skin of NsdhlΔ5/Y mice. Data are represented as mean±SEM from 6 individual animals, (1) p=0.01. (C) LDLR and ABCA1 immunohistochemistry in the newborn skin; 4× magnification. (D) Expression of LDLR and ABCA1 in the interfollicular epidermis of newborn mice and (E) in 4 week-old K14Cre;NsdhlΔ5/+mosaic females. Digital imaging data in (D–E) are represented as mean±SEM of randomly selected tissue sections from 3 skin samples per group; *, p<0.01; **, p<10−9, by Wilcoxon test.
Induction of LXR targets in NSDHL-and SC4MOL-deficient cancer cells
For potential therapeutic exploitation of the LXR signaling, the MAS/LXR relationship outlined above would ideally be consistent in multiple tumor models. We evaluated two additional models. Using a series of SCC61 carcinoma cells in which the expression of NSDHL or SC4MOL was silenced with specific shRNAs, versus vector controls (Sukhanova et al., 2013), we found depletion of these proteins increased levels of cleaved SREBP2 (Radhakrishnan et al., 2008), and expression of ABCA1 (Fig. 4A). The induction of cleaved SREBP2 was particularly pronounced in lipid-depleted serum (LDS) media (Fig. 4A), which is known to increase MAS metabolite production in NSDHL deficiency (Caldas et al., 2005; Cunningham et al., 2015). The mRNA encoding ABCA1 and a second LXR-regulated transporter, ABCG1, were similarly elevated by depletion of SC4MOL and NSDHL in A431 cervical carcinoma cells (Fig. 4B), while LDLR expression was decreased (Fig. 4C). Silencing of CYP51A1, an enzyme acting upstream of NSDHL in the cholesterol synthesis pathway that prevents accumulation of MAS (Sukhanova et al., 2013), reversed the upregulation of ABCA1 in NSDHL-deficient cancer cells (Fig. 4D) supporting the idea that upregulation of MAS metabolites is important in triggering LXR activation. Importantly, the SC4MOL or NSDHL deficiency caused reduction of total cholesterol in tumor cells (Fig. 4E).
Figure 4.
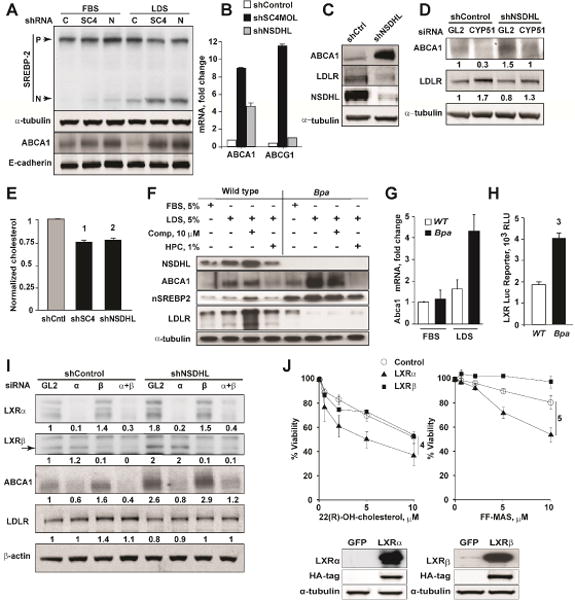
Deficiency of MAS metabolizing enzymes activates LXR targets in cancer cells. (A) Increased nuclear (N) fragments of SREBP2 and membrane-bound ABCA1 expression in SCC61 cells with shRNA-silenced SC4MOL (SC4) and NSDHL (N). (B) qRT-PCR measurement of LXR targets, ABCA1 and ABCG1, in SC4MOL-depleted A431 cells. Results represent mean±SEM from 2 independent RNA samples each analyzed in duplicates. (C) Expression of ABCA1 and loss of LDLR in NSDHL-silenced A431 cells by Western blot. (D) Silencing of an upstream enzyme, CYP51A1, with siRNA abrogates ABCA1 expression and rescues LDLR in NSDHL-deficient A431 cells (numbers, normalized bands density). (E) Reduced total cholesterol levels in SCC61 cells depleted of SC4MOL or NSDHL with shRNA. Data were normalized to total protein and shControl-depleted cells and represented as mean ± St.Dev of 3 independent experiments; (1) and (2), p=0.02 by Kruskal-Wallis test. (F) Pre-senescent mouse embryonic fibroblasts deficient in NSDHL (Bpa) show increased ABCA1 and loss of LDLR expression. This phenotype is exacerbated in cholesterol poor conditions which are used to accelerate the metabolic activity in the cholesterol pathway downstream of SREBP2 (LDS, lipid depleted serum; comp, compactin; HPC, hydroxypropyl-cyclodextrin). Increased mRNA of LXR targets, ABCA1 (G), and activity of LXR-luciferase reporter (H) in pre-senescent Bpa fibroblasts. Data are represented as mean±SEM, n=2 for (G), n=3 for (H); (3) p=0.03. (I) siRNA silencing of LXRα, but not LXRβ, abrogates ABCA1 expression in NSDHL-deficient A431 cells. (J) Lentiviral expression of LXRα but not LXRβ sensitizes SCC61 cells to FF-MAS and 22(R)-hydroxycholesterol. Shown, viability of SCC61 cells cultured in 1%LDS with indicated concentrations of sterols for 72 hours. Data are represented as mean±SEM from 4 experiments; (4) and (5), p=0.002. Insert, expression of LXRα and LXRβ as confirmed by Western blot.
We also investigated the expression of ABCA1 and LDLR proteins in pre-senescent (passage 6) MEFs obtained from NSDHL-deficient bare patches (Bpa1H) mice (Methods and (Cunningham et al., 2005)). As with cancer cell lines, these Bpa1H fibroblasts demonstrated high expression of ABCA1 protein (Fig. 4F) and mRNA (Fig. 4G) relative to control MEFs, particularly in the lipid-poor media. The levels of LDLR were markedly diminished in Bpa1H cells (Fig. 4F). Activity of an LXR-responsive luciferase reporter was significantly elevated in Bpa1H fibroblasts (Fig. 4H).
Human cells express two homologous LXR proteins (Bensinger et al., 2008). The LXRα isoform has been shown to be abundant in the liver and adipose tissue (Peet et al., 1998), while LXRβ is more ubiquitously expressed (Bensinger et al., 2008). In A431 carcinoma cells, depletion of LXRα, but not LXRβ, abrogated the ABCA1 induction associated with NSDHL deficiency (Fig. 4I, S5A). Conversely, overexpression of LXRα in SCC61 cells markedly sensitized these carcinoma cells to the growth suppression with FF-MAS, or another established LXR ligand, 22(R)-hydroxycholesterol (Fig. 4J). Treatment of LXRβ overexpressing SCC61 cells either with 22(R)-hydroxycholesterol or FF-MAS had no effect on the viability in comparison to the control GFP-expressing cells (Fig. 4J) indicating that, in the context of squamous epithelial carcinoma cells, the tested LXR agonists act specifically via LXRα. Together, these results indicate NSDHL to LXR signaling is a consistent feature in both normal and cancer cell models with a selective growth-inhibiting effect against tumors rather than normal cells in vivo.
Accumulation of sterol metabolites in NSDHL-deficient neonatal skin
The effector mechanism of LXR transcriptional activation in the NSDHL-deficiency is poorly understood. The limited data suggests that T-MAS (Janowski et al., 1996) can modulate LXR-responsive genes although no direct interaction of MAS sterols with LXR has been demonstrated. We found that in vitro treatment of A431 cells with several distinct LXR agonists, 22(R)-hydroxycholesterol, GW3965, or FF-MAS produced different effects on LXR targets (Fig. S5B), and it is also possible that additional as yet unidentified metabolites accumulating in NSDHL-null cells also contribute to LXR activation. We therefore conducted detailed gas chromatography-mass spectrometry analysis of sterol metabolites (Table 1 and Fig. S6) using snap frozen skin tissues of K14Cre;Nsdhlflx5/Y versus age-matched Nsdhlflx5/flx5 mice. As expected (Cunningham et al., 2015), an immediate substrate for NSDHL, 4β-carboxy,4α-methyl-cholesta-8,27-dien-3β-ol, was elevated in the Nsdhl-null skin. FF-MAS accumulation was not detected; however, interestingly, a number of “unconventional” C4-methylsterols were also selectively detected in the context of Nsdhl conditional ablation. These likely arise through the metabolic activity of the downstream enzymes altering different carbon moieties of the sterol precursor, e.g. by 24-dehydrocholesterol reductase (DHCR24) eliminating the C24 double bond to produce 4α-methyl-5-cholesta-8-en-3β-ol, and by the C8-sterol isomerase (also known as EBP) to produce 4α-methyl-5-cholesta-7-en-3β-ol and 4α-methyl-5-cholesta-7,24-dien-3β-ol (Fig. S6). Levels of the DHCR24 substrate desmosterol, a known LXR agonist (Yang et al., 2006), were increased >2.5-fold in NSDHL-deficient skin samples suggesting a possible competitive inhibitory effect of some of these metabolites on DHCR24. The abundance of multiple sterol species in NSDHL-deficient skin tissues highlights the complex non-linear metabolic shifts in the sterol metabolome induced by a single enzyme deficiency.
Table 1.
Cholesterol pathway metabolites in the skin tissues (% of total sterols). (See also Figure S6)
| Genotype | Lanosterol | Dihydro- lanosterol |
4β- carboxy,4α- methyl- cholesta- 8,24-dien- 3β-ol |
4α-methyl- 5α- cholesta- 8,24-dien- 3β-ol (ester) |
4α-methyl- 5α- cholest-8- en-3β-ol |
4α-methyl- 5α-cholest- 7-en-3β-ol |
4α-methyl- cholest-7- en-3-one |
Cholest-8- en-3β-ol |
Cholesterol | Desmo- sterol |
7-Dehydro- cholesterol |
Latho- sterol |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Corr. sterols in Fig S6 |
1 | 4 | 8 | 9 | 10 | 11 | 12 | |||||
| Nsdhlflx5/flx5 | 0.023±0.00 | 0.003±0.00 | 0 | 0.003±0.0 | 0.01±0.01 | 0.01±0.0 | 0 | 0.22±0.1 | 95±0.5 | 1.78±0.08 | 0.07±0.01 | 1.8±0.25 |
| K14Cre; NsdhlΔ5/Y | 0.013±0.00 | 0.001±0.00 | + | 2.5±0.91 | 1.93 ±0.4 | 0.5±0.1 | + | 0.21±0.02 | 86±2.7 | 4.92±0.92 | 0.16±0.04 | 1.11±0.03 |
Convergent LXR and EGFR signaling regulates sensitivity to erlotinib in carcinomas
In many solid tumors, the de novo biosynthesis and uptake of cholesterol are under tight regulation by the EGFR signaling cascade (Guo et al., 2009; Guo et al., 2011). Conversely, we previously reported aberrant EGFR signaling in cancer cells deficient in NSDHL or SC4MOL (Astsaturov et al., 2010). We hypothesized that combined inhibition of cholesterol metabolism and EGFR pathway signaling could be synergistic in human carcinomas because of signaling interactions mediated through LXRα. In support of this idea, treatment of carcinoma cell line, A431 and SCC61, with EGF markedly increased levels of LDLR and nuclear SREBP2, while treatment with the EGFR inhibitor erlotinib reduced the expression of these proteins (Fig. 5A, B). In opposing action, treatment of A431 and SCC61 cells with the LXR agonists, 22-hydroxycholesterol and MAS, suppressed LDLR and nuclear fragment of SREBP2 (Fig. 5A).
Figure 5.
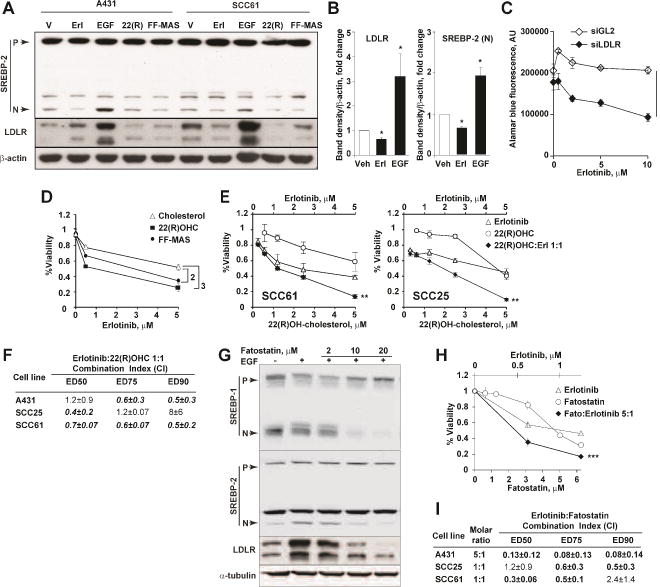
EGFR and LXR signaling co-regulate cholesterol pathway targets in carcinoma. (A) Activation (EGF) or blockade of EGFR (erlotinib) regulates cholesterol metabolism via the expression of nuclear SREBP2 and LDLR in EGFR-positive A431 and SCC61 cells. Conversely, LXR agonists FF-MAS and 22(R)-hydroxycholesterol reduce the expression of nuclear SREBP2 and LDLR. (B) Summary of results quantified from (A) for SCC61 cells; *, p=0.02. (C) Silencing of LDLR with siRNA sensitizes refractory SCC61 cells to erlotinib. Bars, SEM from 3 independent experiments; (1) p=3*10−11. See also Figure S6. (D) Addition of 3 μM FF-MAS or 3 M 22(R)-hydroxycholesterol increased cytotoxicity of erlotinib in SCC25 cells. Data are represented as mean±SEM of 3 repeats; interaction, p=0.003 (2) and p=0.0002 (3), two-way ANOVA test. (E) Combined effects of erlotinib and 22(R)-hydroxycholesterol SCC61 and SCC25 cells; interaction, **, p<0.01. (F) Enumeration of erlotinib and 22(R)-hydroxycholesterol interactions in vitro. Shown are averaged Chou-Talalay CI values ±SEM from at 3 independent repeats. ED, effective dose; CI, combination index. See also Figure S6. (G) Effects of fatostatin on SREBP1, SREBP2 and LDLR expression in A431 cells following treatment of serum-starved cells with EGF (20ng/ml) and/or fatostatin for 18 hours. (H) Combined effects of erlotinib and fatostatin in A431 cells; interaction ***, p<10−3. (I) Enumeration of erlotinib and fatostatin interactions in vitro as in (E).
The PI3K mutation-positive squamous carcinoma cell line SCC61 (Seiwert et al., 2009) is refractory to EGFR inhibitors in vitro. Silencing of LDLR with validated siRNA (Fig. S5C) in these cells markedly sensitized them to the effect of erlotinib (Fig. 5C). Given the established effect of LXR agonists on depleting cancer cells of LDLR (Fig. 5A), predicted to deplete intracellular cholesterol (Fig. 4E and (Joseph et al., 2004)), we tested the combined treatment in erlotinib-refractory SCC25 and SCC61 cells, and in erlotinib-sensitive A431 cells. Both 22(R)-hydroxycholesterol and MAS sensitized the refractory and sensitive carcinoma cell lines to erlotinib (combination index, CI<1, Chou-Talalay method (Chou and Talalay, 1984), Fig. 5D–F, S5D).
The expression of LDLR is, in part, stimulated by activated SREBP1 (Guo et al., 2009). Therefore, inhibition of nuclear activated form of SREBP can potentially be exploited to deplete carcinoma cells of LDLR. We tested an established SREBP inhibitor, fatostatin (Kamisuki et al., 2009), which potently reversed the effect of EGF on the expression of nuclear fragments of SREBP1 and SREBP2, and depleted LDLR in A431 cells in vitro (Fig. 5G). Combined treatment with fatostatin and erlotinib produced synergistic cytotoxicity in the three squamous cell carcinoma lines tested (combination index, CI<1, Chou-Talalay method (Chou and Talalay, 1984), Fig. 5H, I).
Discussion
This study identifies the interaction between NSDHL and LXR as relevant to and targetable in the broad class of tumors with activated EGFR and RAS oncogenes. These results indicate that therapeutic exploitation of NSDHL with potentially little toxicity may be possible in adults despite earlier studies showing hypomorphic variants of SC4MOL (He et al., 2011) and NSDHL (McLarren et al., 2010) cause developmental defects, while alleles eliminating enzymatic activity cause arrest during early embryonic development (Caldas et al., 2005). However, the conditional knockout model of NSDHL employed in this study allowed us to circumvent the severe pre- and perinatal effects of NSDHL deficiency (Cunningham et al., 2015) and has indicated that loss of activity of the enzyme is tolerated in adult tissues, except when these tissues are induced to malignant growth by oncogenic transformation.
Earlier studies of NSDHL and SCMOL deficiency left unresolved the question of whether the MAS accumulation was itself responsible for observed biological effects, or whether these effects occurred as a consequence of changes in cholesterol pools. Reflecting the complexity of biological systems, these may be inseparable issues. An interesting aspect of this study, which defines the nuclear sterol-binding receptor LXR as a critical effector activated by the accumulating sterol metabolites, is that it now provides additional connections between NSDHL activity and cholesterol levels, beyond the straightforward role of C4-methylsterols as biosynthetic intermediates. Activated LXR causes deregulation of cholesterol uptake by increasing degradation of LDLR (Chawla et al., 2001; Guo et al., 2011), and transcriptional induction of ATP-binding cassette (ABC) transporters, known to negatively regulate the growth of hematopoietic (Bensinger et al., 2008; Yvan-Charvet et al., 2010) and tumor cells (Guo et al., 2011). Both in vivo and in carcinoma cell lines, we demonstrated that deficiency of SC4MOL or NSDHL activates canonical LXR targets such as ABCA1, ABCG1, and represses LDLR, which regulate cellular release and uptake of cholesterol, respectively. Our in vivo demonstration of a functional NSDHL-LXR interaction mechanistically links C4-demethylating enzymes to regulation of the growth of normal and malignant cells. Further extending the interest of SC4MOL and NSDHL function in oncogenesis, recent paper (Santori et al., 2015) used elegant biochemical experiments to identify a range of “unusual” SC4MOL- and NSDHL-catabolized derivatives of lanosterol that directly bind another nuclear receptor, the retinoid orphan receptor-γ (RORγ), to regulate maturation of Th17 lymphocytes, which play important roles in regulating tumor growth. In our study, the sterol metabolites accumulating in vivo in NSDHL-deficient cells are distinct from the reported RORγ ligands (Santori et al., 2015) (Table 1, Fig. S6). Our demonstration of LXRα activation (Fig. 4) in SC4MOL or NSDHL-deficient cells will need further determination of the specific sterol species bound to LXRα in the future.
In an earlier study, we showed that loss of NSDHL or SC4MOL affected EGFR signaling in cancer cells (Sukhanova et al., 2013). We here show that activated LXR showed synergy with EGFR antagonists, thus pointing out the cholesterol pathway as a critical target regulating the growth of human carcinomas. These results are interesting in the context of a recent study of glioblastoma in which activated EGFR was shown to positively regulate cholesterol uptake via LDLR and de novo cholesterol synthesis (Guo et al., 2009; Guo et al., 2011). The fact that we observed similar signaling interactions in KRAS-induced skin tumors extends the relevance of NSDHL-LXR signaling to the oncogenic KRAS, a major cancer causing gene currently not amenable to drug therapy. Previous attempts to disrupt cholesterol homeostasis in cancer with inhibitors of HMGCR (statins) have been limited in efficacy, due to extensive compensation for reduced HMGCR activity by increased cholesterol uptake and de novo synthesis (Clendening et al., 2010b; Dong et al., 2010). Arresting the cholesterol pathway more distally at the level of the C4-demethylating enzymes has the potential to create an irrevocable metabolic trap of combined blockade of cholesterol de novo synthesis and LXR-dependent blockade of uptake through LDLR and increased cholesterol efflux via ABCA1 and ABCG1. Detailed analysis of each of these regulatory elements will likely provide novel metabolic targets for cancer therapy.
Experimental Procedures
Cell Lines, Compounds and Antibodies
The A431 and FaDu cells were obtained from the American Type Culture Collection and maintained at the Fox Chase Cancer Center Cell Culture Facility (Philadelphia, PA). The identity of the A431 cell line was confirmed by single tandem repeat DNA profiling (Biosynthesis). The SCC61 and SCC25 cells were kindly provided by Dr. Tanguy Y. Seiwert (University of Chicago, Chicago, IL). All cell lines were mycoplasma free and maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% v/v FBS and L-glutamine with 100ug/ml Penicillin/Streptomycin.
Pre-senescent mouse embryonic fibroblasts (MEFs) were obtained from embryos at dpc13.5. We isolated Bpa1H/+ NSDHL-null MEFs from GFP-positive NSDHL-expressing cells by flow sorting as described (Cunningham et al., 2005). Cells were propagated in DMEM supplemented with 10% v/v FBS and L-glutamine with 100ug/ml Penicillin/Streptomycin and used at passages 4–6.
Erlotinib was obtained from the Fox Chase Cancer Center pharmacy; FF-MAS (follicular fluid meiosis-activating sterol) (14-demethyl-14-dehydrolanosterol, cat. no. 700077) was purchased from Avanti Polar Lipids, Inc.; 22(R)-OH-cholesterol (H9384) and EGF (E9644) were purchased from Sigma; compactin (sc-200853) and 2-Hydroxypropyl-β-cyclodextrin (sc-203461) were purchased from Santa Cruz; fatostatin (#341329) was purchased from Calbiochem (EMD Millipore); LDS was prepared as described in (Cunningham et al., 2005). For Western blot and Immunohistochemistry experiments we used the following antibodies from Abcam: SREBP2 (ab30682), ABCA1 (ab7360), LDLR (ab30531), LXRα (ab41902), LXRβ (ab106473); from Cell Signaling: E-cadherin (#3195P), β-actin (#4967S), α-tubulin (#3873S), HA-tag (#2367S); from Proteintech Group: NSDHL (#15111-2-AP); from Santa Cruz: SREBP1 (sc-13551); from Dako: Ki67 (M7249). Cholesterol in total cellular lysates was determined by Amplex Red kit from Life Technologies.
Quantitative RT-PCR
For evaluation of target gene knockdown, cells were reverse transfected in 6-well plates, and total RNA was extracted using the RNeasy Mini Kit (Qiagen) 48 hours after transfection. Quantitative reverse-transcriptase PCR reactions were conducted using TaqMan probes and primers designed by the manufacturer, using an ABI PRISM 7700 Detection System (Applied Biosystems). The results were analyzed with the comparative Ct method to establish relative expression curves.
Mouse models
Mice carrying the conditional knockout allele of Nsdhl (Cunningham et al., 2015) were kindly provided by Dr. Gail Herman (The Research Institute at Nationwide Children’s Hospital, Columbus, OH). These mice (official name: Nsdhltm1.1Hrm, MGI:5581334, to be designated as Nsdhlflx5 here) are congenic on a C57BL/6J background and were bred and kept under defined-flora pathogen-free conditions at the AAALAC-approved Animal Facility of the Fox Chase Cancer Center, Philadelphia, PA. The 4-week old K5CreERT;LSL-KrasG12D;Nsdhlflx5 mice (Supplemental Methods and Fig. S1A) at their 1st anagen (Nowak et al., 2008) were treated with tamoxifen (Sigma, T5648) dissolved in corn oil (Sigma, C8267) at 40mg/kg by oral gavage for 5 consecutive days. Mice were observed once weekly until first tumors formed. Papilloma volumes were assessed twice a week as (length × width2)/2. Mice were followed for at least 5 months, or until average papilloma volumes exceeded 2,000 mm3, ulceration occurred, or animals showed distress or weight loss more than 10%, per the local Institutional Animal Care and Use Committee guidelines. See also Supplemental Experimental Procedures.
Mouse skin tissue collection and immunohistochemistry
Skin biopsy of the adult animals was taken from the dorsal side of the mouse under anesthesia with Isoflurane and fixed in formalin for the IHC. Newborn pups were sacrificed the same day after the birth by CO2 inhalation and skin samples were collected. Skin samples were cut into 3 parts, where the first one was fixed in formalin for further IHC, RNA was isolated from the second part by using the TRIzol (Life technologies #15596-026) according to the manufacturer’s protocol and for Western blot analysis the third part of tissue sample was homogenized in RIPA buffer (Santa Cruz #24928) with phosphotase and protease inhibitors (Thermo Scientific #1862495, #1861278) on ice and cleared then by centrifugation. Small (1–2mm) pieces from mouse tails were used for genotyping (Supplemental methods). For IHC fixed skin was embedded in paraffin and stained with indicated antibodies diluted per the manufacturer’s instructions. Primary antibody binding was amplified using a Vectastain Elite ABC Kit (Vector Laboratories), including biotinylated anti-rabbit secondary antibody. Antibody binding was visualized using the Liquid DAB+ Substrate Chromogen System (Dako). Samples were counterstained for 1 minute with hematoxylin. Immunostained slides were scanned by an Aperio ScanScope CS scanner (Aperio) and selected regions of interest were outlined manually. Expression levels of NSDHL, ABCA1, LDLR, Ki67, skin thickness and sebaceous glands areas were measured using the ImageScope software.
siRNA Transfections
The siRNA targeting human sterol biosynthesis genes and controls were obtained from Qiagen (Supplemental methods). Cells were transfected in triplicates with siRNA at 5 nmol/L concentrations mixed with HiPerFect Transfection Reagent (Qiagen) according to the manufacturer’s reverse transfection protocol.
In Vitro Viability Assays
For in vitro viability assays cells were plated in 96 well plate ~3000 cells/well and treated with drugs according to experiment conditions. The viability was measured in 96 hours using CellTiter-Blue Viability Assay (Promega). A combination index (CI) was calculated from drug cytotoxicity or growth inhibition curves. Corresponding dilutions of ethanol and DMSO were added as vehicle and did not affect the viability, compared with that of untreated cells. To calculate a CI, the computer software Calcusyn can be used, taking the entire shape of the growth inhibition curve into account for calculating whether a combination is synergistic, additive, or antagonistic.
LXRα luciferase reporter assay
LXRα luciferase reporter assay (Qiagen, Cignal Lenti Reporter Assay LXRα Reporter Assay (luc)) was performed on primary MEFs cell culture. Cells were plated in the 96-well plate in concentration 105 cells per well in 100 μl of the 10%FBS/DMEM/25mM HEPES/L-glutamin medium. In 24 hrs cells were infected with lentivirus containing LXRα luciferase reporter construct (Qiagen, 336851 CLS-7041L-1) with MOI=10. To increase efficiency of infection Polybrene (Santa-Cruz, sc-134220) was added at final concentration 8 μg/ml. In 72 hrs after infection cells were washed with PBS once and lysed in Passive Lysis Buffer (Promega, E194A). Cell lysates were collected then and the level of luciferase expression was evaluated by luminescence intensity in chemical reaction with luciferase substrate (Promega, E151A).
Growth curves of primary MEFs
To determine the growth rate of primary MEFs cells were propagated in 10%FBS/DMEM/25mM HEPES/L-glutamin medium with Penicillin/Strepromycin. Early-passage MEFs (passage 4) were used for growth curve analysis. 3 × 105 cells were cultured and every three days cells were harvested by trypsinization and number of cells was counted using the cell counter, 3 × 105 cells was plated back again every time. Cumulative amount of cells was estimated by multiplying the number of cells from the previous passage by the ratio between the number of harvested and plated cells.
Senescence-Associated β-Galactosidase
SA-β-Gal activity was detected with Senescence beta – Galactosidase Staining Kit (Cell Signaling #9860S) according to the manufacturer’s protocol. Briefly, cells, grown on the glass coverslips, were washed with PBS and fixed in glutaraldehyde fixative solution for 15 min. Then cells were washed again in PBS and stained in X-gal solution overnight at 37°C.
LXRα and LXRβ expression
LXRα and LXRβ coding plasmid (DNASU Plasmid Repository, NR1H3 and NR1H2 cDNA in pDONR221 vector, HsCD00296058 and HsCD00042671 clones, respectively) were cloned in pLEX-HA-MYC plasmid vector (Thermo Scientific Open Biosystems, OHS4492) by using Gateway cloning system (Invitrogen) according to the manufacturer’s protocol. Then lentiviruses, containing NR1H3 or NR1H2, were generated and expanded in HEK293T cells by using Trans-Lentiviral Packaging Mix (Thermo Scientific, TLP4606) and Express-In Transfection Reagent (Thermo Scientific Open Biosystems, ETR4620-4623). SCC61 cells were treated with obtained viruses and infected clones were selected then with 2 μg/ml Puromycin (Santa Cruz #108071). For control cell line, pLEX-JRed-TurboGFP Control Vector (Thermo Scientific Open Biosystems, OHS4625) was packed in the same lentivirus (Trans-Lentiviral Packaging Mix, Thermo Scientific, TLP4606)). SCC61 cells were treated with obtained GFP-coding virus and infected clones were selected with 2 μg/ml Puromycin. LXRα overexpression was confirmed by Western Blot analysis of cell lysates.
Sterols gas chromatography-mass spectrometry
Sterol analysis was performed using ion-ratio GC/MS on an Agilent 6390N/5973 GC/MS system as previously described (Kelley, 1995) with modifications to the GC/MS method to include ions for additional intermediates in the cholesterol biosynthetic pathway between lanosterol and cholesterol.
Statistical Analysis
For analysis of skin samples for biomarkers expression, skin thickness and sebaceous glands proliferation, papilloma growth and the level of NSDHL expression in the tumors we used Wilcoxon test. Growth curves, unless otherwise indicated, were modeled using linear regression with interactions between exposure and concentration. All clustered or repeated data was accounted for using linear regression with Generalized Estimating Equations (Liang and Zeger, 1986).
Supplementary Material
Acknowledgments
We are grateful to Catherine Reiner for technical assistance with histological analyses and immunohistochemistry experiments and to Dr. Marc Poirot (University of Toulouse, France) for critical comments on the work.
Grant Support: This work was supported by NIH core grant CA-06927, by the Pew Charitable Fund, and by a generous gift from Mrs. Concetta Greenberg to Fox Chase Cancer Center. Some of the authors were supported by NIH R01 CA188430, K22 CA160725, R21 CA164205, a career development award from Genentech; by Tobacco Settlement funding from the State of Pennsylvania (IA), and by a grant from the Bucks County Board of Associates (LG, IA); by NIH R01 HD38572 (GEH) and R01 CA63366 (EAG); and by the Program of Competitive Growth of Kazan Federal University (LG); cloning experiments were supported by the grant from Russian Scientific Foundation (project 15-15-20032) to IA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions: L.G., D.R., E.G. and I.A. designed research; L.G., A.G., K.M., E.H., A.K.S., K.Q.C., D.R., L.E.K, and D-H.Y. performed research; L.E.K, D-H.Y., D.C. and G.E.H. contributed new reagents/analytical tools; L.G., A.G., D-H. Y., A. K-S., L.E.K., E.H. and I.A. analyzed data; L.G., G.E.H, E.G. and I.A. wrote the paper.
References
- Acimovic J, Rozman D. Steroidal triterpenes of cholesterol synthesis. Molecules. 2013;18:4002–4017. doi: 10.3390/molecules18044002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, Devarajan K, Silverman JS, Tikhmyanova N, Skobeleva N, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Science signaling. 2010;3:ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA, Jamieson BD, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byskov AG, Andersen CY, Nordholm L, Thogersen H, Xia G, Wassmann O, Andersen JV, Guddal E, Roed T. Chemical structure of sterols that activate oocyte meiosis. Nature. 1995;374:559–562. doi: 10.1038/374559a0. [DOI] [PubMed] [Google Scholar]
- Caldas H, Cunningham D, Wang X, Jiang F, Humphries L, Kelley RI, Herman GE. Placental defects are associated with male lethality in bare patches and striated embryos deficient in the NAD(P)H Steroid Dehydrogenase-like (NSDHL) Enzyme. Molecular genetics and metabolism. 2005;84:48–60. doi: 10.1016/j.ymgme.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molecular cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, Martirosyan A, Hakem A, Hakem R, Jurisica I, et al. Dysregulation of the mevalonate pathway promotes transformation. Proceedings of the National Academy of Sciences of the United States of America. 2010a;107:15051–15056. doi: 10.1073/pnas.0910258107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clendening JW, Pandyra A, Li Z, Boutros PC, Martirosyan A, Lehner R, Jurisica I, Trudel S, Penn LZ. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood. 2010b;115:4787–4797. doi: 10.1182/blood-2009-07-230508. [DOI] [PubMed] [Google Scholar]
- Cunningham D, DeBarber AE, Bir N, Binkley L, Merkens LS, Steiner RD, Herman GE. Analysis of hedgehog signaling in cerebellar granule cell precursors in a conditional Nsdhl allele demonstrates an essential role for cholesterol in postnatal CNS development. Human molecular genetics. 2015;24:2808–2825. doi: 10.1093/hmg/ddv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham D, Swartzlander D, Liyanarachchi S, Davuluri RV, Herman GE. Changes in gene expression associated with loss of function of the NSDHL sterol dehydrogenase in mouse embryonic fibroblasts. Journal of lipid research. 2005;46:1150–1162. doi: 10.1194/jlr.M400462-JLR200. [DOI] [PubMed] [Google Scholar]
- Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. Journal of lipid research. 2010;51:1486–1495. doi: 10.1194/jlr.M003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, Lin KY, Huang TT, Akhavan D, Hock MB, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Science signaling. 2009;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, et al. An LXR agonist promotes GBM cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer discovery. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Kratz LE, Michel JJ, Vallejo AN, Ferris L, Kelley RI, Hoover JJ, Jukic D, Gibson KM, Wolfe LA, et al. Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay. The Journal of clinical investigation. 2011;121:976–984. doi: 10.1172/JCI42650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:11603–11607. doi: 10.1073/pnas.90.24.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel M, Cunningham D, Mullett CJ, Kelley RI, Herman GE. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. American journal of medical genetics Part A. 2003;122A:246–251. doi: 10.1002/ajmg.a.20248. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’Connell R M, Cheng G, Saez E, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chemistry & biology. 2009;16:882–892. doi: 10.1016/j.chembiol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Kelley RI. Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clinica chimica acta; international journal of clinical chemistry. 1995;236:45–58. doi: 10.1016/0009-8981(95)06038-4. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Angel JM, Sano S, DiGiovanni J. Constitutive activation and targeted disruption of signal transducer and activator of transcription 3 (Stat3) in mouse epidermis reveal its critical role in UVB-induced skin carcinogenesis. Oncogene. 2009a;28:950–960. doi: 10.1038/onc.2008.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DJ, Kataoka K, Sano S, Connolly K, Kiguchi K, DiGiovanni J. Targeted disruption of Bcl-xL in mouse keratinocytes inhibits both UVB- and chemically induced skin carcinogenesis. Molecular carcinogenesis. 2009b;48:873–885. doi: 10.1002/mc.20527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KY, Zeger SL. Longitudinal Data-Analysis Using Generalized Linear-Models. Biometrika. 1986;73:13–22. [Google Scholar]
- Liu XY, Dangel AW, Kelley RI, Zhao W, Denny P, Botcherby M, Cattanach B, Peters J, Hunsicker PR, Mallon AM, et al. The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase. Nature genetics. 1999;22:182–187. doi: 10.1038/9700. [DOI] [PubMed] [Google Scholar]
- McLarren KW, Severson TM, du Souich C, Stockton DW, Kratz LE, Cunningham D, Hendson G, Morin RD, Wu D, Paul JE, et al. Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome. American journal of human genetics. 2010;87:905–914. doi: 10.1016/j.ajhg.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S. Lipid rafts: elusive or illusive? Cell. 2003;115:377–388. doi: 10.1016/s0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- Nes WD. Biosynthesis of cholesterol and other sterols. Chem Rev. 2011;111:6423–6451. doi: 10.1021/cr200021m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak JA, Polak L, Pasolli HA, Fuchs E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell stem cell. 2008;3:33–43. doi: 10.1016/j.stem.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Su YQ, Ariga M, Law E, Jin SL, Conti M. EGF-like growth factors as mediators of LH action in the ovulatory follicle. Science. 2004;303:682–684. doi: 10.1126/science.1092463. [DOI] [PubMed] [Google Scholar]
- Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell. 2014;156:986–1001. doi: 10.1016/j.cell.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Pitroda SP, Khodarev NN, Beckett MA, Kufe DW, Weichselbaum RR. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5837–5841. doi: 10.1073/pnas.0812029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell metabolism. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santori FR, Huang P, van de Pavert SA, Douglass EF, Jr, Leaver DJ, Haubrich BA, Keber R, Lorbek G, Konijn T, Rosales BN, et al. Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell metabolism. 2015;21:286–297. doi: 10.1016/j.cmet.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiwert TY, Jagadeeswaran R, Faoro L, Janamanchi V, Nallasura V, El Dinali M, Yala S, Kanteti R, Cohen EE, Lingen MW, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer research. 2009;69:3021–3031. doi: 10.1158/0008-5472.CAN-08-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvente-Poirot S, Poirot M. Cancer. Cholesterol and cancer, in the balance. Science. 2014;343:1445–1446. doi: 10.1126/science.1252787. [DOI] [PubMed] [Google Scholar]
- Sukhanova A, Gorin A, Serebriiskii IG, Gabitova L, Zheng H, Restifo D, Egleston BL, Cunningham D, Bagnyukova T, Liu H, et al. Targeting C4-demethylating genes in the cholesterol pathway sensitizes cancer cells to EGF receptor inhibitors via increased EGF receptor degradation. Cancer discovery. 2013;3:96–111. doi: 10.1158/2159-8290.CD-12-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, Sanvito F, Ponzoni M, Valentinis B, Bregni M, et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nature medicine. 2010;16:98–105. doi: 10.1038/nm.2074. [DOI] [PubMed] [Google Scholar]
- Wang X, Zinkel S, Polonsky K, Fuchs E. Transgenic studies with a keratin promoter-driven growth hormone transgene: prospects for gene therapy. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:219–226. doi: 10.1073/pnas.94.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC, et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. The Journal of biological chemistry. 2006;281:27816–27826. doi: 10.1074/jbc.M603781200. [DOI] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.