

Abstract
β-Diketiminates are widely used supporting ligands for building a range of metal complexes with different oxidation states, structures, and reactivities. This Perspective summarizes the steric and electronic influences of ligand substituents on these complexes, with an eye toward informing the design of new complexes with optimized properties. The backbone and N-aryl substituents can give significant steric effects on structure, reactivity and selectivity of reactions. The electron density on the metal can be tuned by installation of electron withdrawing or donating groups on the β-diketiminate ligand as well. Examples are shown from throughout the transition metal series to demonstrate different types of effects attributable to systematic variation of β-diketiminate ligands.
Keywords: β-diketiminate, steric, electronic
Graphical Abstract
We summarize steric and electronic influences on structure, spectroscopy, and reactivity in transition metal β-diketiminate complexes.

1. Introduction
The properties and reactions of metal complexes are highly dependent on the choice of supporting ligand, and this choice is one of the keys to successful coordination chemistry. Since its introduction in 1968,1-3 the β-diketiminate (often called “nacnac” because of its addition of two nitrogen atoms to the common acac ligand) has gained great popularity as a supporting ligand. Unlike acetylacetonate (acac), the β-diketiminate ligand scaffold offers steric protection at the metal center through the choice of N-substituents; this makes β-diketiminates less labile and more suitable as spectator ligands. β-Diketiminate ligands are typically synthesized from condensation of a β-diketone and an amine, and chemists have only scratched the surface of the thousands of potential combinations.4
N-aryl β-diketiminate ligands have been most widely used, and they support a variety of metals in many oxidation states. Complexes of N-aryl β-diketiminates have shown great reactivity and selectivity for a variety of methodologies,4, 5 including polymerization and functionalization of alkenes and cross-coupling reactions. In addition, late transition metal β-diketiminate complexes have been used to build low coordinate metal centers, mimicking the active sites of metalloproteins.6-14 A vast number of ligand variations and different coordination modes have been reported, and some examples are shown in Figure 1.1. In this Perspective, the focus will be solely on complexes of the type shown in Figure 1.1 with d block transition metals in a η2 binding mode. We summarize trends from systematic variations in these complexes with examples, though we make no claim that our coverage is complete. This Perspective is intended to serve as a guide to chemists who are interested in tuning the properties of β-diketiminate complexes to achieve their specific goals. We also refer the interested reader to another Perspective by Budzelaar which gives more depth on N-aryl β-diketiminate complexes of Ru, Os, Rh, Ir, Pd, and Pt.15
Figure 1.1.
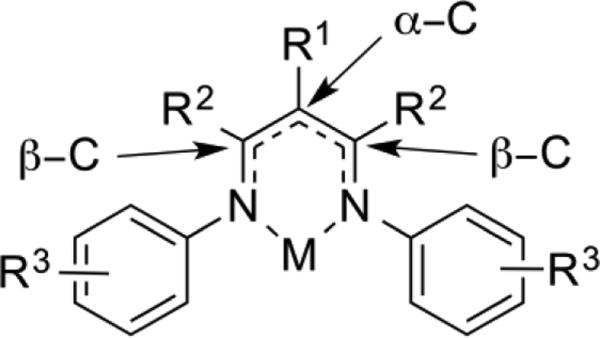
Substituent patterns in β-diketiminate ligands.
2. Nomenclature
In this Perspective, the ligand abbreviation R1LR2,R3 is used to specify the substituents on a β-diketiminate ligand. R1 refers to the substituent on the central backbone carbon (α-C), R2 refers to the substituents on the nitrogen-bearing carbon atoms (β-C), and R3 refers to the substituents on the N-aryl group. For the R3 aryl substituents meta- and para- substitutions of N-aryl are specified as m- and p-, respectively, while the common ortho-substituents are given without the o- abbreviation for convenience. Some other abbreviations can be found in Chart 1.1.
Chart 1.1.
Abbreviations used in this Perspective
| Dipp | 2,6-diisopropylphenyl |
| Tipp | 2,4,6-triisopropylphenyl |
| Dep | 2,6-diethylphenyl |
| Mes | 2,4,6-trimethylphenyl |
| An | 1-anthracenyl |
| ArF | 3,5-bis(trifluoromethyl)phenyl |
| Tbt | 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl |
3. Steric effects on β-diketiminates
The steric demands of β-diketiminate ligands can be tuned by substitution of functional groups on the backbone (β-C) or the N-aryl substituents. Typical backbone (β-C) substituents are tert-butyl, phenyl, trifluoromethyl and methyl; unsubstituted (β-dialdiminate) ligands are also known. Two approaches can be used to tune the sterics of the N-aryl groups: first, to change the size of ortho-substituents on the N-aryl; or second, to relocate the substituents from ortho- position to the meta- or para- position.
The modification of β-diketiminate steric hindrance can bring changes in the structure and reactivity. The structural differences include changes on the coordination number, bond angles and bond lengths, geometry and conformation of metal complexes. We highlight three types of reactivity differences: different structures of β-diketiminate complexes, different outcomes of stoichiometric reactions of β-diketiminate complexes, and different activity in catalytic reactions.
3.1. Steric effects on structural properties
Generally, using smaller substituents on the β-C and N-aryl, or relocation of the N-aryl substituents farther from the metal center, reduces the overall steric coverage of the metal coordination sphere. As a result, dimeric/polymeric metal complexes are more often formed with less sterically hindered β-diketiminate ligands. For example, comparisons with more hindered monomeric analogues were reported for [LScCl2]n (LtBu,iPr, 16 n=1; LMe,iPr,17 n=2), [LSc(CH3)2]n (LtBu,iPr, 16 n=1; LMe,iPr,17 n=2), [LFeCl]n (LtBu,iPr,18 n=1; LMe,iPr,19 MeLMe,Me,20 n=2), [LFeF]n (LtBu,iPr, n=1; LMe,iPr, n=2),21 [LCoCl]n (LtBu,iPr,22 n=1; LMe,iPr,23 n=2), [LNiCl]n (LtBu,iPr,22 n=1; LMe,iPr,24 LMe,Me,25 n=2), [LNi(CO)]n, (LtBu,iPr,26 LMe,iPr,27 n=1; LMe,Me,28 n=2), [LR,iPrCuCl]n (LMe,iPr,29 ClLMe,iPr,29 n=1; PhLH,iPr,30 LMe,Cl,31 n=2), and [LPd(μ-OAc)]n (LMe,iPr,32 n=1; LMe,H32 ClLMe,H,33 n=2). The angle between the two β-diketiminate ligand planes in dimeric metal complexes is often influenced by the different substituents on the ligand (Table 3.1.1). However, there is no clear correlation between the substituent size and the angle, indicating that this angle is dependent on the bonding at the metal as well as steric interactions between the ligands on the two sides.
Table 3.1.1.
Selected examples of steric effects on ligand plane orientation of bimetallic complexes complexes
| Complex | Ligand | Dihedral angle between two ligand planes | Reference |
|---|---|---|---|
| [LV]2 | LMe,An | 65.59° | 34 |
| LMe,Et | 0° | 34 | |
| LMe,Me | 0° | 34 | |
| [LCr(μ-Cl)]2 | LtBu,iPr | 32.41° | 35 |
| LMe,iPr | 0° | 36 | |
| LMe,Me | 0° | 37 | |
| LCr(η5-Cp)(μ-O)Cr(η5-Cp)L | LMe,Me | 9.14° | 38 |
| LMe,m-TIPP | 17.27° | 38 | |
| [LFe(μ-H)]2 | LtBu,iPr3 | 66.70° | 39 |
| LtBu,iPr | 68.92° | 40 | |
| LMe,iPr | 71.15° | 21 | |
| MeLMe,Me | 82.38° | 20 | |
| LFe(tBuPy)(NN)Fe(tBuPy)L | LtBu,iPr | 81.68° | 41 |
| LMe,iPr | 50.04° | 41 | |
| LFeNNFeL | LtBu,iPr | 87.18° | 6 |
| LMe,iPr | 0.00° | 41 | |
| [LFeNNFeL]K2 | LtBu,iPr | 35.7° | 6 |
| LMe,iPr | 34.3° | 41 | |
| LNi(P4)NiL | LMe,iPr | 39.96° | 42 |
| LMe,Et | 51.24° | 42 | |
| [LCu(μ-Cl)]2 | LMe,Et | 0.00° | 43 |
| LMe,Cl | 81.37° | 31 | |
| ClLMe,Me | 74.96° | 43 | |
| [LCu(μ-OH)]2 | LCF3,Me | 60.03° | 44 |
| LMe,Me | 0.00° | 45 | |
| CNLH,Et | 0.00° | 46 | |
| CNLH,Me3 | 11.34° | 43 | |
| NO2LH,Me3 | 40.86° | 30 |
One trend that emerges is that higher coordination numbers can be achieved with smaller β-diketiminate supporting ligands. For example, more solvent molecules (THF, arene, etc.) and neutral ligands (CO, PPh3, etc.) can be coordinated to a metal center with less sterically hindered β-diketiminate in LScCl2(THF)n (LtBu,iPr,16 n=0; LMe,iPr,47 n = 1) LSc(CH3)2(THF)n (LtBu,iPr,16 n=0; LMe,iPr,16 n = 1), LSc(Cl)(NHAr)(THF)n LtBu,iPr,48 n=0; LMe,iPr,49 n = 1), [LSc(CH3)(arene)n]+ (LtBu,iPr,50 n=0; LMe,iPr,50 n = 1), LTiCl2(THF)n(LtBu,iPr,51 LtBu,Me3,52 LMe,Tbt/Me3,53 n=0; LMe,iPr,54 n=1; LMe,H,55 n=2), LVCl2(THF)n(LMe,iPr,52, 56 LMe,Et,34LMe,Me3,34 LPh,iPr,34 n=0; LMe, H,55 n=2), [LCr(μ-Cl)(Solvent)n]2 (LtBu,iPr,35 n=0; LMe,iPr,36 LMe,Me,37 n = 1; Solvent = THF, benzene), LFe(NHdipp)(THF)n (LtBu,iPr,19 n=0; LMe,iPr,28 n = 1), and LCu(PPh3)n (PhLH,iPr,57 LMe,Me,58LMe,iPr,59LMe,Me3,60 n=1; PhLH,Me,57 LCF3,m-CF3,61 n=2). Steric conflict between N-aryl substituents and metal can also push the metal center out of the β-diketiminate ligand plane in some metal complexes, especially for early transition metals (Table 3.1.2). However, exceptions can be found in LR,MesTiCl2,52 LMe,RCr(η5-Cp),62, 63 LR,iPrFeNNFeL,6, 41 [LMe,RNi(μ-Cl)]2,24, 25 LMe,RCu(OAc),64, 65 [LCu(μ-OH)]2,44-46 [LCu(μ-S)]2,66, 67 and LR,iPr Cu(CO).68
Table 3.1.2.
Selected examples of steric effect on distance of metal to ligand plane
| Complex | Ligand | Distance from M to ligand plane (Å) | Reference |
|---|---|---|---|
| LScCl2(THF)n | LtBu,iPr | 1.295 | 16 |
| LMe,iPr | 0.694 | 47 | |
| LSc(alkyl)2 | LtBu,iPr | 1.154 | 16 |
| LMe,iPr | 1.116 | 16 | |
| LMe,m-tBu | 0.489 | 69 | |
| LMe,m-Tipp | 0.204 | 69 | |
| LZrCl3 | LtBu,iPr | 1.650 | 70 |
| LMe,iPr | 0.820 | 71 | |
| LVCl2(THF)n | LMe,Me | 0.528 | 52 |
| LMe,H | 0.227 | 55 | |
| LCr(Cp)(Me) | LMe,iPr | 0.702 | 62 |
| LMe,Et | 0.699 | 72 | |
| LMe,Me | 0.650 | 72 | |
| LCr(Cp)(Cl) | LMe,iPr | 0.719 | 62 |
| LMe,Et | 0.751 | 72 | |
| LMe,Me | 0.680 | 63 | |
| LMe,H | 0.087 | 73 | |
| LCr(Cp)(μ-O)Cr(Cp)L | LMe,Me | 0.858, 0.848 | 38 |
| LMe,m-TIPP | 0.771, 0.726 | 38 | |
| [LCr(μ-Cl)(THF)]2 | LMe,iPr | 0.668 | 36 |
| LMe,Me | 0.554 | 37 | |
| [LFe(μ-H)]2 | LtBu,iPr | 0.565 | 40 |
| LMe,iPr | 0.540 | 21 | |
| MeLMe,Me | 0.260 | 20 | |
| LFe(μ-H)2B(Et)2 | LtBu,iPr | 0.093 | 74 |
| LMe,iPr | 0.000 | 74 | |
| LFe(μ-Cl)2Li(THF)2 | LMe,iPr | 0.381 | 18 |
| LMe,Me3 | 0.000 | 20 | |
| LFe(F)(tBuPy) | LtBu,iPr | 0.339 | 21 |
| LMe,iPr | 0.294 | 21 | |
| LFe(tBuPy)(NN)Fe(tBuPy)L | LtBu,iPr | 0.394, 0.553 | 41 |
| LMe,iPr | 0.250, 0.250 | 41 | |
| LFe-(η3-N3Ad) | LtBu,iPr | 0.762 | 75 |
| LMe,iPr | 0.753 | 75 | |
| [LFeNNFeL]K2 | LtBu,iPr | 0.290, 0.111 | 6 |
| LMe,iPr | 0.072, 0.004 | 41 | |
| LFe-alkyl | LtBu,iPr | 0.065 | 76 |
| LMe,iPr | 0.019 | 77 | |
| LFe-alkyne | LtBu,iPr | 0.097 | 78 |
| LMe,iPr | 0.008 | 79 | |
| LCo(μ-Cl)2Li(THF)2 | LtBu,iPr | 0.362 | 22 |
| LMe,iPr | 0.314 | 80 | |
| LNi(P4)NiL | LMe,iPr | 0.184, 0.184 | 42 |
| LMe,Et | 0.215, 0.030 | 42 | |
| LCu(CNAr) | LMe,iPr | 0.342 | 29 |
| LMe,Me | 0.144 | 81 | |
| [LCu(μ-S)]2 | PhLH,iPr | 0.349 | 67 |
| PhLH,Et | 0.302 | 67 | |
| ArFLH,iPr | 0.271 | 67 | |
| ArFLH,Me | 0.002 | 67 | |
| LCu(NCCH3) | LtBu,iPr | 0.046 | 8 |
| LCF3,iPr | 0.028 | 82 | |
| LCF3/Me,iPr | 0.022 | 82 | |
| LRu(Cl)(η6-Benzene) | LCF3,Me | 0.624 | 83 |
| LMe,Me | 0.635 | 84 | |
| LCF3, m-CF3 | 0.246 | 83 | |
| LMe, m-Me | 0.207 | 85 | |
| LMe,H | 0.048 | 83 | |
| LRu(Cl)(η5-Cp*) | LMe,Me | 0.628 | 86 |
| LMe,m-Me | 0.343 | 86 |
When the backbone (β-C) substituent size increases (H < Me < CF3 < tBu, Ph), the steric conflict between backbone (β-C) substituents and N-aryl groups escalates, pushing the N-aryl rings closer to the metal and forcing them into a more rigid configuration. As a consequence of this “buttressing effect,” the metal center often moves deeper into the β-diketiminate binding pocket. This brings three changes to the structure: it typically increases the N-M-N bite angle, increases the C(aryl)-N-C(β) bond angle, and shortens the N-M bond length (see Table 3.1.3). Bulky substituents on the N-aryl may also affect the bonding to other ligands (see Table 3.1.4). Exceptions to this trend, however, are seen with LTiCl2,52 LZrCl3,70, 87 [LCr(μ-Cl)]2, and K2[LFeNNFeL],6, 41 due to cation coordination or conformational changes at the metal center. The distances from the metal to the non-diketiminate co-ligand can also be affected by the backbone substituents (see ESI for details).
Table 3.1.3.
Steric effects of backbone (β-C) substituents on structural properties
| Complex | Ligand | N-M-N Bite angle | C(aryl)-N-C(β) bond angle | M-N distance (Å) | Reference |
|---|---|---|---|---|---|
| LScCl2(THF)n | LtBu,iPr | 95.9° | 125.3° 126.9° |
2.046 2.099 |
16 |
| LMe,iPr | 86.8° | 116.9° 117.8° |
2.107 2.175 |
47 | |
| LSc(alkyl)2 | LtBu,iPr | 93.5° | 125.5° 126.2° |
2.091 2.144 |
16 |
| LMe,iPr | 90.7° | 120.1° 120.8° |
2.113 2.133 |
16 | |
| LFe(μ-H)2BEt2 | LtBu,iPr | 97.35° | 127.80° 129.28° |
1.971 1.969 |
74 |
| LMe,iPr | 95.91° | 120.58° | 1.971 | 74 | |
| LFeX | LtBu,iPr | 96.35° | 128.39° | 1.946 | 18 |
| LMe,iPr | 94.50° | 116.61° 116.72° |
2.002 2.006 |
19 | |
| LFe(F)(tBuPy) | LtBu,iPr | 97.80° | 124.80° 126.43° |
2.015 2.007 |
21 |
| LMe,iPr | 95.00° | 118.38° 119.53° |
2.012 2.009 |
21 | |
| LFe(tBuPy)(NN) Fe(tBuPy)L |
LtBu,iPr | 99.23° 97.33° |
123.02° 124.13° 124.22° 124.76° |
2.005 2.000 |
41 |
| LMe,iPr | 95.86° | 118.59° 119.99° |
2.005 1.993 |
41 | |
| LFe(N3Ad) | LtBu,iPr | 98.84° | 123.88° 123.39° |
2.043 2.018 |
75 |
| LMe,iPr | 97.95° | 118.34° 117.40° |
2.021 2.016 |
75 | |
| LFeNNFeL | LtBu,iPr | 96.01° | 129.11° 127.00° |
1.965 1.970 |
6 |
| LMe,iPr | 94.78° | 121.57° 118.66° |
1.945 1.984 |
41 | |
| LFeiPr | LtBu,iPr | 94.25° | 126.33° 128.11° |
1.990 1.989 |
76 |
| LMe,iPr | 92.78° | 119.84° 120.60° |
1.983 1.983 |
77 | |
| LFe-(η2-PhCΞCH) | LtBu,iPr | 96.16° | 123.65° 124.62° |
1.975 2.005 |
78 |
| LMe,iPr | 93.67° | 119.31° 118.57° |
1.973 1.990 |
79 | |
| LCo(μ-Cl)2Li(THF)2 | LtBu,iPr | 99.42° | 124.78° 125.81° |
1.968 1.961 |
22 |
| LMe,iPr | 98.19° | 120.23° 120.38° |
1.957 1.962 |
80 | |
| LCo(alkyl) | LtBu,iPr | 97.68° | 127.59° 125.04° |
1.960 1.950 |
88 |
| LMe,iPr | 95.60° | 119.70° 118.82° |
1.948 1.946 |
89 | |
| LNi(CO) | LtBu,iPr | 98.85° | 126.33° 129.40° |
1.924 1.856 |
26 |
| LMe,iPr | 96.41° | 119.89° 122.58° |
1.917 1.868 |
27 | |
| LCu(η2-OAc) | CNLMe,iPr | 96.63° | 119.68° 120.45° |
1.905 1.914 |
90 |
| CNLH,iPr | 94.79° | 116.9° 116.9° |
1.944 1.944 |
46 | |
| [LCu(μ-OH)]2 | LCF3,Me | 95.28° | 122.69° 122.87° |
1.940 1.943 |
44 |
| LMe,Me | 94.83° | 117.36° 117.61° |
1.937 1.945 |
45 | |
| LCu(NCCH3) | LtBu,iPr | 102.33° | 128.75° 127.68° |
1.936 1.931 |
8 |
| LCF3,iPr | 98.98° | 124.74° 125.00° |
1.940 1.935 |
68 | |
| LMe,iPr | 98.98° | 118.94° 119.21° |
1.940 1.942 |
8 | |
| PhLH,iPr | 97.25° | 118.46° 116.59° |
1.964 1.950 |
8 | |
| LRu(Cl)( η5-Cp*) | LCF3,m-Me | 90.18° | 118.55° 118.42° |
2.069 2.055 |
86 |
| LMe,m-Me | 87.83° | 116.43° 115.98° |
2.050 2.051 |
86 | |
| LCF3,m-CF3 | 89.67° | 117.47° 118.21° |
2.070 2.071 |
86 | |
| LMe,m-CF3 | 87.99° | 114.91° 115.46° |
2.071 2.071 |
86 | |
| LRu(η5-Cp*) | LCF3,m-Me | 90.08° | 116.95° 117.42° |
2.050 2.050 |
86 |
| LMe,m-Me | 87.92° | 115.62° 115.29° |
2.060 2.063 |
86 | |
| LCF3,m-CF3 | 89.55° | 116.09° 116.53° |
2.055 2.056 |
86 | |
| LMe,m-CF3 | 87.37° | 114.08° 114.07° |
2.045 2.040 |
86 |
Table 3.1.4.
Steric effects of N-aryl substituents on structural properties
| Complex | Ligand | N-M-N bite angle | M-N Distance (Å) | C(aryl)-N-C(β) bond angle | Selected bond length (Å) | Reference |
|---|---|---|---|---|---|---|
| LSc(CH2TMS)2 | LMe,iPr | 90.7° | 2.113 2.133 |
120.1° 120.8° |
Sc-C: 2.244 2.194 |
16 |
| LMe,m-tBu | 83.1° | 2.128 2.128 |
121.6° 122.1° |
Sc-C: 2.210 2.215 |
69 | |
| LMe,m-Tipp | 84.9° | 2.127 2.123 |
120.4° 119.2° |
Sc-C: 2.203 2.202 |
69 | |
| [LV]2 | LMe,Et | 88.69° | 2.066 2.041 |
115.84° 114.05° |
V-arene: 1.422 | 34 |
| LMe,Me | 88.73° | 2.057 2.034 |
115.98° 113.22° |
V-arene: 1.411 | 34 | |
| LMe,An | 88.83° | 2.025 2.020 |
117.05° 117.01° |
V-arene: 1.744 | 34 | |
| LCr(Cl)(η5-Cp) | LMe,iPr | 89.9° | 2.036 2.036 |
117.3° 117.3° |
Cr-Cp: 1.929 | 62 |
| LMe,Et | 90.3° | 2.022 2.016 |
118.0° 117.9° |
Cr-Cp: 1.901 | 72 | |
| LMe,Me | 90.5° | 2.019 2.018 |
117.7° 119.0° |
Cr-Cp: 1.897 | 63 | |
| LCr(Cp)(alkyl) | LMe,iPr | 90.7° | 2.039 2.039 |
118.3° 118.8° |
Cr-Cp: 1.972 | 62 |
| LMe,Et | 90.2° | 2.029 2.017 |
118.7° 118.3° |
Cr-Cp: 1.963 | 72 | |
| LMe,Me | 90.7° | 2.024 2.026 |
116.9° 117.6° |
Cr-Cp: 1.966 | 72 | |
| LFe(μ-Cl)2Li(THF)2 | LMe,iPr | 93.22° | 2.021 2.006 |
120.27° 118.59° |
Fe-Cl: 2.338 2.324 |
18 |
| MeLMe,Me | 93.19° | 1.983 1.983 |
119.19° 119.19° |
Fe-Cl: 2.325 2.325 |
91 | |
| [LNi(μ-Cl)2 | LMe,iPr | 93.66° | 1.946 1.938 |
117.11° 116.42° |
Ni-Cl: 2.350 2.325 |
24 |
| LMe,Me | 94.7° | 1.915 1.913 |
117.88° 117.30° |
Ni-Cl: 2.313 2.300 |
25 | |
| LNi(μ-P4)NiL | LMe,iPr | 94.98° | 1.947 1.968 |
117.74° 116.94° |
Ni-P: 2.339, 2.217, 2.195 | 42 |
| LMe,Et | 96.44° | 1.931 1.928 |
119.86° 115.87° |
Ni-P: 2.203, 2.329, 2.167 | 42 | |
| [LCu(μ-S)]2 | LMe,Et | 99.30° | 1.907 1.910 |
118.43° 118.18° |
Cu-S: 2.197 2.193 |
66 |
| LMe,Me | 99.43° | 1.899 1.896 |
119.65° 119.17° |
Cu-S:2.184 2.187 |
67 | |
| PhLH,iPr | 96.95° | 1.913 1.905 |
116.70° 115.97° |
Cu-S:2.205 2.198 |
67 | |
| PhLH,Et | 96.92° | 1.911 1.909 |
116.96° 117.21° |
Cu-S:2.195 2.194 |
67 | |
| ArFLH,iPr | 97.07° | 1.921 1.905 |
115.47° 116.00° |
Cu-S:2.194 2.206 |
67 | |
| ArFLH,Me | 98.07° | 1.906 1.912 |
115.21° 117.26° |
Cu-S:2.198 2.198 |
67 | |
| [LCu(μ-OH)]2 | CNLH,Et | 93.63° | 1.955 1.943 |
115.90° 115.44° |
Cu-O: 1.926 1.926, 1.909 |
46 |
| CNLH,Me3 | 93.35° | 1.962 1.958 1.946 |
117.62° 117.29° |
Cu-O: 1.922 1.920, 1.904 |
46 | |
| LRu(Cl)(η6-Benzene) | LMe,Me | 86.56° | 2.099 2.099 |
116.80° 116.80° |
Ru-Cl: 2.521 Ru-Benzene:1.688 |
84 |
| LMe,m-Me | 88.21° | 2.098 2.091 |
117.53° 117.38° |
Ru-Cl:2.453 Ru-Benzene:1.683 |
85 | |
| LRu(Cl)(η5-Cp*) | LMe,Me | 87.51° | 2.089 2.075 |
114.98° 115.14° |
Ru-Cl: 2.461 Ru-Cp*:1.889 |
86 |
| LMe,m-Me | 87.83° | 2.050 2.051 |
116.43° 115.98° |
Ru-Cl:2.451 Ru-Cp*:1.869 |
86 | |
| LRu(η5-Cp*) | LMe,Me | 87.23° | 2.070 2.060 |
114.36° 113.70° |
Ru-Cp*:1.819 | 86 |
| LMe,m-Me | 87.92° | 2.060 2.063 |
115.62° 115.29° |
Ru-Cp*:1.809 | 86 | |
| LMe,H | 87.68° | 2.053 2.046 |
113.89° 113.74° |
Ru-Cp*:1.800 | 92 | |
| [LPd(μ-Cl)]2 | LMe,iPr | 91.78° | 2.023 2.013 |
118.65° 117.87° |
Pd-Cl: 2.366 2.354 |
93 |
| LMe,m-CF3 | 90.93° | 2.006 1.989 |
118.57° 118.97° |
Pd-Cl: 2.350 2.352 |
93 | |
| LMe,H | 91.30° | 2.000 2.001 |
118.20° 120.61° |
Pd-Cl: 2.342 2.356 |
33 | |
| LPd(Cl)(Py) | LMe,iPr | 91.70° | 2.031 2.014 |
118.19° 116.65° |
Pd-Cl: 2.315 Pd-Py: 2.078 |
93 |
| LMe,m-CF3 | 90.08° | 2.026 2.013 |
119.46° 120.11° |
Pd-Cl: 2.302 Pd-Py: 2.039 |
93 |
The choice of N-aryl substituent has a smaller influence on the bite angle, C(aryl)-N-C(β) bond angle and N-M bond length in most cases. However, changing N-aryl substituents can build up steric bulk above and below the N-M-N plane, which can significantly influence the distance from the metal to the other ligands. In general, more hindered N-aryl substituents lead to a longer M-L bonds (Table 3.1.4).
Other modifications of β-diketiminate ligands, including installation of functional groups on the backbone α-C, or on the para-position of the N-aryl substituents, have little influence on the core structural parameters of β-diketiminate metal complexes.
The geometry and conformation of metal complexes can also be changed with modification of the supporting β-diketiminate ligand. The zirconium center in LMe,RZr(CH2Ph)3 (R = iPr, p-Me)94 adopts a square pyramidal geometry with a crystallographic mirror plane passing through it. However, the relative orientation of the ligand planes shows differences (Figure 3.1.1). Without ortho-substitution on N-aryl, the β-diketiminate ligand plane in LMe,pMeZr(CH2Ph)3 forms an angle of 67.7(3)° with the least squares plane defined by C(Bn)-C(Bn)-N-N. In contrast, the angle between the ligand planes in LMe,MeZr(CH2Ph)3 is only 7.0(3)°. Presumably, this difference is due to steric conflict between the benzyl and N-aryl substituents. N-Aryloxy-β-diketiminate zirconium complexes also showed a different orientation depending on steric bulk (Scheme 3.1.1).95 Bridged aryloxides were observed with one meta-tBu on the N-aryl, but the presence of a second meta-tBu group gave steric conflict that resulted in the isolation of a [LZrCl2]2 dimer instead. In the same system, the L2Zr complexes also showed conformational differences where the bulkier ligand adopted a trigonal prismatic geometry (Figure 3.1.2).
Figure 3.1.1.

Structural influence of sterically different aryl groups on the conformation of Zr complexes.
Scheme 3.1.1.

Figure 3.1.2.

Structural differences between bis(ligand) complexes on zirconium, with different ortho substituents.
The solution structure of the metal complex can be affected by different steric bulk as well. For example, two sets of peaks were observed in 1H NMR and 125Te NMR spectra of LtBu,iPrSc(TeCH2TMS)2,96 suggesting exo and endo tellurolates that are static on the NMR time scale. In contrast, the two tellurolate groups are equivalent for LMe,iPrSc(TeCH2TMS)2,96 indicating rapid endo/exo flipping. Thus, larger groups create more difficulty for Sc(TeR)2 to flip through the channel restricted by the N-aryl groups. In another example, 1H NMR peaks of a molybdenum imido alkylidene supported by LMe,m-Me was broadened compared with that of its LMe,Me analogue, suggesting the relatively free rotation of N-aryl in the less sterically hindered meta-substituted ligand.
3.2. Steric effects on reactivity and product formation
Here, we highlight other cases where different choices of steric bulk of the supporting β-diketiminate ligand give structurally different products under the same reaction conditions. In general, bulkier groups restrict the available conformations. For example, treatment of LtBu,iPrScCl2 or [LMe,iPrScCl(μ-Cl)]2 with LiNHtBu in hexanes generated different products (Scheme 3.2.1).48, 49 The authors proposed that the less sterically hindered LMe,iPr allows the formation of a dimeric transition state that is necessary for ligand exchange and disproportionation.
Scheme 3.2.1.

Extrusion of Te(CH2TMS)2from LR,iPrSc(TeCH2TMS)2 (R = tBu, Me) under photolysis formed different products depending on R (Scheme 3.2.2).96 Crossover between (LSc(TeCH2SiMe3)2 and LSc(TeCH2CMe3)2) showed that the product came from a bimolecular process. It is likely that the tellurolate-telluride (LSc(TeCH2TMS))2(μ-Te) is an intermediate on the way to the bridging telluride complex. However, the greater steric bulk of LtBu,iPr stabilized the tellurolate-telluride species, preventing the loss of a second molecule of Te(CH2TMS)2.
Scheme 3.2.2.

Reduction of LMe,RVCl2 (R = Me, Et, anthracenyl) with 2 equivalents of KC8 in THF gave dimeric vanadium(I) complexes, while reaction of LPh,iPrVCl2 gave extrusion of the imido fragment from diketiminate under the same conditions (Scheme 3.2.3).34 This was not only from having an available arene for binding, because reduction of LMe,iPrVCl2 in toluene gave an inverted sandwich complex. Rather, the authors surmised that the steric conflict between N-aryl and backbone phenyl group twisted the N-aryl group, destabilizing the LV intermediate and bringing about the reductive C-N bond cleavage of the ligand.
Scheme 3.2.3.

In another example, oxidation of a chromium(II) complex gave a highly reactive chromium oxo complex. However, the attempt to generate a chromium oxo complex gave different products depending on the steric bulk of different β-diketiminate ligands (Scheme 3.2.4).38 Reaction of LMe,MeCrCp or LMe,m-TIPPCrCp with pyridine N-oxide gave a μ-oxo dimer, while the bulkier LMe,EtCr-Cp generated a product from hydrogen atom transfer. The sterically more hindered ortho-ethyl substituents may prevent the μ-oxo dimer from forming, and rather the highly reactive terminal oxo (LMe,Et(Cp)Cr=O) can abstract a hydrogen atom from its own ligand, ultimately generating a new C-C bond.
Scheme 3.2.4.

Upon addition of O2, copper(I) complexes supported by different β-diketiminate ligands form different products (Scheme 3.2.5). More sterically hindered LtBu,iPrCu(NCCH3) and LMe,iPrCu(NCCH3) formed a copper(II) peroxo LCu(O2) while less bulky R’LH,RCu (R = iPr, Me, Et; R’ = H, Ph) complexes gave a bis(μ-oxo)dicopper(III) complex.8, 30 These reactivity differences between the two systems were attributed to the steric effect of the backbone (β-C) substituents, which rigidify the N-aryl substituents and prevent the dimer from forming.
Scheme 3.2.5.

The dinitrogen ligand in LR,iPrFeNNFeLR,iPr (R = tBu, Me) can be replaced by other neutral ligands like carbon monoxide or isocyanide.41 When exposing with excess CO, LMe,iPrFeNNFeL converted to square pyramidal LMe,iPrFe(CO)3, while the LtBu,iPr analogue gave a mixture of LtBu,iPrFe(CO)3 and LtBu,iPrFe(CO)2. Since the two N-dipp substituents are closer in LtBu,iPr, binding the third axial CO may bring steric tension between iPr and CO, which explains the formation of square planar LtBu,iPrFe(CO)2. Similarly, N2 exchange in LR,iPrFeNNFeLR,iPr is much more rapid with R=Me than R=tBu, implying that transient species with axial N2 are also accessible but only with the smaller R = Me.41 In a more deep-seated difference in reactivity, attempts to make analogous MeLMe,MeFeNNFeMeLMe,Me complexes gave N2 cleavage to a tetra-iron bis(nitride) complex, with complete cleavage of the N-N bond (Scheme 3.2.6).20 The authors proposed that the smaller supporting ligand allows access to an intermediate in which three LFe units can interact simultaneously with the same molecule of N2.
Scheme 3.2.6.

3.3. Steric effect on activity of metal complexes
Varying the steric bulk of the β-diketiminate ligand has a significant effect on activity of metal complexes in both stoichiometric and catalytic reactions. In most cases, a more sterically hindered β-diketiminate ligand builds up steric tension in transition states or intermediates, which raises the activation barrier and slows the reaction rates. However, the added steric bulk has advantages because it can enable the isolation of transient intermediates.
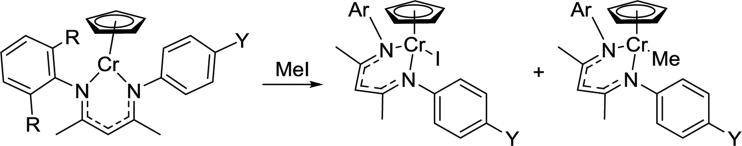
The single-electron oxidative addition of organic halides to chromium(II) complexes (Scheme 3.3.1) illustrated the steric effect of ortho-substituents on the N-aryl group.62, 72, 97 The less hindered asymmetric LMe,iPr/p-YCr(Cp) gave a rate constant of 0.5-1.0 M−1s−1 (depending on the electronic properties of Y; see section 4.2 below),97 whereas LMe,iPrCr(Cp) and its LMe,Me, LMe,Mes, and LMe,Et analogues gave rate constants that were more than an order of magnitude smaller, ranging from 0.02-0.03 M−1s−1.72 Thus, removing the ortho-alkyl groups from one of the N-aryl groups greatly enhanced the reactivity of chromium(II) by increasing the accessibility of methyl iodide.
Scheme 3.3.1.
Catalytic 1-hexene isomerization and dimerization was reported with [LMe,RNiBr]2 (R = iPr, Me), where the less sterically hindered [LMe,MeNiBr]2 gave higher conversions under the same conditions.98 The authors proposed that a β-diketiminate nickel hydride complex was the active catalyst, which would proceed through insertion, β-hydride elimination and chain walking to generate internal alkenes. This makes sense if β-hydride elimination is the rate-limiting step, because larger β-diketiminate substituents would prevent the increase in coordination number. In a demonstration of this idea in a stoichiometric reaction, LtBu,iPrFe-tBu isomerized to LtBu,iPrFe-CH2iBu only at elevated temperatures, while LMe,iPrFe-tBu isomerized at room temperature to LMe,iPrFe-CH2iBu (Scheme 3.3.2).76
Scheme 3.3.2.

The mechanism of alkyne insertion was also studied in detail with isolated β-diketiminate iron hydride complexes. The rate of alkyne insertion was first order in [FeH] and zero order in [alkyne], with kobs = 1.7(2) × 10−3 s−1 for [LMe,iPrFeH]299 and 5.0(5) × 10−4 s−1 for [LtBu,iPrFeH]2;40 again the less hindered complex had higher reactivity. In a related B-C bond cleavage reaction, two mechanisms were proposed: the less hindered iron complex undergoes single iron-hydride opening followed by insertion, while the more hindered LtBu,iPr system can completely dissociate to a reactive monomer.74
β-Diketiminate iron imido complexes are prone to hydrogen atom transfer (HAT) from the ortho isopropyl substituents of the supporting ligand. To solve the problem, LMe,Ph3Fe=NR was prepared.100 The second-order rate constants for hydrogen atom transfer to LFe=NAd from 1,4-cyclohexadiene in C6D6 were 2.0(2) × 10−2 M−1 s−1 for LMe,Ph3Fe=NAd, 1.4(2) × 10−4 M−1 s−1 for LMe,iPrFe=NAd and ~0 for LtBu,iPrFe=NAd (Scheme 3.3.3). Clearly the most bulky LtBu,iPrFe=NAd gave the slowest HAT reactivity. However, the relative sizes of LMe,iPr and LMe,Ph3 were not obvious. The authors measured the size using the G parameter, which estimates the fraction of the metal overshadowed by the ligand.101 The results indicated very similar G parameter for LMe,iPrFe=NAd (G = 63.8%) over LMe,Ph3Fe=NAd (G = 62.2%), but different shapes (Figure 3.3.1). The different orientation of N-aryl with respect to the ligand backbone shows more opening above the imido nitrogen, which results in a larger binding pocket for hydrocarbon substrates (Figure 3.3.2).
Scheme 3.3.3.

Figure 3.3.1.

Differences in ligand coverage in LMe,iPr vs. LMe,Ph3 in iron(III) imido complexes. The G parameter quantifies the ligand coverage, as describe in ref. 100. Thus, even though the overall coverage is similar between the two ligands, the shape of the coverage is different.
Figure 3.3.2.

Side view of the complexes in Fig. 4, showing the greater access to the Fev=N bond when using LMe,Ph3.
Increasing the steric bulk of the β-diketiminate can also prevent formation of certain metal complexes due to steric blocking. In an example, β-diketiminate zirconium tribenzyl complex (LMe,p-MeZr(CH2Ph)3) can be synthesized through alkane elimination between tetra-alkyl zirconium (IV) and β-diketimines. For its bulkier analogue LMe,iPrZr(CH2Ph)3, sterically hindered iPr groups prevent Zr(CH2Ph)4 from accessing the β-diketiminate binding pocket. Therefore, it was necessary to develop a different synthetic method for LMe,iPrZr(CH2Ph)3 involving salt metathesis of LLi and ZrCl4 followed by alkylation (Scheme 3.3.4).94 In another example, LMe,iPrFeNNFeLMe,iPr releases the labile dinitrogen ligand immediately in aromatic solvents forming LMe,iPrFe(η6-C6H6). However, the more sterically hindered LtBu,iPrFeNNFeLtBu,iPr retains its structure in C6H6 up to 100 °C, without coordination of benzene.41
Scheme 3.3.4.

However, more sterically hindered metal complexes are favored in some cases because a sterically crowded environment can facilitate intramolecular reactions or increase the concentration of key unsaturated species. An example comes in reactions where metalation of ligand C-H bonds involves intramolecular C-H insertion. Upon heating in aromatic solvent, the four-coordinate dialkyl complexes LR,iPrScR’2 (R = tBu, Me; R’ = alkyl) (Scheme 3.3.5) underwent C-H metalation and eliminated alkane. The half-life of LMe,iPrScR2 in metalation was significantly longer than its LtBu,iPr analogue, suggesting lower reactivity with the less sterically hindered metal complex.16
Scheme 3.3.5.

LR,R’NiBr, LR,R’NiPh(PPh3) and LMe,R’Ni(alkyl) (R = CF3, Me; R’ = iPr, Me) were reported to be active catalysts for ethylene,102, 103 styrene,104 norbornene105, 106 polymerization and their copolymerization.107, 108 The polymer yield was significantly higher with more hindered ligand systems. Presumably, alkyl insertion into coordinated alkene is greatly facilitated by the more sterically hindered coordination environment.105
Reductive elimination is another process facilitated by a crowded coordination environment. With a β-diketiminate-supported Pd(II) methyl phosphine complex, catalytic Castro-Stephens coupling,109 Stille coupling110 and Hiyama coupling111 were more rapid with a more sterically hindered β-diketiminate ligand (LMe,Me vs. LMe,H) which gave faster reductive elimination.
In addition, homolysis is influenced by ligand size. Since chromium(III) alkyl mediated radical polymerization often involves homolysis of the Cr-C bond to gain chain growth, more sterically hindered β-diketiminate ligand increases the Cr-C bond distances (see Table 3.1.4), giving a lower BDE, and increasing the rate of homolysis and thus rate of polymerization.112, 113
Catalytic carbodiimide formation from isocyanide and organic azide with a diketiminate-iron(I) catalyst gave significantly higher yields with a more sterically bulky catalyst (LtBu,iPr > LMe,Ph3 > LMe,iPr). The proposed mechanism involves loss of one molecule of coordinated isocyanide before turning over the catalytic cycle. Not surprisingly, more hindered complexes favor a lower coordination number, which facilitates the loss of isocyanide, production of an active site, and turnover of the catalytic reaction.114
LCrCp catalyzed oxygen atom transfer reaction38 (eq 3.3.1) and LCu(2-methylpyridine)-catalyzed alkene azirdination115 (Scheme 3.3.6) are also more rapid with more hindered complexes because the smaller catalysts have more rapid rates for corresponding side reactions. Upon formation of catalytically active [LCr=O] intermediate, LMe,MeCr-Cp generates LMe,MeCr(Cp)(μ-O)Cr(Cp)LMe,Me which is inactive towards catalytic oxygen atom transfer from O2 to PPh3. In contrast, more hindered LMe,EtCr(Cp)=O is less reactive towards formation of the μ-oxo complex and more catalytically active. Under catalytic aziridination conditions, smaller LMe,MeCu(2-methylpyridine) underwent a side reaction generating TsNH2, which lowered the reactivity and yield of aziridination compared with LMe,Me/iPrCu(2-methylpyridine).
Scheme 3.3.6.

Ethylene polymerization with L2TiCl2 complexes supported by different ligands have been studied. LMe,iPr2TiCl2 amd LCF3,iPr2TiCl2 showed significantly higher activity than their corresponding LMe,Me,LMe,H and LCF3,Me analogues. In this case, it is possible that bulky N-aryl substituents can prohibit β-hydride elimination and thus maintain chain growth.116 In contrast, LTiMe2 showed a different steric effect, where the less hindered LMe,Me3TiMe2 was an order of magnitude more reactive than its more hindered LtBu,Me3TiMe2 and LMe,iPrMe2 analogues.52
The steric effect for C-P cross-coupling catalyzed by LCrCp complex is another interesting example, because the influence is different depending on the relative rate of oxidative addition and Cr-C homolysis.117 For more reactive alkyl bromide substrates, more hindered LMe,MeCrCp or LMe,MeCr(Cp)Br gave higher yields than less hindered asymmetric LMe,iPr/p-MeCrCp and LMe,iPr/p-MeCr(Cp)Br. Because these substrates undergo rapid single electron oxidative addition, the rate determining step is homolysis of the Cr-C bond. As previously mentioned, the Cr-C BDE is lower with more hindered ligands, so these ligands speed the catalytic rate. On the other hand, for less active substrates like Cy-Cl, oxidative addition is rate limiting, and the rate is faster with a less sterically hindered coordination environment.
3.4. Steric effects on selectivity of metal complexes
Changing steric bulk can also influence the selectivity of reactions of β-diketiminate complexes. This is due to the conformational differences in the energy of the intermediate/transition state with different steric hindrance. In one example, a vanadium(I) β-diketiminate complex catalyzed cyclotrimerization of terminal alkynes at room temperature to give trisubstituted benzenes, with a mixture of isomers.34 Catalysis with [LMe,MeV]2 gave a 65:35 ratio of 1,3,5-trisubstituted benzene over 1,2,4-trisubstituted benzene, whereas the more sterically hindered [LMe,iPrV]2 gave a slightly lower yield with 80:20 regioselectivity. The steric restrictions in the transition states or intermediates apparently can prevent formation of products with adjacent substituents.
As mentioned in section 3.3.3, changing the steric bulk can affect the reactivity of alkene polymerization and isomerization catalyzed by [LNiBr]2. Less bulky supporting ligands lead to more rapid β-hydride elimination, giving polyethylene with more branching. In alkene isomerization, the steric hindrance of the ligand can have important influences on the selectivity between cis and trans alkene products. More sterically hindered [LMe,iPrNiBr]2 gave more cis product (44%) compared with [LMe,MeNiBr]2 (28%).98 It is believed that the crowded coordination environment restricted the rotation of C-C bond in Ni-alkyl complex, hindering the formation of trans-transition states. A bulkier LtBu,iPrCo-alkyl complex isomerized alkenes with much higher cis selectivity, often greater than 6:1 cis/trans, but the LMe,iPrCo analogue gave poor selectivity. In this cobalt(II) system, the preference of the LtBu,iPr complex for isomerization of terminal alkenes to only the 2 position was also attributed to the bulk of the ligand above and below the N2Co plane.88
4. Electronic effects on β-diketiminate complexes
To tune the electronic properties of β-diketiminate ligands, various groups have been installed on the backbone (α-C and β-C) or on the N-aryl substituents. These modify the electron density at the metal center, which can affect the redox potential, IR frequency of other ligands, UV-Vis absorption maxima, and NMR chemical shifts. In addition, these electronic changes can also affect the reactivity through perturbation of the energy of transition states or intermediates. It should be borne in mind that many of the substituents used to change the electronic effects can also influence sterics as well, particularly on the backbone (β-C) and ortho positions of N-aryl groups.
4.1. Electronic effects on electron density and core structure of the metal center
Changes in electron density on the metal center can be monitored by various methods. Often, electron-withdrawing groups lead to more positive redox potentials, lower field chemical shifts in NMR spectra, and less backbonding into coordinated ligands, consistent with less electron density at the metal ion.
Copper and nickel complexes supported by β-diketiminate ligands bearing different electronic properties have been studied with cyclic voltammetry (Table 4.1.1). Judging from the redox potentials in Table 4.1.1, NO2 and CF3 have the strongest electronic effect, followed by CN and 3,5-bis(trifluroromethyl)phenyl substituents. In addition, greater electronic effects result from substitutions on α-C and β-C, and less with N-aryl substituents. This is reasonable because the aryl ring is roughly perpendicular to the MN2C3 plane, and thus there is little conjugation of the π-systems. In contrast, backbone substituents are in the plane of the ligand backbone, and thus can have a greater impact on the electron density of the metal center. The exception is the relatively small electronic effect from 3,5-bis(trifluoromethyl)phenyl substituents on the backbone (α-C), which is presumably again from lack of conjugation between the perpendicular π-systems. However, the electronic influence of N-aryl substituents is not negligible. For example, alkyl substituents on the N-aryl behaved as electron-donating groups when PhLH,iPr-supported copper complexes had a more negative redox potential than PhLH,Me and PhLH,Et (Table 4.1.1).30
Table 4.1.1.
Dependence of Reduction Potential on Substituents
| Complex | Ligand | Reduction potentiala (V) | Reference |
|---|---|---|---|
| LCu(NCCH3)b | PhLH,iPr | 0.384 | 30 |
| Ar-CF3LH,iPr | 0.449 | 30 | |
| PhLH,Et | 0.420 | 30 | |
| Ar-CF3LH,Et | 0.428 | 30 | |
| PhLH,Me | 0.388 | 30 | |
| CF3LH,Me | 0.400 | 30 | |
| NO2LH,Mes | 0.520 | 30 | |
| LCu(NCCH3)c | LMe,iPr | −0.096 | 68 |
| LMe/CF3, iPr | 0.11 | 68 | |
| LCF3, iPr | 0.411 | 68 | |
| LCu(OAc)b | LMe,iPr | −1.29 | 118 |
| LMe,iPr/iPr-CN | −1.26 | 118 | |
| LMe,iPr/Et-CN | −1.24 | 118 | |
| L2Cuc | MeLH,H | −1.62 | 46 |
| HLH,H | −1.46 | 46 | |
| CNLH,H | −0.97 | 46 | |
| NO2LH,H | −0.68 | 46 | |
| L2Nic | MeLH,H | −2.42 | 119 |
| HLH,H | −2.16 | 119 | |
| BrLH,H | −1.89 | 119 | |
| CNLH,H | −1.64 | 119 | |
| NO2LH,H | −1.28 | 119 |
Bu4NPF6 was used as electrolyte.
All values reported with Fc/Fc+ in CH3CN.
All values reported with Fc/Fc+ in THF.
Another consequence of the changing redox potentials is the relative stability of certain oxidation levels. In L2Cu complexes, irreversible reductions were observed with MeLH,H and HLH,H while reversible redox couples were observed in CNLH,H and NO2LH,H, suggesting that the reduced Cu(I) state of the bis(β-diketiminate) complex is unstable in the complexes with more electron rich ligands. In contrast, with LCu(NCCH3) complexes, the Cu(II) state was less stable with a more electron withdrawing group.46 Ruthenium(II) complexes of LCF3,m-CF3Ru(Cl)(Ar) (Ar = arene ligand) were studied to determine the electronic effects of the supporting ligand on the metal and the other coordinating ligands in comparison to analogous complexes with the LMe,m-Me supporting ligand.85 Interestingly, there was no clear trend between the RuII/RuIII redox potentials from the cyclic voltammograms through the series LMe,Me, LMe,m-Me, LCF3,m-Me, and LCF3,m-CF3, indicating that other factors also play a role.86
Electronic modification can also have an impact on the positions of the maxima in electronic absorption (UV-Vis) spectra. β-Diketiminate complexes typically have a π→π* transition in the 300-400 nm region, which shifts to shorter wavelength with more electron-withdrawing substituents in LCu(NCCH3).30 This suggests that electron-withdrawing groups lower the energy of the π orbital more than they do the π* orbital. The positions of d-d transitions was also studied in L2Cu complexes, where the d-d absorption bands shift toward shorter wavelength with electron withdrawing backbone substituents (α-C) and shift to longer wavelength with more electron donating substituents on the N-aryl group.46 It is proposed that the ligand field was enhanced with electron donating substituents and thus affected the UV-Vis absorptions.
IR and Raman peaks on coordinated diatomic ligands is another traditional method for quantifying the relative electron density of a metal center. The ν(CO) in LCu(CO) complexes and ν(OO) in LCu(O2) each shift to higher frequency when electron withdrawing CF3 groups were installed on the backbone β-C.68 This is attributable to a less electron rich metal center that has weaker back-donation into ligand antibonding orbitals. The influence of m-CF3 groups on the N-aryl substituents was less, again indicating a smaller influence from N-aryl substitution.
Due to the shielding or deshielding effect of substituents, the chemical shift in NMR spectra also indicates the electron density on metal center. For example, the chemical shift of the backbone (α-C) proton shifted downfield when CF3 was substituted for CH3 on backbone and for meta- positions on the N-aryl.85 This is correlated to the deshielding effect with more electron withdrawing groups attached directly to the π system.
Though the introduction of electron withdrawing groups hardly affects the metal ligand core structure, it can affect the coordination number as well as bonding properties in some cases. For example, when NO2 was installed on backbone (α-C) of LCu-OAc, one molecule of methanol coordinated to the metal center, but no coordinated methanol was observed with CNLH,iPr and PhLH,iPr. This is consistent with the stronger Lewis acidity of metal center when its supporting ligand has an electron withdrawing NO2 substituent.90 Ru-Cl bond lengths and Ru-arene distances in LRu(Cl)(η6-arene) are shorter with LCF3,m-CF3 compared with LMe,m-Me, suggesting an increase in Lewis acidity of the metal with more electron-withdrawing substitutents.85
4.2. Electronic effects on reactivity of metal complex
Changes of electron density on the metal center can have a significant effect on reactivity of metal complexes. For example, the oxidative addition of methyl iodide to mixed-aryl LCrCp complexes (Scheme 3.3.1) is affected by electronic substituents on para-N-aryl (OMe, Me, H, CF3).97 There was a correlation between the para-substituent and the rate constant, with the rate constant decreasing two-fold from most electron-donating (para-OMe, kobs=(9.80±0.3) × 10−1 M−1 s−1) to most electron-withdrawing (para-CF3, kobs=(4.96±0.3) × 10−1 M−1 s−1) substituent. Even though the solid structures indicate that the N-aryl planes are aligned roughly perpendicular to the metal-ligand plane, the authors noted that the lack of ortho-substituents may allow the N-aryl to rotate closer to the diketiminate plane in solution, enabling some conjugation. In this way, the more electron-donating substituents can stabilize the chromium(III) product, which could lower the barrier if Hammond's postulate holds.
In another example, catalytic oxidation of alkanes to alcohols and ketones was reported with LCu(OAc) as a catalyst.90 When LCu(OAc) was supported by a more electron-withdrawing β-diketiminate ligand, the catalytic reactivity was higher. The results were rationalized through a mechanistic model where the reactions proceed through a metal-based oxidant, based on the observed kinetic isotope effect and regioselectivity.120 Thus, more electron withdrawing groups would give more unstable and energetic high-valent copper intermediates that are more reactive toward the alkane.
Atom transfer radical addition (ATRA) and atom transfer radical cyclization (ATRC) are particularly interesting for organic synthesis. Using β-diketiminate ruthenium complexes (LRu(Cp*)Cl and LRu(Cp*)), lower conversions were observed with LMe,Me, LMe,m-Me, and LMe,m-CF3, while the addition of electron-withdrawing substituents in LCF3,m-Me and LCF3,m-CF3 gave higher reactivity.86 No simple correlation between catalytic reactivity and redox potential of the ruthenium complexes was observed, but the addition of the CF3 groups also rendered the complexes air-stable in solution and solid state. Likewise, in the copper(I) complexes mentioned above, LMe,iPrCu(NCMe) and LCF3/Me,iPrCu(NCMe) react with O2, but LCF3,iPrCu(NCMe) does not react with O2. This agrees with the more positive redox potential with an electron-withdrawing group.68
The previously mentioned nickel catalyzed polymerization of styrene and norbornene (see section 3.3) showed a strong influence of the β-diketiminate ligand electronic properties. The substitution of backbone methyl with trifluoromethyl significantly improved the catalytic reactivity.104, 105, 121 This can be explained if the more electrophilic nickel center has a lower activation energy for alkene insertion during rate-limiting chain growth.
5. Conclusions
The examples in this Perspective support the idea that β-diketiminate ligands have great tunability in terms of both steric and electronic effects, and they point future chemists in the directions that could benefit their own chemistry. The β-C and N-aryl ortho substituents are most important for steric effects, whereas the α-C and β-C positions are most influential for electronic effects. N-aryl groups can have a small electronic influence, but this has been best documented when there are no ortho-substituents and the N-aryl group can rotate closer to planarity with the ligand backbone. In contrast, the steric effects are more varied, because they can change the structure and transition states in different ways depending on the specific coordination number, reaction, and co-ligands. However, the ability of relatively small changes to cause structural, spectroscopic, and reactivity differences suggests that further tuning will uncover multitudes of new chemistry. We note particularly that chiral substituents have only been used in β-diketiminate ligands with N-benzyl substituents,122-125 and incorporation of chiral anilines should be a fruitful area for preparation of C1 and C2 symmetric complexes.
Supplementary Material
Scheme 4.2.1.

Scheme 4.2.2.

6. Acknowledgments
Research on β-diketiminate complexes in the Holland laboratory has been supported by the National Institutes of Health (GM065313), the National Science Foundation (CHE-0112658 and CHE-0911314), the A.P. Sloan Foundation, the Petroleum Research Fund (44942-AC), and by the U.S. Department of Energy, Office of Basic Energy Sciences (DE-FG02-09ER16089). We thank the University of Rochester and Yale University for financial and other support, and K. Cory MacLeod for thoughtful comments.
Biography
 Chi Chen received his Bachelor of Science degree at Peking University in 2009 and did additional research at the University of Texas - Arlington before starting graduate research at the University of Rochester in 2011. In a joint project with Daniel Weix and Patrick Holland, he is developing and studying new β-diketiminate supported cobalt catalysts for alkene transformations such as isomerization and hydrosilylation. In 2013, he moved to Yale University where he is completing his PhD research.
Chi Chen received his Bachelor of Science degree at Peking University in 2009 and did additional research at the University of Texas - Arlington before starting graduate research at the University of Rochester in 2011. In a joint project with Daniel Weix and Patrick Holland, he is developing and studying new β-diketiminate supported cobalt catalysts for alkene transformations such as isomerization and hydrosilylation. In 2013, he moved to Yale University where he is completing his PhD research.
 Sarina Bellows received her Bachelor of Science degree from Syracuse University in 2008, and pursued PhD research at the University of Rochester with Patrick Holland. In her research, she synthesized iron complexes of new β-diketiminate ligands, and also performed computations to explain the mechanisms of their reactions. Since receiving her PhD in 2014, she has been a postdoctoral fellow at Rochester with Thomas Cundari and William Jones through the Center for Enabling New Technologies through Catalysis.
Sarina Bellows received her Bachelor of Science degree from Syracuse University in 2008, and pursued PhD research at the University of Rochester with Patrick Holland. In her research, she synthesized iron complexes of new β-diketiminate ligands, and also performed computations to explain the mechanisms of their reactions. Since receiving her PhD in 2014, she has been a postdoctoral fellow at Rochester with Thomas Cundari and William Jones through the Center for Enabling New Technologies through Catalysis.
 Patrick Holland completed an AB at Princeton University, and a PhD at UC Berkeley with Richard Andersen and Robert Bergman. In postdoctoral work at Minnesota with William Tolman, he learned to love β-diketiminates through the synthesis of copper complexes. In his independent career, he has explored the use of β-diketiminate complexes of iron, cobalt and nickel, as applied to N2 reduction, C-H oxidation, redox-active ligands, new bonding environments, and novel reactivity. He was on the faculty at the University of Rochester from 2000-2013, and is now a Professor of Chemistry at Yale University.
Patrick Holland completed an AB at Princeton University, and a PhD at UC Berkeley with Richard Andersen and Robert Bergman. In postdoctoral work at Minnesota with William Tolman, he learned to love β-diketiminates through the synthesis of copper complexes. In his independent career, he has explored the use of β-diketiminate complexes of iron, cobalt and nickel, as applied to N2 reduction, C-H oxidation, redox-active ligands, new bonding environments, and novel reactivity. He was on the faculty at the University of Rochester from 2000-2013, and is now a Professor of Chemistry at Yale University.
References
- 1.McGeachin SG. Can. J. Chem. 1968;46:1903–1912. [Google Scholar]
- 2.Bonnett R, Bradley DC, Fisher KJ. Chem. Commun. 1968:886–887. [Google Scholar]
- 3.Parks JE, Holm RH. Inorg. Chem. 1968;7:1408–1416. [Google Scholar]
- 4.Bourget-Merle L, Lappert MF, Severn JR. Chem. Rev. 2002;102:3031–3066. doi: 10.1021/cr010424r. [DOI] [PubMed] [Google Scholar]
- 5.Tsai Y. Coord. Chem. Rev. 2012;256:722–758. [Google Scholar]
- 6.Smith JM, Lachicotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Rodgers KR, Holland PL. J. Am. Chem. Soc. 2001;123:9222–9223. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]
- 7.Holland PL, Tolman WB. J. Am. Chem. Soc. 1999;121:7270–7271. [Google Scholar]
- 8.Spencer DJE, Aboelella NW, Reynolds AM, Holland PL, Tolman WB. J. Am. Chem. Soc. 2002;124:2108–2109. doi: 10.1021/ja017820b. [DOI] [PubMed] [Google Scholar]
- 9.Aboelella NW, Lewis EA, Reynolds AM, Brennessel WW, Cramer CJ, Tolman WB. J. Am. Chem. Soc. 2002;124:10660–10661. doi: 10.1021/ja027164v. [DOI] [PubMed] [Google Scholar]
- 10.Aboelella NW, Gherman BF, Hill LMR, York JT, Holm N, Young VG, Cramer CJ, Tolman WB. J. Am. Chem. Soc. 2006;128:3445–3458. doi: 10.1021/ja057745v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL. J. Am. Chem. Soc. 2004;126:4522–4523. doi: 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]
- 12.Tonzetich ZJ, Do LH, Lippard SJ. J. Am. Chem. Soc. 2009;131:7964–7965. doi: 10.1021/ja9030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Randall DW, George SD, Holland PL, Hedman B, Hodgson KO, Tolman WB, Solomon EI. J. Am. Chem. Soc. 2000;122:11632–11648. [Google Scholar]
- 14.Brown EC, York JT, Antholine WE, Ruiz E, Alvarez S, Tolman WB. J. Am. Chem. Soc. 2005;127:13752–13753. doi: 10.1021/ja053971t. [DOI] [PubMed] [Google Scholar]
- 15.Zhu D, Budzelaar PHM. Dalton Trans. 2013;42:11343–11354. doi: 10.1039/c3dt50715g. [DOI] [PubMed] [Google Scholar]
- 16.Hayes PG, Piers WE, Lee LWM, Knight LK, Parvez M, Elsegood MRJ, Clegg W. Organometallics. 2001;20:2533–2544. [Google Scholar]
- 17.Hayes PG, Piers WE, Parvez M. J. Am. Chem. Soc. 2003;125:5622–5623. doi: 10.1021/ja034680s. [DOI] [PubMed] [Google Scholar]
- 18.Smith JM, Lachicotte RJ, Holland PL. Chem. Commun. 2001:1542–1543. doi: 10.1039/b103635c. DOI: 10.1039/B103635C. [DOI] [PubMed] [Google Scholar]
- 19.Eckert NA, Smith JM, Lachicotte RJ, Holland PL. Inorg. Chem. 2004;43:3306–3321. doi: 10.1021/ic035483x. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780–783. doi: 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vela J, Smith JM, Yu Y, Ketterer NA, Flaschenriem CJ, Lachicotte RJ, Holland PL. J. Am. Chem. Soc. 2005;127:7857–7870. doi: 10.1021/ja042672l. [DOI] [PubMed] [Google Scholar]
- 22.Holland PL, Cundari TR, Perez LL, Eckert NA, Lachicotte RJ. J. Am. Chem. Soc. 2002;124:14416–14424. doi: 10.1021/ja025583m. [DOI] [PubMed] [Google Scholar]
- 23.Wei Gao YM, Li Guang-hua, Liu Xiao-ming, Su Qing, Yao Wei. Chem. Res. Chin. Univ. 2005;21:240. [Google Scholar]
- 24.Eckert NA, Bones EM, Lachicotte RJ, Holland PL. Inorg. Chem. 2003;42:1720–1725. doi: 10.1021/ic025986n. [DOI] [PubMed] [Google Scholar]
- 25.Wiencko HL, Kogut E, Warren TH. Inorg. Chim. Acta. 2003;345:199–208. [Google Scholar]
- 26.Horn B, Pfirrmann S, Limberg C, Herwig C, Braun B, Mebs S, Metzinger R. Z. Anorg. Allg. Chem. 2011;637:1169–1174. [Google Scholar]
- 27.Eckert NA, Dinescu A, Cundari TR, Holland PL. Inorg. Chem. 2005;44:7702–7704. doi: 10.1021/ic0510213. [DOI] [PubMed] [Google Scholar]
- 28.Wiese S, Aguila MJB, Kogut E, Warren TH. Organometallics. 2013;32:2300–2308. [Google Scholar]
- 29.Jazdzewski BA, Holland PL, Pink M, Young VG, Spencer DJE, Tolman WB. Inorg. Chem. 2001;40:6097–6107. doi: 10.1021/ic010615c. [DOI] [PubMed] [Google Scholar]
- 30.Spencer DJE, Reynolds AM, Holland PL, Jazdzewski BA, Duboc-Toia C, Le Pape L, Yokota S, Tachi Y, Itoh S, Tolman WB. Inorg. Chem. 2002;41:6307–6321. doi: 10.1021/ic020369k. [DOI] [PubMed] [Google Scholar]
- 31.Wiese S, Badiei YM, Gephart RT, Mossin S, Varonka MS, Melzer MM, Meyer K, Cundari TR, Warren TH. Angew. Chem. Int. Ed. 2010;49:8850–8855. doi: 10.1002/anie.201003676. [DOI] [PubMed] [Google Scholar]
- 32.Hadzovic A, Song D. Inorg. Chem. 2008;47:12010–12017. doi: 10.1021/ic801557c. [DOI] [PubMed] [Google Scholar]
- 33.Hadzovic A, Song D. Organometallics. 2008;27:1290–1298. [Google Scholar]
- 34.Chang K-C, Lu C-F, Wang P-Y, Lu D-Y, Chen H-Z, Kuo T-S, Tsai Y-C. Dalton Trans. 2011;40:2324–2331. doi: 10.1039/c0dt01061h. [DOI] [PubMed] [Google Scholar]
- 35.Fan H, Adhikari D, Saleh AA, Clark RL, Zuno-Cruz FJ, Sanchez Cabrera G, Huffman JC, Pink M, Mindiola DJ, Baik M-H. Journal of the American Chemical Society. 2008;130:17351–17361. doi: 10.1021/ja803798b. [DOI] [PubMed] [Google Scholar]
- 36.Gibson Vernon C., Newton C, Redshaw C, Solan Gregory A., White Andrew J. P., Williams David J. Eur. J. Inorg. Chem. 2001;2001:1895–1903. [Google Scholar]
- 37.Charbonneau F, Oguadinma PO, Schaper F. Inorg. Chim. Acta. 2010;363:1779–1784. [Google Scholar]
- 38.MacLeod KC, Patrick BO, Smith KM. Inorg. Chem. 2012;51:688–700. doi: 10.1021/ic202233f. [DOI] [PubMed] [Google Scholar]
- 39.Sadique AR, Gregory EA, Brennessel WW, Holland PL. J. Am. Chem. Soc. 2007;129:8112–8121. doi: 10.1021/ja069199r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith JM, Lachicotte RJ, Holland PL. J. Am. Chem. Soc. 2003;125:15752–15753. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]
- 41.Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J. Am. Chem. Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 42.Yao S, Xiong Y, Milsmann C, Bill E, Pfirrmann S, Limberg C, Driess M. Chem. Eur. J. 2010;16:436–439. doi: 10.1002/chem.200902820. [DOI] [PubMed] [Google Scholar]
- 43.Spencer DJE, Reynolds AM, Holland PL, Jazdzewski BA, Duboc-Toia C, Le Pape L, Yokota S, Tachi Y, Itoh S, Tolman WB. Inorganic Chemistry. 2002;41:6307–6321. doi: 10.1021/ic020369k. [DOI] [PubMed] [Google Scholar]
- 44.Hong S, Hill LMR, Gupta AK, Naab BD, Gilroy JB, Hicks RG, Cramer CJ, Tolman WB. Inorg. Chem. 2009;48:4514–4523. doi: 10.1021/ic9002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai X, Warren TH. Chem. Commun. 2001:1998–1999. doi: 10.1039/b105244f. DOI: 10.1039/B105244F. [DOI] [PubMed] [Google Scholar]
- 46.Shimokawa C, Yokota S, Tachi Y, Nishiwaki N, Ariga M, Itoh S. Inorg. Chem. 2003;42:8395–8405. doi: 10.1021/ic0344766. [DOI] [PubMed] [Google Scholar]
- 47.Lee LWM, Piers WE, Elsegood MRJ, Clegg W, Parvez M. Organometallics. 1999;18:2947–2949. [Google Scholar]
- 48.Knight LK, Piers WE, Fleurat-Lessard P, Parvez M, McDonald R. Organometallics. 2004;23:2087–2094. [Google Scholar]
- 49.Basuli F, Tomaszewski J, Huffman JC, Mindiola DJ. Organometallics. 2003;22:4705–4714. [Google Scholar]
- 50.Hayes PG, Piers WE, Parvez M. Chem. Eur. J. 2007;13:2632–2640. doi: 10.1002/chem.200601087. [DOI] [PubMed] [Google Scholar]
- 51.Basuli F, Bailey BC, Watson LA, Tomaszewski J, Huffman JC, Mindiola DJ. Organometallics. 2005;24:1886–1906. [Google Scholar]
- 52.Budzelaar PHM, van Oort AB, Orpen AG. Eur. J. Inorg. Chem. 1998;1998:1485–1494. [Google Scholar]
- 53.Hamaki H, Takeda N, Tokitoh N. Organometallics. 2006;25:2457–2464. [Google Scholar]
- 54.Basuli F, Bailey BC, Tomaszewski J, Huffman JC, Mindiola DJ. J. Am. Chem. Soc. 2003;125:6052–6053. doi: 10.1021/ja034786n. [DOI] [PubMed] [Google Scholar]
- 55.Kim W-K, Fevola MJ, Liable-Sands LM, Rheingold AL, Theopold KH. Organometallics. 1998;17:4541–4543. [Google Scholar]
- 56.Tsai Y-C, Wang P-Y, Lin K-M, Chen S-A, Chen J-M. Chem. Commun. 2008:205–207. doi: 10.1039/b711816c. DOI: 10.1039/B711816C. [DOI] [PubMed] [Google Scholar]
- 57.Li X, Ding J, Jin W, Cheng Y. Inorg. Chim. Acta. 2009;362:233–237. [Google Scholar]
- 58.York JT, Young VG, Tolman WB. Inorg. Chem. 2006;45:4191–4198. doi: 10.1021/ic060050q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reynolds AM, Lewis EA, Aboelella NW, Tolman WB. Chem. Commun. 2005:2014–2016. doi: 10.1039/b418939f. DOI: 10.1039/B418939F. [DOI] [PubMed] [Google Scholar]
- 60.Badiei YM, Warren TH. J. Organomet. Chem. 2005;690:5989–6000. [Google Scholar]
- 61.Carrera N, Savjani N, Simpson J, Hughes DL, Bochmann M. Dalton Trans. 2011;40:1016–1019. doi: 10.1039/c0dt01422b. [DOI] [PubMed] [Google Scholar]
- 62.Doherty JC, Ballem KHD, Patrick BO, Smith KM. Organometallics. 2004;23:1487–1489. [Google Scholar]
- 63.Champouret Y, MacLeod KC, Baisch U, Patrick BO, Smith KM, Poli R. Organometallics. 2010;29:167–176. [Google Scholar]
- 64.Inosako M, Kunishita A, Shimokawa C, Teraoka J, Kubo M, Ogura T, Sugimoto H, Itoh S. Dalton Trans. 2008:6250–6256. doi: 10.1039/b808678h. DOI: 10.1039/b808678h. [DOI] [PubMed] [Google Scholar]
- 65.Yokota S, Tachi Y, Itoh S. Inorg. Chem. 2002;41:1342–1344. doi: 10.1021/ic0156238. [DOI] [PubMed] [Google Scholar]
- 66.Brown EC, Aboelella NW, Reynolds AM, Aullón G, Alvarez S, Tolman WB. Inorg. Chem. 2004;43:3335–3337. doi: 10.1021/ic049811k. [DOI] [PubMed] [Google Scholar]
- 67.Brown EC, Bar-Nahum I, York JT, Aboelella NW, Tolman WB. Inorg. Chem. 2007;46:486–496. doi: 10.1021/ic061589r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hill LMR, Gherman BF, Aboelella NW, Cramer CJ, Tolman WB. Dalton Trans. 2006:4944–4953. doi: 10.1039/b609939d. DOI: 10.1039/b609939d. [DOI] [PubMed] [Google Scholar]
- 69.Kenward AL, Ross JA, Piers WE, Parvez M. Organometallics. 2009;28:3625–3628. [Google Scholar]
- 70.Basuli F, Kilgore UJ, Brown D, Huffman JC, Mindiola DJ. Organometallics. 2004;23:6166–6175. [Google Scholar]
- 71.Kakaliou L, Scanlon WJ, Qian BX, Baek SW, Smith MR, Motry DH. Inorganic Chemistry. 1999;38:5964–5977. doi: 10.1021/ic981364j. [DOI] [PubMed] [Google Scholar]
- 72.MacLeod KC, Conway JL, Tang L, Smith JJ, Corcoran LD, Ballem KHD, Patrick BO, Smith KM. Organometallics. 2009;28:6798–6806. [Google Scholar]
- 73.Huang Y-B, Jin G-X. Dalton Transactions. 2009:767–769. doi: 10.1039/b820798b. DOI: 10.1039/B820798B. [DOI] [PubMed] [Google Scholar]
- 74.Yu Y, Brennessel WW, Holland PL. Organometallics. 2007;26:3217–3226. doi: 10.1021/om7003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cowley RE, Elhaïk J, Eckert NA, Brennessel WW, Bill E, Holland PL. J. Am. Chem. Soc. 2008;130:6074–6075. doi: 10.1021/ja801375g. [DOI] [PubMed] [Google Scholar]
- 76.Vela J, Vaddadi S, Cundari TR, Smith JM, Gregory EA, Lachicotte RJ, Flaschenriem CJ, Holland PL. Organometallics. 2004;23:5226–5239. [Google Scholar]
- 77.Vela J, Smith JM, Lachicotte RJ, Holland PL. Chem. Commun. 2002:2886–2887. doi: 10.1039/b209389h. DOI: 10.1039/B209389H. [DOI] [PubMed] [Google Scholar]
- 78.Stoian SA, Yu Y, Smith JM, Holland PL, Bominaar EL, Munck E. Inorg. Chem. 2005;44:4915–4922. doi: 10.1021/ic050321h. [DOI] [PubMed] [Google Scholar]
- 79.Yu Y, Smith JM, Flaschenriem CJ, Holland PL. Inorg. Chem. 2006;45:5742–5751. doi: 10.1021/ic052136+. [DOI] [PubMed] [Google Scholar]
- 80.Panda A, Stender M, Wright RJ, Olmstead MM, Klavins P, Power PP. Inorg. Chem. 2002;41:3909–3916. doi: 10.1021/ic025552s. [DOI] [PubMed] [Google Scholar]
- 81.Oguadinma PO, Schaper F. Inorg. Chim. Acta. 2009;362:570–574. [Google Scholar]
- 82.Hill LMR, Gherman BF, Aboelella NW, Cramer CJ, Tolman WB. Dalton Transactions. 2006:4944–4953. doi: 10.1039/b609939d. DOI: 10.1039/B609939D. [DOI] [PubMed] [Google Scholar]
- 83.Phillips AD, Zava O, Scopelitti R, Nazarov AA, Dyson PJ. Organometallics. 2010;29:417–427. [Google Scholar]
- 84.Phillips AD, Laurenczy G, Scopelliti R, Dyson PJ. Organometallics. 2007;26:1120–1122. [Google Scholar]
- 85.Schreiber DF, Ortin Y, Müller-Bunz H, Phillips AD. Organometallics. 2011;30:5381–5395. [Google Scholar]
- 86.Phillips AD, Thommes K, Scopelliti R, Gandolfi C, Albrecht M, Severin K, Schreiber DF, Dyson PJ. Organometallics. 2011;30:6119–6132. [Google Scholar]
- 87.Kakaliou L, Scanlon, Qian B, Baek SW, Smith MR, Motry DH. Inorg. Chem. 1999;38:5964–5977. doi: 10.1021/ic981364j. [DOI] [PubMed] [Google Scholar]
- 88.Chen C, Dugan TR, Brennessel WW, Weix DJ, Holland PL. J. Am. Chem. Soc. 2014;136:945–955. doi: 10.1021/ja408238n. [DOI] [PubMed] [Google Scholar]
- 89.Young J, Yap GA, Theopold K. J. Chem. Crystallogr. 2009;39:846–848. [Google Scholar]
- 90.Shimokawa C, Teraoka J, Tachi Y, Itoh S. J. Inorg. Biochem. 2006;100:1118–1127. doi: 10.1016/j.jinorgbio.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 91.Bernoud E, Oulié P, Guillot R, Mellah M, Hannedouche J. Angew. Chem. Int. Ed. 2014;53:4930–4934. doi: 10.1002/anie.201402089. [DOI] [PubMed] [Google Scholar]
- 92.Huang H, Hughes RP, Rheingold AL. Polyhedron. 2008;27:734–738. [Google Scholar]
- 93.Annibale VT, Tan R, Janetzko J, Lund LM, Song D. Inorg. Chim. Acta. 2012;380:308–321. [Google Scholar]
- 94.Qian B, Scanlon, Smith MR, Motry DH. Organometallics. 1999;18:1693–1698. [Google Scholar]
- 95.Dulong F, Thuéry P, Ephritikhine M, Cantat T. Organometallics. 2013;32:1328–1340. [Google Scholar]
- 96.Knight LK, Piers WE, McDonald R. Chem. Eur. J. 2000;6:4322–4326. doi: 10.1002/1521-3765(20001201)6:23<4322::aid-chem4322>3.3.co;2-s. [DOI] [PubMed] [Google Scholar]
- 97.Zhou W, Tang L, Patrick BO, Smith KM. Organometallics. 2011;30:603–610. [Google Scholar]
- 98.Zhang J, Gao H, Ke Z, Bao F, Zhu F, Wu Q. J. Mol. Catal. A: Chem. 2005;231:27–34. [Google Scholar]
- 99.Yu Y, Sadique AR, Smith JM, Dugan TR, Cowley RE, Brennessel WW, Flaschenriem CJ, Bill E, Cundari TR, Holland PL. J. Am. Chem. Soc. 2008;130:6624–6638. doi: 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cowley RE, Holland PL. Inorg. Chem. 2012;51:8352–8361. doi: 10.1021/ic300870y. [DOI] [PubMed] [Google Scholar]
- 101.Guzei IA, Wendt M. Dalton Trans. 2006:3991–3999. doi: 10.1039/b605102b. DOI: 10.1039/B605102B. [DOI] [PubMed] [Google Scholar]
- 102.Zhang J, Ke Z, Bao F, Long J, Gao H, Zhu F, Wu Q. J. Mol. Catal. A: Chem. 2006;249:31–39. [Google Scholar]
- 103.Li Y, Wang L, Gao H, Zhu F, Wu Q. J. Appl. Organomet. Chem. 2006;20:436–442. [Google Scholar]
- 104.Li Y, Gao M, Wu Q. J. Appl. Organomet. Chem. 2008;22:659–663. [Google Scholar]
- 105.Li Y, Gao M, Wu Q. J. Appl. Organomet. Chem. 2007;21:965–969. [Google Scholar]
- 106.Li Y, Jiang L, Wang L, Gao H, Zhu F, Wu Q. J. Appl. Organomet. Chem. 2006;20:181–186. [Google Scholar]
- 107.Li Y, Wu Q, Shan M, Gao M. J. Appl. Organomet. Chem. 2012;26:225–229. [Google Scholar]
- 108.Li Y, Gao M, Gao H, Wu Q. Eur. Polym J. 2011;47:1964–1969. [Google Scholar]
- 109.Lee D-H, Kwon Y-J, Jin M-J. Adv. Synth. Catal. 2011;353:3090–3094. [Google Scholar]
- 110.Lee D-H, Qian Y, Park J-H, Lee J-S, Shim S-E, Jin M-J. Adv. Synth. Catal. 2013;355:1729–1735. [Google Scholar]
- 111.Lee D-H, Jung J-Y, Jin M-J. Chem. Commun. 2010;46:9046–9048. doi: 10.1039/c0cc03535a. [DOI] [PubMed] [Google Scholar]
- 112.Champouret Y, Baisch U, Poli R, Tang L, Conway JL, Smith KM. Angew. Chem. Int. Ed. 2008;47:6069–6072. doi: 10.1002/anie.200801498. [DOI] [PubMed] [Google Scholar]
- 113.MacLeod KC, Conway JL, Patrick BO, Smith KM. J. Am. Chem. Soc. 2010;132:17325–17334. doi: 10.1021/ja1083392. [DOI] [PubMed] [Google Scholar]
- 114.Cowley RE, Golder MR, Eckert NA, Al-Afyouni MH, Holland PL. Organometallics. 2013;32:5289–5298. [Google Scholar]
- 115.Amisial LD, Dai X, Kinney RA, Krishnaswamy A, Warren TH. Inorg. Chem. 2004;43:6537–6539. doi: 10.1021/ic048968+. [DOI] [PubMed] [Google Scholar]
- 116.Li Y, Gao H, Wu Q. J. Polym. Sci., Part A: Polym. Chem. 2008;46:93–101. [Google Scholar]
- 117.Zhou W, MacLeod KC, Patrick BO, Smith KM. Organometallics. 2012;31:7324–7327. [Google Scholar]
- 118.Rajendran NM, Maheswari K, Reddy ND. Polyhedron. 2014;81:329–334. [Google Scholar]
- 119.Takaichi J, Morimoto Y, Ohkubo K, Shimokawa C, Hojo T, Mori S, Asahara H, Sugimoto H, Fujieda N, Nishiwaki N, Fukuzumi S, Itoh S. Inorg. Chem. 2014;53:6159–6169. doi: 10.1021/ic5006693. [DOI] [PubMed] [Google Scholar]
- 120.Costas M, Chen K, Que L., Jr Coord. Chem. Rev. 2000;200–202:517–544. [Google Scholar]
- 121.Gao H, Pei L, Li Y, Zhang J, Wu Q. J. Mol. Catal. A: Chem. 2008;280:81–86. [Google Scholar]
- 122.Oguadinma PO, Schaper F. Organometallics. 2009;28:4089–4097. [Google Scholar]
- 123.El-Zoghbi I, Latreche S, Schaper F. Organometallics. 2010;29:1551–1559. [Google Scholar]
- 124.Binda PI, Abbina S, Du G. Synthesis. 2011;2011:2609–2618. [Google Scholar]
- 125.Ellis WC, Jung Y, Mulzer M, Di Girolamo R, Lobkovsky EB, Coates GW. Chem. Sci. 2014;5:4004–4011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.