

Abstract
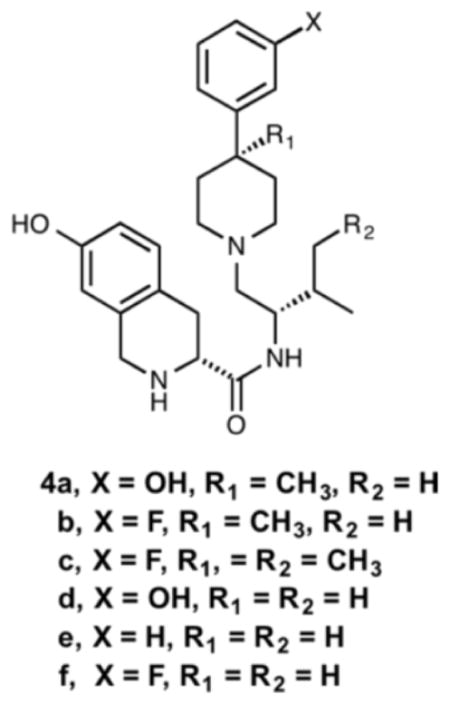
The design and discovery of JDTic as a potent and selective kappa opioid receptor antagonist used the N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine pharmacophore as the lead structure. In order to determine if the 3-methyl or 4-methyl groups were necessary in JDTic and JDTic analogs for antagonistic activity, compounds 4a–c, and 4d–f which have either the 3-methyl or both the 3- and 4-methyl groups removed, respectively, from JDTic and analogs were synthesized and evaluated for their in vitro opioid receptor antagonist activities using a [35S]GTPγS binding assay. Other ADME properties were also assessed for selected compounds. These studies demonstrated that neither the 3-methyl or 3,4-dimethyl groups present in JDTic and analogs are required to produce potent and selective κ opioid receptor antagonists.
Keywords: JDTic, opioids, kappa antagonist, ADME properties
Graphical Abstract
1. Introduction
The design and discovery of JDTic as a potent and selective kappa opioid receptor antagonist used the N-substituted trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine (1) pharmacophore as the lead structure.[1, 2] (Fig. 1) In a recent study we reported that N-phenylpropyl-4-methyl-4-(3-hydroxyphenyl)-piperidine (2a) where the 3-methyl group was removed from 1 and N-phenylpropyl-4-(3-hydroxyphenyl)-piperidine (2b), where both the 3- and 4-methyl groups were removed from 1 were still pure opioid receptor antagonists.[3] In another recent study we reported that the replacement of the hydroxy group in the 4-(3-hydroxyphenyl) moiety of JDTic with a hydrogen or fluoro group gave compounds 3a and 3b both of which were potent and selective kappa opioid receptor antagonists.[4] (Fig. 1) As a continuation of our structural activity relationship (SAR) studies directed toward the kappa opioid receptor as well as the development of potential pharmacotherapies for CNS disorders, compounds 4a–f were synthesized and evaluated in vitro using a [35S]GTPγS binding assay at the μ, δ, and κ opioid receptors and their ADME properties were established. (Fig. 1) A comparison of their in vitro antagonist activity and ADME properties of selected compounds to those of JDTic and compounds 3a and 3b are herein presented. In addition, since the synthesis and [35S]GTPγS in vitro properties of compounds 4d were recently reported, we compare the results from the two studies.[5]
Figure 1.
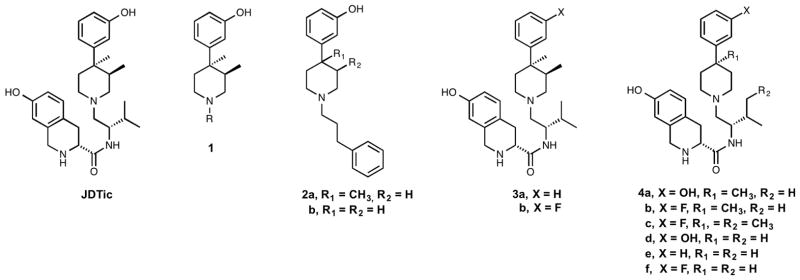
Structures of JDTic, 2a–b, 3a–b, and 4a–f.
2. Chemistry
Compound 4a was prepared according to Scheme 1. 3-Bromiosopropoxybenzene was prepared and distilled from the corresponding phenol and isopropyl bromide to be used as substrate for the lithium-halogen exchange. Addition of the resulting aryl lithium to N-methylpiperidione 5 gave the alcohol 6 in 42% yield. Dehydration of 6 with 2 eq. p-toluenesulfonic acid in toluene gave tetrahydropyridine 7 in 79% yield. Methylation according to the method reported by Werner et al.[6] was followed by NaBH4 reduction of the enamine intermediate to afford 8. Treatment of 8 with 1-chloroethyl chloroformate (ACE-Cl) in refluxing dichloroethane followed by methanolysis yielded piperidine 9. Reductive amination of amine 9 with Boc-L-Valinal[7] using sodium cyanoborohydride in trifluoroethanol followed by Boc-deprotection afforded 10. HBTU coupling of 10 with Boc-7-hydroxy-D-Tic-OH followed by deprotection with hydrogen bromide in acetic acid afforded 4a.
Scheme 1.

Reagents and Conditions: a) 3-isopropoxyphenyl lithium, THF; b) TsOH, toluene; c) 1. n-BuLi, THF, −15 °C; 2. Me2SO4, −50 °C; 3. NaBH4, CH3OH; d) 1. 1-chloroethyl chloroformate, DCE; 2) CH3OH, reflux; e) 1. Boc-L-valinal, NaBH3CN; 2. TFA, DCM; f) 1. Boc-7-hydroxy-D-Tic-OH, HBTU, NEt3; 2. HBr, AcOH.
Compound 4b was prepared as outlined in Scheme 2. 1-Methyl-4-(3-fluorophenyl)-1,2,5,6-tetrahydropyridine (11) was prepared according to the method of Nagai et al.[8] The deprotonation and methylation proceeded analogously to the method described by Werner.[6] The resulting enamine was reduced with sodium borohydride in methanol. Demethylation with ACE-Cl followed by heating at reflux in methanol to hydrolyze the intermediate carbamate afforded 4-(3-fluorophenyl)-4-methylpiperidine (12). Piperidine 12 was coupled with Boc-L-valine using HBTU in acetonitrile. The tert-butyloxycarbonyl protecting group was removed with aqueous hydrogen chloride in methanol so that the amide could be reduced cleanly with borane dimethylsulfide at reflux in tetrahydrofuran. The resulting amine (13) was coupled with Boc-7-hydroxy-D-Tic-OH using 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (EDC • HCl) in dichloromethane followed by treatment with hydrogen chloride in dioxane and acetonitrile to afford 4b.
Scheme 2.
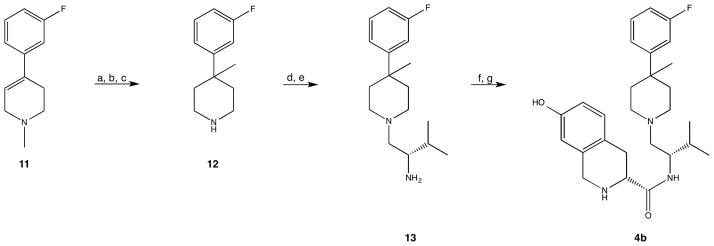
Reagents and conditions: a) 1. n-BuLi, THF, −78 °C; 2. Me2SO4; b) NaBH4, CH3OH; c) 1. ACE-Cl, DCE; 2. CH3OH, reflux; d) HBTU, NEt3, Boc-L-valine, CH3CN; e) 1. HCl; 2. BH3 • SMe2; f) Boc-7-hydroxy-D-Tic-OH, EDC • HCl, HOBt, DIPEA, THF; g) HCl, CH3CN, dioxane.
Compound 4c was prepared from 12 via a procedure analogous to that used for 4b by substituting Boc-L-isoleucine for Boc-L-valine as outlined in Scheme 3. Piperidine 12 was first coupled to Boc-L-isoleucine using EDC and HOBt in acetonitrile followed by removal of the Boc protecting group present with aqueous hydrogen chloride in methanol so that the amide could be reduced cleanly with borane dimethylsulfide at reflux in tetrahydrofuran to give 14. The resulting amine (14) was coupled with Boc-7-hydroxy-D-Tic-OH using EDC•HCl in dichloromethane followed by treatment with hydrogen chloride in dioxane and acetonitrile to afford 4c.
Scheme 3.
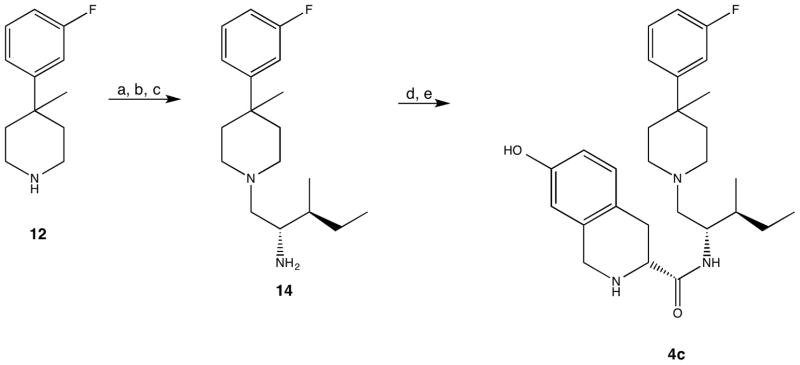
Reagents and conditions: a) Boc-L-isoleucine, HOBt, EDC, DIPEA, CH3CN; b) HCl, CH3OH; c) BH3 • S(CH3)2; d) Boc-7-hydroxy-D-Tic-OH, EDC • HCl, DIPEA, HOBt, THF; e) HCl, CH3CN, dioxane.
The synthesis of 4f is outlined in Scheme 4. Coupling of N-Boc-L-valine with 4-(3-fluorophenyl)piperidine 15 using HBTU in acetonitrile followed by cleavage of the tert-butyloxycarbonyl protecting group in intermediate tert-butyl [(1S)-1-{[4-(3-fluorophenyl)piperidin-1-yl]carbonyl}-2-methylpropyl]carbamate 16 using concentrated hydrochloric acid in methanol gave 17. Reduction of 17 with diborane in tetrahydrofuran gave 18. Coupling of 18 with Boc-7-hydroxy-D-Tic-OH using EDC and catalytic amount of N-hydroxybenzotriazole (HOBt) in dichloromethane provided tert-butyl (3R)-3-{[(1S)-1-{[4-(3-fluorophenyl)piperidin-1-yl]methyl}-2-methylpropyl]carbamoyl}-7-hydroxy-3,4-dihydroisoquinoline-2(1H)-carboxylate 19. Compound 4f was obtained by treating 19 with concentrated hydrochloric acid in methanol.
Scheme 4.

Reagents: a) N-Boc-L-valine, HBTU, CH3CN, TEA; b) HCl, MeOH; c) B2H6, THF; d) EDC, HOBt, TEA, Boc-7-hydroxy-D-Tic-OH, DCM.
Compounds 4d and 4e were synthesized by procedures analogous to the reported synthesis.[5]
3. Results and Discussion
In order to determine if the 3-methyl group on the piperidine ring of JDTic was required to obtain a pure antagonist, we synthesized and tested 4a for its opioid antagonism properties. We found that 4a had no agonist activity at 10 μM in the [35S]GTPγS binding assay and was indeed a pure opioid receptor antagonist (Table 1). With Ke values of 67.5, 927, and 0.69 nM at the μ, δ, and κ opioid receptors, respectively, it was a potent and selective kappa opioid receptor antagonist. However, it was 35-times less potent than JDTic as a kappa opioid receptor antagonist (Table 1).
Table 1.
Inhibition of Agonist-Stimulated [35S]GTPγS Binding by Compounds in Cloned Human μ, δ, and κ Opioid Receptors
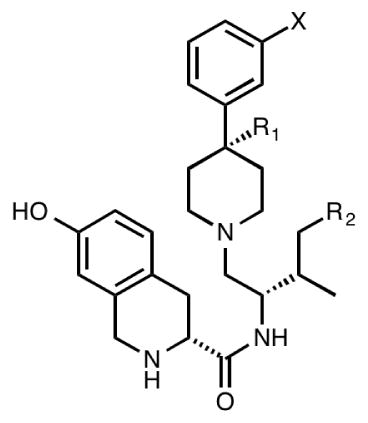
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | Ke (nM)a | |||||||
|
| ||||||||
| X | R1 | R2 | μ, DAMGO | δ, DPDPE | κ, U69,593 | μ/κ | δ/κ | |
| nor-BNI | — | — | — | 26 | 29 | 0.05 | 520 | 580 |
| JDTic | — | — | — | 25 ± 4 | 74 ± 2 | 0.02 | 1255 | 3800 |
| 3a | — | — | — | 8.9 | 442 | 0.024 | 370 | 18,400 |
| 3b | — | — | — | 14.8 | 249 | 0.01 | 1480 | 24,900 |
| 4a | OH | CH3 | H | 67.5 ± 10.4 | 927 ± 350 | 0.69 ± 0.21 | 98 | 1344 |
| 4b | F | CH3 | H | 38.0 ± 6.4 | 1480 ± 350 | 0.18 ± 0.04 | 211 | 8211 |
| 4c | F | CH3 | CH3 | 11.3 ± 4.3 | 903 ± 200 | 0.033 ± 0.01 | 342 | 27,363 |
| 4d | OH | H | H | 12.3 ± 1.0 | 240 ± 73 | 0.10 ± 0.03 | 123 | 2400 |
| 4e | H | H | H | 3.96 ± 1.1 | 281 ± 44 | 0.051 ± 0.01 | 77 | 5500 |
| 4f | F | H | H | 3.55 ± 1.1 | 128 ± 36 | 0.023 ± 0.01 | 154 | 5600 |
Ke values are the mean ±SEM of at least three independent experiments performed in duplicate.
Replacement of the 3-OH in 4a with a fluoro group gave 4b which was also a pure opioid antagonist in the [35S]GTPγS test (Table 1). Compound 4b, which has a Ke = 0.18 nM at the κ receptor is only 9- and 18-fold less potent than JDTic and 3b as a κ opioid antagonist (Table 1). With Ke values of 38 and 1480 nM at the μ and δ receptors, respectively, the compound 4b remains highly selective for the κ relative to the μ and δ opioid receptors. Compound 4c, which has the isopropyl group in 4b replaced with an isobutyl group, has Ke values of 11.3, 903, and 0.033 nM at the μ, δ, and κ receptors, respectively. Thus, 4c is 6-fold more potent than 4b as a κ opioid receptor antagonist.
Compounds 4d, 4e, and 4f have both the 3- and 4-methyl groups removed from the piperidine ring, relative to JDTic, 3a and 3b, respectively (Table 1). Compound 4d with a Ke = 0.1 nM at the κ receptor is 5-times less potent than JDTic as a κ antagonist in this assay (Table 1). Having Ke values of 12.3 and 240 nM at the μ and δ receptors, respectively, 4d is 123-fold and 2400-fold selective for the κ receptor relative to the μ and δ receptors, respectively. Compound 4e also has the 3-OH removed. With a Ke = 0.051 nM at the κ receptor, this compound is only 2.6- and 2-times less potent as a κ antagonist than JDTic and 3a, respectively. Compound 4e has Ke values of 3.96 and 281 nM at the μ and δ receptors, respectively, and is 77- and 5500-fold selective for the κ receptor relative to the μ and δ receptors, respectively. Compound 4f like 3b has a 3-fluoro substituent in place of the 3-OH in JDTic and 4d. Similar to JDTic, 4f with a Ke = 0.023 nM at the κ receptor is a potent κ antagonist. Compound 4f has a Ke = 3.55 and 128 nM at the μ and δ receptors, respectively. Thus, 4f is 7- and 4-times more potent as a μ antagonist than JDTic and 3b, respectively, and is 154- and 5600-fold selective for the κ receptor relative to the μ and δ receptors, respectively.
Compound 4d has a structure identical to that of a compound named AT-076 reported by Zaveri and coworkers.[5] These authors report Ke values of 0.48, 24.95, and 4.3 nM at the μ, δ, and κ opioid receptors for AT-076 compared to Ke values of 12.3, 240, and 0.1 nM determined in this study for 4d using a [35S]GTPγS binding assay. Although the Zaveri et al Ke values for the mu and delta receptors are lower than those for 4d, both compounds show approximately the same relative selectivity for these receptors, but their Ke value is 43-fold higher for the κ receptor than that determined for 4d. Differences in opioid receptor-effector coupling efficiency cannot explain this difference because the Ke is a ratiometric number that is calculated from the rightward shift in the agonist EC50 in the presence of antagonist.[9] However, differences in our Ke assay methods could affect receptor conformation altering the manner in which this ligand interacts with these receptors.
In order to determine if the compounds would be predicted to cross the blood/brain barrier, their topological polar surface area (TPSA), clogP, and derived logBB values were calculated and compared to JDTic, 3a and 3b. In general CNS compounds that have TPSA values less than 76Å2[10], clogP values in the range of 2–4[11], and derived logBB values greater than −1[12] are predicted to cross the blood/brain barrier. Compounds 3b, 4f and 4c have TPSA values less than 76Å2. JDTic and 4a with TPSA values of 84.83Å2 are a little greater than 76Å2. JDTic, 4a, and 4b have clogP values in the range of 2–4. Compounds 3b and 4c have clogP values a little above 4. All of the compounds have logBB values greater than −1 and thus, would be predicted to penetrate the brain.
Compound 4d has a calculated TPSA value of 84.83 Å2, which is identical to that of JDTic. Compounds 4e and 4f have TPSA values of 64.60 Å2, which are identical to those of 3a and 3b. Compounds 4d, 4e and 4f all have clogP values less than 4 and greater than 2. Compounds 4d, 4e and 4f with logBB values of greater than −1 would be predicted to have good brain penetration.
ADME properties of these compounds were assessed (Table 2). Compounds that interact with the human ether-a-go-go gene (hERG) product, which is a potassium channel, can produce QT prolongation and cardiotoxic effects. The hERG Ki values of 4a–f were compared to the Ki value for JDTic, 3a and 3b (Table 2). Compound 4a had a Ki = >10 μM which is larger than the Ki values for JDTic (Ki = 8.820 μM). All the other compounds (4b–f) had Ki values lower than JDTic.
Table 2.
In vitro ADME data for 3a, 3b, and 4 a–f
| Compd | hERG (Ki, μM) | MDCK-mdr1 (% Transported, A to B) | Solubility (μM)
|
Plasma Stability (% of Parent) | S9 Stability (% of Parent) | PAMPA (% Transported)
|
TPSA | cLogP | LogBB | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pH 7.4 | pH 3 | pH 7.4 | pH 5.5 | ||||||||
| JDTic | 8.820 | 11 | 11 | 34 | 97 | 76 | 26.8 | 57.9 | 84.83 | 3.60 | −0.57 |
| 3a | 7.048 | 27 | 11 | 42 | 67.0 | 82.0 | 91.4 | 67.3 | 64.60 | 3.89 | −0.23 |
| 3b | 6.251 | 6 | 10 | 47 | 49.6 | 77.5 | 19.6 | 3 | 64.60 | 4.15 | −0.19 |
| 4a | >10 | <1 | 44 | 101 | 62.5 | 62.5 | 1 | 2.6 | 84.83 | 3.43 | −0.59 |
| 4b | 4.237 | 1.3 | 64.60 | 3.97 | −0.21 | ||||||
| 4c | 1.666 | 64.60 | 4.38 | −0.15 | |||||||
| 4d | 1.732 | 84.83 | 3.23 | −0.63 | |||||||
| 4e | 0.777 | 64.6 | 3.53 | −0.28 | |||||||
| 4f | 0.436 | 64.60 | 3.75 | −0.25 | |||||||
Compounds that have >5% permeability in the Madin Derby Canine Kidney cells stably expressing the human multidrug resistant gene (mdr1) product P-glycoprotein (MDCK-mdr1) monolayer permeability assay, >50% stability in the plasma and hepatic S9 stability assay, >25% transported in the Parallel Artificial Membrane Permeability Assay (PAMPA), and >20 μM solubility are considered desirable for further development by our group. The MDCK-mdr1 permeability, solubility, plasma, S9 stability and PAMPA permeability properties of 4a were determined and compared to the properties of JDTic (Table 2). The permeability of 4a is negligible across MDCK-mdr1 monolayers compared to 11% for JDTic. However, 4a with solubility of 44 and 101 μM at pH 7.4 and 3, respectively, is more soluble than JDTic. With plasma and S9 stabilities of 62.5% following incubation in both assays, it is not as metabolically stable as JDTic but is above the 50% threshold considered to be desirable. The percent transported values of 1 and 2.6% at pH 7.4 and 5.5 in the PAMPA assay are well below the 26.8 and 57.9 percent values for JDTic. Due to the low Ki values for 4b–f in the hERG assay the ADME properties of the compounds were not determined.
4. Conclusions
In conclusion, JDTic analogs 4a–c and 4d–f which have the 3-methyl or both the 3- and 4-methyl groups removed, respectively, from JDTic and analogs were synthesized and evaluated for their ability to antagonize [35S]GTPγS binding at the μ, δ, and κ opioid receptors. The data showed that neither the 3- or 3,4-dimethyl groups present in JDTic and analogs are required to obtain potent and selectivity kappa opioid receptor antagonists. Compound 4c with a Ke = 0.033 nM at the κ receptor and 342- and 27,360-fold selectivity for the κ relative to the μ and δ receptors, respectively, was the most potent and κ selective compound having only the 3-methyl group removed. Compound 4f with a Ke = 0.023 nM at the κ receptor and 154- and 5600-fold selectivity for the κ relative to the μ and δ receptors was the most potent and κ selective compound having both the 3- and 4-methyl groups removed. However, all of the compounds 4a–f have subnanomolar potency for the κ receptor and are selective for the κ receptor relative to the μ and δ receptors and may be useful pharmacological tools for studying the κ opioid receptor.
5. Experimental
Melting points were determined using a MEL-TEMP II capillary melting point apparatus and are uncorrected. Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were obtained on a Varian Avance DPX-300 MHz NMR spectrometer or a Bruker Unity Inova 500 MHz NMR spectrometer. Chemical shifts are reported in parts per million (ppm) with reference to internal solvent. Mass spectra (MS) were run on a Perkin-Elmer Sciex AP1 150 EX mass spectrometer equipped with APCI (atmospheric pressure chemical ionization) or ESI (turbospray) sources or on a Hewlett Packard 5989A instrument by electron impact. Elemental analyses were performed by Atlantic Microlab Inc., Atlanta, GA. Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research. Analytical thin-layer chromatography (TLC) was carried out using EMD silica gel 60 F254 TLC plates. TLC visualization was achieved with a UV lamp or in an iodine chamber. Flash column chromatography was done on a CombiFlash Companion system using ISCO prepacked silica gel columns or using EM Science silica gel 60A (230–400 mesh). Solvent system: CMA80=80:18:2 CHCl3-MeOH-conc. NH4OH. Unless otherwise stated, reagent-grade chemicals were obtained from commercial sources and were used without further purification. All moisture- and air-sensitive reactions and reagent transfers were carried out under dry nitrogen.
(3R)-7-Hydoxy-N-[(1S)-1-{[4-(3-hydroxyphenyl)-4-methylpiperidin-1-yl]methyl}-2-methylpropyl]-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (4a) dihydrochloride
The amine 10 and Boc-7-hydroxy-D-Tic-OH (1.1 eq.) were combined with HBTU (1.1 eq.) and NEt3 (4 eq.) in DCM (30 mL). The reaction mixture was stirred overnight then concentrated. The residue was subjected to chromatography using EtOAc as the eluent to afford the protected product. The residue resulting from concentration of the desired fractions was refluxed in 1:1 48% HBr:glacial AcOH overnight. The resulting solution was concentrated and the resulting residue was subjected to a gradient of CMA80:CH2Cl2 through silica gel to afford 128 mg of 4a free base (21% over two steps): 1H NMR (CDCl3) δ 7.16 (t, 1H, J = 7.9 Hz), 6.95 (d, 1H, J = 8.1 Hz), 6.85 – 6.78 (m, 2H), 6.69 – 6.62 (m, 2H), 6.54 – 6.49 (m, 1H), 4.07 – 3.97 (m, 1H), 3.96 – 3.90 (m, 1H), 3.10 – 2.95 (m, 1H), 2.83 – 2.29 (m, 6H), 2.17 – 2.00 (m, 2H), 1.86 – 1.67 (m, 2H), 1.22 – 1.16 (m, 3H), 0.96–0.80 (m, 6H); 13C NMR (CDCl3) δ 174.3, 173.7, 156.9, 155.1, 155.0, 136.3, 136.2, 130.2, 129.9, 129.5, 124.8, 124.7, 117.2, 114.0, 113.0, 112.7, 112.3, 112.1, 60.1, 59.8, 57.3, 56.9, 47.3, 47.1, 36.5, 36.4, 36.3, 36.0, 31.3, 31.2, 30.4, 19.8, 17.7, 17.5; MS (ESI) m/z 452.6 (M + H)+; The free base was converted into the dihydrochloride salt: mp 180–184 °C (fusion); [α]25 D +45.6° (c 0.50, CH3OH). Anal. Calcd for C27H39Cl2N3O3 • 1.5 H2O: C, 58.80; H, 7.68; N, 7.62. Found: C, 59.08; H, 7.34; N, 7.34.
(3R)-N-[(1S)-1-{[4-(3-fluorophenyl)-4-methylpiperidin-1-yl]methyl}-2-methylpropyl]-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (4b) dihydrochloride
(2S)-1-[4-(3-Fluorophenyl)-4-methylpiperidin-1-yl]-3-methylbutan-2-amine (13) (274 mg, 0.98 mmol), Boc-7-hydroxy-D-Tic (300 mg, 1.02 mmol), hydroxybenzotriazole hydrate (20 mg, 0.12 mmol), and EDC • HCl(390 mg, 2.0 mmol) were combined in THF (10 mL). Diisopropylethylamine (0.75 mL, 4.3 mmol) was slowly added. The resulting solution was stirred at room temperature for 12 h then concentrated. The residue was dissolved in CH2Cl2 (15 mL) then washed with saturated aqueous NaHCO3 (5 mL). The aqueous layer was extracted once with EtOAc (15 mL). The combined organic layers were washed with brine (5 mL), dried (Na2SO4), and concentrated. The residue was purified by chromatography on silica gel using a gradient up to 40% CMA80 in CH2Cl2. The product containing fractions were combined and concentrated then dissolved in acetonitrile (5 mL) to which HCl in dioxane (4 N, 5 mL) was added. The resulting solution was evaporated under a stream of N2 to afford a white powder. The powder dissolved in a minimum of MeOH was purified by chromatography on silica gel using a gradient up to 50% CMA80 in CH2Cl2 to afford 4b as the free base: 1H NMR (300 MHz, CDCl3) d 7.22 – 7.32 (m, 1H), 7.03 – 7.14 (m, 2H), 6.99 (td, J = 2.03, 11.21 Hz, 1H), 6.82 – 6.92 (m, 2H), 6.55 (dd, J = 2.35, 8.19 Hz, 1H), 6.42 (d, J = 2.26 Hz, 1H), 4.12 – 4.25 (m, 1H), 3.57 – 3.77 (m, 2H), 3.24 (dd, J = 5.18, 11.40 Hz, 1H), 2.78 – 2.98 (m, 2H), 2.46 – 2.69 (m, 3H), 2.06 – 2.43 (m, 5H), 1.73 – 1.91 (m, 3H), 1.19 (s, 3H), 0.84 – 0.96 (m, 6H); 19F NMR (282 MHz, CDCl3) d −112.84 (s, 1F); 13C NMR (75 MHz, CDCl3) δ 173.4, 163.1 (d, J = 245 Hz), 155.0, 151.2 (broad), 137.3, 130.7, 129.9 (d, J = 8.3 Hz), 125.1, 121.3 (d, J = 1.8 Hz), 113.8, 112.9 (d, J = 25.6 Hz), 112.6 (d, J = 24.8 Hz), 112.2, 59.9, 56.7, 50.5, 50.0, 49.8, 48.2, 36.2, 36.1, 31.6, 29.5, 19.0, 17.9. The free base was converted to the dihydrochloride salt, affording 142 mg (26% over two steps) of a white powder: MS (ESI) m/z 454.4 (M + H)+, mp 205–209 °C (fusion), [α]25 D +122 (c 0.1, CH3OH). Anal. Calcd for C27H38Cl2FN3O2•1.5H2O: C, 58.59; H, 7.47; N, 7.59. Found: C, 58.73; H, 7.43, N, 7.30.
(3R)-N-[(1S,2S)-1-{[4-(3-Fluorophenyl)-4-methylpiperidin-1-yl]methyl}-2-methylbutyl]-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (4c) dihydrochloride
(2S,3S)-1-[4-(3-Fluorophenyl)-4-methylpiperidin-1-yl]-3-methylpentan-2-amine (14) (266 mg, 0.91 mmol), Boc-7-hydroxy-D-Tic (293 mg, 1.00 mmol), hydroxybenzotriazole hydrate (20 mg, 0.12 mmol), and EDC • HCl(390 mg, 2.0 mmol) were combined in 10 mL THF. Diisopropylethylamine (0.75 mL, 4.3 mmol) was slowly added. The resulting solution was stirred at room temperature for 12 h then concentrated. The residue was dissolved in CH2Cl2 (15 mL) then washed with saturated aqueous NaHCO3 (5 mL). The aqueous layer was extracted once with EtOAc (15 mL). The combined organic layers were washed with brine (5 mL), dried (Na2SO4), and concentrated. The residue was purified by chromatography on silica gel using a gradient up to 40% CMA80 in CH2Cl2. The product containing fractions were combined and concentrated then dissolved in acetonitrile (10 mL) to which aq. HCl (6 N, 2 mL) was added. The resulting solution was evaporated under a stream of N2 to afford a powder. The powder dissolved in a minimum of MeOH was purified by chromatography on silica gel using a gradient up to 50% CMA80 in CH2Cl2 to afford 4c as the free base: 1H NMR (300 MHz, CDCl3) δ 7.22 – 7.33 (m, 1H), 7.14 (d, J = 9.04 Hz, 1H), 7.06 (d, J = 8.10 Hz, 1H), 6.98 (td, J = 2.03, 11.21 Hz, 1H), 6.83 – 6.92 (m, 2H), 6.50 (dd, J = 2.45, 8.29 Hz, 1H), 6.40 (d, J = 2.26 Hz, 1H), 4.26 (t, J = 9.98 Hz, 1H), 3.52 – 3.75 (m, 2H), 3.20 (dd, J = 5.18, 11.40 Hz, 1H), 2.89 (dd, J = 5.18, 16.48 Hz, 2H), 2.45 – 2.74 (m, 3H), 2.05 – 2.43 (m, 5H), 1.74 – 1.95 (m, 2H), 1.55 – 1.71 (m, 1H), 1.42 (ddd, J = 4.90, 7.54, 13.00 Hz, 1H), 1.17 – 1.23 (m, 3H), 1.04 – 1.17 (m, 1H), 0.83 – 0.96 (m, 6H); 19F NMR (282 MHz, CDCl3) δ −112.82 (s, 1F); 13C NMR (75 MHz, CDCl3) δ 173.2, 163.1 (d, J = 245 Hz), 154.9, 151.1 (broad), 137.5, 130.8, 129.9 (d, J = 8.4 Hz), 125.3, 121.3 (d, J = 2.0 Hz), 113.6, 112.9 (d, J = 21.9 Hz), 112.6 (d, J = 21.2 Hz), 112.1, 112.1, 58.8, 56.6, 50.5, 50.1, 48.8, 48.3, 38.3, 36.2, 36.1, 29.2, 25.7, 14.8, 11.9. The free base was converted to the dihydrochloride salt, affording 216 mg (38% over two steps) of a white powder: MS (ESI) m/z 468.3 (M + H)+, mp 191–195 °C (fusion), [α]25 D +65.2 (c 0.50, CH3OH). Anal. Calcd for C28H40Cl2FN3O2•1.25H2O: C, 59.73; H, 7.61; N, 7.46. Found: C, 59.76; H, 7.69; N, 7.21.
N-[(1S)-1-{[4-(3-Hydroxyphenyl)piperidin-1-yl]methyl}-2-methylpropyl]-3-methyl-4-(3-methylphenoxy)benzamide (4d) dihydrochloride
Compound 4d was synthesized by a procedure analogous to the reported synthesis.[7] 1H NMR (300 MHz, CHLOROFORM-d) δ 6.94 – 7.11 (m, 2H), 6.75 (d, J = 8.29 Hz, 1H), 6.45 – 6.62 (m, 4H), 6.38 (s, 1H), 4.08 (d, J = 9.61 Hz, 1H), 3.75 (d, J = 16.58 Hz, 1H), 3.61 (d, J = 16.39 Hz, 1H), 3.33 (t, J = 7.06 Hz, 1H), 3.09 (d, J = 9.98 Hz, 1H), 2.86 (d, J = 10.36 Hz, 1H), 2.74 (br s, 1H), 2.58 (t, J = 11.59 Hz, 1H), 2.24 (d, J = 10.74 Hz, 2H), 2.08 (br s, 1H), 1.72 (dd, J = 6.31, 11.96 Hz, 2H), 1.60 (br s, 2H), 1.38 (br s, 2H), 0.86 (t, J = 6.69 Hz, 6H); 13C NMR (75 MHz, CHLOROFORM-d) δ 173.4, 171.6, 156.7, 154.7, 147.7, 136.2, 132.7, 130.4, 129.5, 124.7, 119.0, 114.5, 113.5, 112.7, 60.2, 56.4, 52.4, 50.2, 49.3, 46.8, 42.0, 32.7, 32.1, 31.7, 29.5, 19.3, 17.9; ESI MS (M + H)+ 438.6. The product was converted to the dihydrochloride salt by adding 2M HCl in ether to solution of the free base in dichloromethane: mp >220 °C (dec), [α]D = + 58.5 (c 0.62, MeOH). Anal. Calcd for C26H37Cl2N3O3•1.25H2O: C, 58.59; H, 7.47; N, 7.88. Found C, 58.45; H, 7.26; N, 7.78.
(3R)-7-Hydroxy-N-{(1S)-2-methyl-1-[(4-phenylpiperidin-1-yl]methyl]propyl}-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (4e) dihydrochloride
Compound 4e was synthesized by the reported procedure.[7] 1H NMR (300 MHz, CHLOROFORM-d) d 7.29 – 7.24 (m, 2H), 7.20 – 7.10 (m, 4H), 6.91 (d, J = 8.3 Hz, 1H), 6.61 (dd, J = 2.5, 8.2 Hz, 1H), 6.45 (d, J = 2.5 Hz, 1H), 4.26 (br. s., 1H), 3.71 (q, J = 15.5 Hz, 2H), 3.45 (d, J = 10.9 Hz, 1H), 3.26 (dd, J = 5.2, 11.2 Hz, 1H), 3.07 (d, J = 11.7 Hz, 1H), 2.94 (dd, J = 5.2, 16.5 Hz, 1H), 2.70 (t, J = 12.1 Hz, 1H), 2.61 – 2.48 (m, 1H), 2.40 – 2.17 (m, 3H), 2.06 (m, 1H), 1.99 – 1.76 (m, 5H), 0.95 (d, J = 7.0 Hz, 6H); 13C NMR (75 MHz, CHLOROFORM-d) d 173.3, 154.8, 145.6, 137.6, 130.8, 128.5, 126.7, 126.3, 125.5, 113.7, 112.1, 60.0, 56.6, 56.2, 52.6, 49.8, 48.3, 42.2, 32.9, 32.4, 31.7, 29.4, 19.0, 18.0; ESI MS (M + H)+ 422.5. The free base was converted to the dihydrochloride salt by adding 2M HCl in ether to solution of product in dichloromethane: [α]D = + 62.7 (c 1.01, MeOH). Anal. Calcd for C26H37Cl2N3O2•1.5H2O: C, 59.88; H, 7.73; N, 8.06. Found C, 59.67; H, 7.40; N, 8.01.
(3R)-N-[(1S)-1-{[4-(3-Fluorophenyl)piperidin-1-yl]methyl}-2-methylpropyl]-7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (4f) dihydrochloride
To a solution of tert-butyl (3R)-3-{[(1S)-1-{[4-(3-fluorophenyl)piperidin-1-yl]methyl}-2-methylpropyl]carbamoyl}-7-hydroxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (19) (150 mg, 0.28 mmol) in 4 mL methanol was added 4 mL conc. HCl and the reaction mixture stirred for overnight. Evaporation of solvents led to a white solid that was purified by chromatography on silica gel with CMA 80: chloroform (1:1) as the eluent to provide 98 mg (79%) of the title compound as a white solid. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 (s, 1H), 7.08 – 7.16 (m, 1H), 7.03 (d, J = 9.61 Hz, 1H), 6.72 – 6.90 (m, 4H), 6.52 (dd, J = 2.45, 8.10 Hz, 1H), 6.37 (d, J = 2.07 Hz, 1H), 4.08 – 4.23 (m, 1H), 3.53 – 3.74 (m, 2H), 3.30 (d, J = 11.11 Hz, 1H), 3.20 (dd, J = 5.18, 11.21 Hz, 1H), 2.97 (d, J = 11.30 Hz, 1H), 2.86 (dd, J = 5.09, 16.58 Hz, 1H), 2.59 (t, J = 12.06 Hz, 1H), 2.38 – 2.52 (m, 1H), 2.21 – 2.36 (m, 2H), 2.06 – 2.20 (m, 1H), 1.90 – 2.03 (m, 1H), 1.60 – 1.88 (m, 5H), 0.87 (d, J = 6.78 Hz, 6H); 13C NMR (75 MHz, CHLOROFORM-d) δ 173.4, 164.6, 161.3, 154.9, 148.4, 148.3, 137.3, 130.6, 129.9, 129.8, 125.4, 122.4, 122.3, 113.9, 113.8, 113.5, 113.2, 112.9, 112.2, 60.0, 56.7, 55.9, 52.7, 50.0, 48.1, 42.0, 32.7, 32.4, 31.5, 29.6, 19.1, 17.9; ESI MS (M + H)+ 440.5. The free base was converted to the dihydrochloride salt by adding 2M HCl in ether to solution of product in dichloromethane: [mp 215 – 219 °C, α]D = + 61.6 (c 1.04, MeOH). Anal. Calcd for C26H36Cl2FN3O2•H2O: C, 58.87; H, 7.22; N, 7.92. Found C, 59.20; H, 7.39; N, 7.70.
1-Methyl-4-[3-(1-methylethoxy)phenyl]piperidin-4-ol (6)
A solution of n-butyl lithium (2.5 M in hexanes, 8.7 mL, 22 mmol) was slowly added to a solution of 1-bromo-3-isopropoxybenzene (5.2 g, 24 mmol) in THF (14 mL) at −78 °C. After 30 minutes, N-methyl-4-piperidinone (5) (2.49 g, 22.0 mmol) was added dropwise. The solution warmed to r.t. overnight. Hydrochloric acid (6 M, 8 mL) was added and the resulting biphasic mixture was extracted with hexanes. The organic layer was discarded and the aqueous layer was adjusted to pH 10 with NH4OH (2 M). Extraction with hexanes, drying with Na2SO4 and concentration to afford 2.29 g (42%) of crude 6. 1H NMR (CDCl3) δ 7.25 (t, J = 7.9 Hz, 1H), 7.01 – 7.10 (m, 2H), 6.74 – 6.83 (m, 1H), 4.56 (spt, J = 6.0 Hz, 1H), 2.75 (d, J = 11.3 Hz, 1H), 2.38 – 2.54 (m, 2H), 2.35 (s, 3H), 2.17 (dt, J = 4.5, 13.0 Hz, 1H), 1.69 – 1.81 (m, 1H), 1.33 (d, J = 6.0 Hz, 6H).
1-Methyl-4-[3-(1-methylethoxy)phenyl]-1,2,3,6-tetrahydropyridine (7)
Crude 6 was refluxed in toluene (15 mL) with TsOH • H2O (2 eq.) for 3 h. The product was extracted into water. The aqueous layer was adjusted to pH 10 with NaOH (2 M) and then extracted with hexanes. The combined organic layer was washed with NaOH (2 M) and dried (Na2SO4). Concentration afforded 1.67 g (79%) of crude 7. 1H NMR (300 MHz, CDCl3) δ 7.14 – 7.25 (m, 2H), 6.96 (d, J = 7.9 Hz, 1H), 6.87 – 6.93 (m, 1H), 6.77 (dd, J = 2.5, 8.1 Hz, 1H), 6.00 – 6.08 (m, 1H), 4.55 (spt, J = 6.1 Hz, 1H), 3.10 (q, J = 2.8 Hz, 2H), 2.62 – 2.70 (m, 2H), 2.51 – 2.62 (m, 2H), 2.40 (s, 3H), 2.36 (s, 1H), 1.67 (s, 1H), 1.33 (d, J = 6.0 Hz, 7H).
1,4-Dimethyl-4-[3-(1-methylethoxy)phenyl]piperidine (8)
Crude 7 in THF (18 mL) was treated at −15 °C with butyl lithium (2.5 M, 4.5 mL) to afford a blood-red solution. After cooling to −50 °C, dimethylsulfate (0.8 mL) was cautiously added. After 30 minutes, NH4OH (2 M, 10 mL) was added and the resulting biphasic mixture was extracted with hexanes. The combined organic layer was washed with water, dried (Na2SO4), and concentrated to a residue which was dissolved in MeOH (20 mL) and treated at 0 °C with NaBH4 (0.42 g). After warming to room temperature the solution was concentrated and subjected to silica gel chromatography using a gradient of CMA80 in CH2Cl2 as the eluent to afford 1.44 g (81%) of 8. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 – 7.31 (m, 1H), 7.01 – 7.10 (m, 2H), 6.73 – 6.83 (m, 1H), 4.56 (spt, J = 5.97 Hz, 1H), 2.75 (d, J = 11.30 Hz, 2H), 2.38 – 2.51 (m, 2H), 2.35 (s, 3H), 2.17 (dt, J = 4.52, 13.00 Hz, 2H), 1.68 – 1.81 (m, 2H), 1.33 (d, J = 6.03 Hz, 6H).
4-methyl-4-[3-(1-methylethoxy)phenyl]piperidine (9)
The N-methyl amine 8 was concentrated from toluene then dissolved in 1,2-dichloroethane (9 mL) and treated with freshly distilled 1-chloroethyl chloroformate (1.8 mL, 3.0 eq.). The resulting dark solution was refluxed 12 h then concentrated. The residue was dissolved in methanol (10 mL), refluxed 24 h and then concentrated. The residue was subjected to chromatography on silica gel using a gradient of CMA80 in CH2Cl2 as the eluent to afford 0.677 g (50%) of 9. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.14 – 7.31 (m, 1H), 6.96 (d, J = 7.91 Hz, 1H), 6.87 – 6.93 (m, 1H), 6.77 (dd, J = 2.45, 8.10 Hz, 1H), 5.99 – 6.10 (m, 1H), 4.55 (spt, J = 6.06 Hz, 1H), 3.10 (q, J = 2.83 Hz, 2H), 2.61 – 2.70 (m, 2H), 2.52 – 2.61 (m, 2H), 2.40 (s, 3H), 1.30 – 1.36 (m, 6H).
(2S)-3-Methyl-1-{4-methyl-4-[3-(1-methylethoxy)phenyl]piperidin-1-yl}butan-2-amine (10)
Boc-L-Valinal was prepared by LAH reduction of the Weinreb amide.[7] The aldehyde (319 mg, 1.6 mmol) was combined with amine 9 (572 mg, 2.5 mmol) in trifluoroethanol (12 mL) and stirred for 15 min prior to addition of NaCNBH3 (154 mg, 2.5 mmol). The resulting mixture was stirred overnight, concentrated, and the residue partitioned between 2 M NH4OH and EtOAc. The aqueous layer was extracted further with EtOAc. The dried (Na2SO4) combined organics were concentrated and the residue subjected to chromatography on silica gel using a gradient of CMA80 in CH2Cl2 as the eluent. 1H NMR (CDCl3) δ 7.23 (t, J = 8.0 Hz, 1H), 6.82 – 6.93 (m, 2H), 6.72 (dd, J = 1.9, 8.1 Hz, 1H), 4.74 (bs, 1H), 4.54 (spt, J = 6.0 Hz, 1H), 3.55–3.74 (m, 1H), 2.75 (bs, 1H), 2.58 (bs, 2H), 2.26 – 2.51 (m, 3H), 2.05 – 2.24 (m, 2H), 1.70 – 1.92 (m, 3H), 1.45 (s, 9H), 1.34 (d, J = 6.0 Hz, 6H), 1.21 (s, 3H), 0.81 – 0.99 (m, 6H). The isolated product was dissolved in CH2Cl2 (5 mL) and treated with TFA (10 mL) at r.t. for 12 h. The concentrated residue was partitioned between CHCl3 and 2 M NH4OH. The organic layer was dried (Na2SO4) and concentrated to afford 439 mg of 10 (1.38 mmol, 56% over two steps), which was used in the next step without further purifications.
4-Methyl-4-(3-fluorophenyl)piperidine (12)
A solution of n-BuLi (7.2 mL, 2.5 M in hexanes, 18 mmol) was added dropwise to a solution of 11 (2.46 g, 12.9 mmol) in THF (22 mL) maintained between −10 and −20 °C. After 15 min, the solution was cooled to −50 °C and dimethyl sulfate (1.75 mL, 18.5 mmol) was slowly and cautiously added. The reaction mixture was stirred an additional 30 min, then 2M NH4OH (10 mL) was added. The resulting mixture was extracted with hexanes. The combined organic layer was washed with water, dried (Na2SO4), and concentrated to a residue. The residue was dissolved in CH3OH (20 mL), cooled in an ice bath, and treated with an excess of NaBH4 (0.5 g, 13 mmol). The reaction mixture was stirred 3 h at room temperature and then was quenched with the addition of acetone and saturated NaHCO3. The concentrated residue was dissolved in water and EtOAc. The aqueous layer was extracted again with EtOAc before the combined organic layer was washed with water then concentrated to afford 1.72 g (8.3 mmol, 64%) of the 1,4-dimethylpiperidine intermediate. This residue was concentrated thrice from toluene then dissolved in 1,2-dichloroethane (12 mL). A freshly distilled aliquot of 1-chloroethyl chloroformate (1.3 mL, 12 mmol) was added under inert atmosphere and the resulting solution was heated at reflux overnight. The concentrated residue was then dissolved in CH3OH and refluxed 1 h. The concentrated residue was dissolved in 2 M NaOH and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), concentrated, and subjected to chromatography on silica gel using a gradient of CMA80 in DCM as the eluent to afford 0.74 g (30% from 11) of 12. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.22 – 7.36 (m, 1H), 7.12 (d, J = 7.91 Hz, 1H), 7.04 (td, J = 2.14, 11.35 Hz, 1H), 6.88 (dt, J = 2.17, 8.05 Hz, 1H), 2.74 – 3.00 (m, 4H), 1.93 – 2.10 (m, 2H), 1.70 (ddd, J = 3.39, 7.25, 13.09 Hz, 3H), 1.20 – 1.30 (m, 3H).
(2S)-1-[4-(3-Fluorophenyl)-4-methylpiperidin-1-yl]-3-methylbutan-2-amine (13)
A solution of amine 12 (740 mg, 3.8 mmol) and Boc-L-valine (0.86 g, 4.0 mmol) in acetonitrile (20 mL) was cooled in an ice-bath, then treated with HBTU (1.51 g, 4.0 mmol) and diisopropylethylamine (2.1 mL, 12 mmol). The flask was removed from the ice bath, and the reaction mixture was stirred overnight. The solution was concentrated then partitioned between concentrated aqueous NaHCO3 and EtOAc. The mixture was extracted three times with EtOAc (25 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and evaporated to leave a residue which was dissolved in acetonitrile (20 mL) and treated with HCl in dioxane (20 mL). The solvent was concentrated under a stream of nitrogen. The remaining residue was subjected to chromatography on silica gel using a gradient of CMA80 in DCM as the eluent to afford the intermediate amide. This amide was dissolved in THF (20 mL) and treated with borane dimethylsulfide (3 mL, 30 mmol). The solution was stirred at reflux overnight and then quenched with methanol. The residue was treated with HCl (6 M, 10 mL) and stirred for 1 h. Solid NaHCO3 was added to adjust the solution to a pH of 8, and the mixture was extracted with CH2Cl2 (3 × 25 mL), washed with brine, and dried over MgSO4. The concentrated residue was subjected to chromatography on silica gel eluting with a gradient of CMA80 in DCM to afford 0.64 g (61% from 12) of 13: 1H NMR (300 MHz, CHLOROFORM-d) δ 7.22 – 7.33 (m, 1H), 7.11 (d, J = 8.10 Hz, 1H), 7.03 (td, J = 2.14, 11.35 Hz, 1H), 6.82 – 6.92 (m, 1H), 2.45 – 2.74 (m, 3H), 1.98 – 2.44 (m, 5H), 1.65 – 1.87 (m, 5H), 1.51 (qd, J = 6.66, 12.97 Hz, 1H), 1.18 – 1.23 (m, 3H), 0.83 – 0.95 (m, 6H); 19F NMR (282 MHz, CHLOROFORM-d) δ −133.33.
(2S)-1-[4-(3-Fluorophenyl)-4-methylpiperidin-1-yl]-3-methylpentan-2-amine (14)
A solution of amine 12 (233 mg, 1.2 mmol) and Boc-L-isoleucine (275 g, 1.2 mmol) in acetonitrile (7 mL) was cooled in an ice-bath, then treated with HBTU (0.50 g, 1.3 mmol) and diisopropylethylamine (0.65 mL, 3.7 mmol). The flask was removed from the ice bath, and the reaction was stirred overnight. The solution was concentrated then partitioned between concentrated aqueous NaHCO3 and EtOAc. The mixture was extracted three times with EtOAc (25 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and evaporated to leave a residue which was dissolved in methanol (10 mL) and treated with HCl (6 M, 10 mL). The solution was concentrated to a solid from toluene. The remaining residue was subjected to chromatography on silica gel using a gradient of CMA80 in DCM to afford the intermediate amide. This amide was dissolved in THF (15 mL) and treated with borane dimethylsulfide (2.5 mL, 25 mmol). The solution was stirred at reflux overnight, then quenched with and concentrated from methanol. The residue was treated with HCl (6 M, 10 mL) and methanol (10 mL), stirred for 1 h, and then concentrated. The residue was combined with 2 M NaOH (10 mL) and extracted with EtOAc (3 × 25 mL). The combined extracts were washed with brine, and dried (MgSO4). The concentrated residue was subjected to chromatography on silica gel eluting with a gradient of CMA80 in DCM to afford 0.27 g (77% from 12) of the intermediate amine 14: 1H NMR (300 MHz, CHLOROFORM-d) δ 7.22 – 7.33 (m, 1H), 7.08 – 7.14 (m, 1H), 7.03 (td, J = 2.14, 11.35 Hz, 1H), 6.87 (ddt, J = 0.75, 2.45, 8.29 Hz, 1H), 2.71 – 2.84 (m, 1H), 2.44 – 2.67 (m, 2H), 1.96 – 2.43 (m, 5H), 1.57 – 1.83 (m, 6H), 1.48 (ddd, J = 3.96, 7.58, 12.95 Hz, 1H), 1.24 – 1.37 (m, 1H), 1.18 – 1.24 (m, 3H), 0.78 – 0.95 (m, 6H).
tert-Butyl [(1S)-1-{[4-(3-fluorophenyl)piperidin-1-yl]carbonyl}-2-methylpropyl]carbamate (16)
To a solution of 4-(3-fluorophenyl)piperidine (15) (5.0 mg, 27.9 mmol) in dry acetonitrile (150 mL) was added Boc-L-valine (6.5 g, 30 mmol) followed by 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (11.4 g, 30 mmol) and triethylamine (8. 3 g, 82.5 mmol). The reaction mixture was stirred overnight then concentrated. The resulting solid was treated with 50 mL saturated NaHCO3. The aqueous layer was extracted with EtOAc (2 × 50 mL). Combined organic layers were dried (Na2SO4), filtered then concentated. The resulting residue was purified by column chromatography on silica gel with hexanes: ethyl acetate (1:1) to provide 7.93 g (75%) of the title compound. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.14 – 7.24 (m, 1H), 6.78 – 6.92 (m, 2H), 5.32 (d, J = 9.04 Hz, 1H), 4.70 (d, J = 12.81 Hz, 1H), 4.44 (d, J = 6.97 Hz, 1H), 3.90 – 4.21 (m, 1H), 3.03 – 3.17 (m, 1H), 2.55 – 2.75 (m, 2H), 1.77 – 2.04 (m, 3H), 1.43 – 1.68 (m, 2H), 1.31 – 1.40 (m, 9H), 0.69 – 0.96 (m, 6H); 13C NMR (75 MHz, CHLOROFORM-d) δ 170.6, 164.7, 161.4, 155.9, 147.5, 130.1, 130.0, 122.4, 113.8, 113.7, 113.5, 113.3, 79.4, 54.8, 46.5, 46.1, 42.8, 42.6, 42.2, 33.7, 33.6, 32.8, 31.8, 31.5, 28.4, 19.8, 19.6, 17.2, 17.1; ESI MS (M + H)+ 379.6.
(2S)-2-Amino-1-[4-(3-fluorophenyl)piperidin-1-yl]-3-methylbutan-1-one (17)
To a solution of tert-butyl [(1S)-1-{[4-(3-fluorophenyl)piperidin-1-yl]carbonyl}-2-methylpropyl]carbamate (16) (7.93 g, 21.0 mmol) in methanol (100 mL) was added conc. HCl (8 mL) and the reaction mixture stirred for 4 h at ambient temperature. Evaporation of solvents led to a white solid that was purified by chromatography on silica gel with CMA 80: chloroform (1:1) as the eluent to provide 6.05 g (92%) of the title compound as a white solid. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.43 (br. s., 2H), 7.11 – 7.23 (m, 2H), 6.76 – 6.97 (m, 2H), 4.70 (d, J = 13.19 Hz, 1H), 4.51 (br. s., 1H), 3.76 – 4.12 (m, 1H), 3.09 (br. s., 1H), 2.67 (d, J = 9.04 Hz, 2H), 2.09 – 2.29 (m, 1H), 1.98 (s, 2H), 1.66 – 1.81 (m, 1H), 1.47 (br. s., 1H), 0.89 – 1.19 (m, 5H); 13C NMR (75 MHz, CHLOROFORM-d) δ 167.4, 166.9, 164.6, 161.3, 147.7, 147.6, 147.3, 130.1, 130.0, 129.9, 122.8, 122.4, 114.1, 113.8, 113.7, 113.4, 113.1, 55.5, 55.2, 47.2, 46.4, 43.7, 43.0, 42.9, 41.8, 33.2, 32.5, 30.2, 30.1, 19.3, 18.9, 17.8, 17.5.
(2S)-1-[4-(3-Fluorophenyl)piperidin-1-yl]-3-methylbutan-2-amine (18)
To a cooled (0 °C) solution of (2S)-2-amino-1-[4-(3-fluorophenyl)-1-piperidin-1-yl]-3-methyl-butan-1-one (17) hydrochloride (6.05 g, 19.2 mmol) in THF (40 mL) was added borane dimethyl sulfide complex (3.8 mL, 38 mmol) (10 M as BH3). The reaction mixture was warmed to ambient temperature and stirred overnight. The reaction mixture was refluxed for 3 h, cooled in ice, quenched with methanol and stirred for 1 h at ambient temperature. The reaction mixture was cooled in ice bath and treated with 2M HCl in ether and refluxed for 2 h. The solvent was evaporated and the resulting material purified by chromatography on silica gel using chloroform/CMA80 gradient to provide 2.5 g (49%) of the desired product as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.68 (br. s, 2H), 7.17 – 7.25 (m, 1H), 6.93 – 7.02 (m, 2H), 6.82 – 6.89 (m, 1H), 3.01 – 3.12 (m, 3H), 2.44 – 2.66 (m, 3H), 2.31 – 2.37 (m, 1H), 2.03 – 2.23 (m, 2H), 1.75 – 1.93 (m, 4H), 1.17 (d, J = 7 Hz, 3H), 1.09 (d, J = 7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.5, 161.3, 148.9, 148.8, 129.8, 129.7, 122.6, 122.5, 113.9, 113.6, 113.0, 112.7, 57.8, 55.5, 54.9, 53.1, 42.1, 33.3, 32.8, 29.3, 19.4, 18.5; ESI MS (M + H)+ 265.5.
tert-Butyl (3R)-3-{[(1S)-1-{[4-(3-fluorophenyl)piperidin-1-yl]methyl}-2-methylpropyl]carbamoyl}-7-hydroxy-3,4-dihydroisoquinoline-2(1H)-carboxylate (19)
To a solution of (2S)-1-[4-(3-fluorophenyl)piperidin-1-yl]-3-methylbutan-2-amine (70 mg, 0.27 mmol) (18) in dichloromethane (15 mL) was added Boc-7-hydroxy-D-Tic-OH (117 mg, 0.40 mmol) and triethylamine (80 mg, 0.8 mmol) followed by EDC • HCl (153 mg, 0.8 mmol) and HOBt (5.4 mg, 0.04 mmol). The reaction mixture was stirred overnight at ambient temperature. The reaction mixture was treated with saturated NaHCO3 and extracted with dichloromethane. Combined organic layers were dried (Na2SO4), filtered then evaporated to obtain crude product. Purification of the crude product by chromatography on silica gel with CMA 80: chloroform (1:1) as the eluent provided 150 mg (74%) of the title compound as a white solid. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.09 – 7.31 (m, 2H), 6.68 – 6.97 (m, 4H), 6.37 – 6.65 (m, 2H), 5.66 – 5.95 (m, 1H), 4.55 – 4.94 (m, 1H), 4.25 – 4.53 (m, 2H), 3.76 (br. s., 1H), 3.15 (dd, J = 2.92, 15.35 Hz, 1H), 2.88 (dd, J = 6.03, 15.26 Hz, 1H), 2.66 (br. s., 2H), 2.22 – 2.43 (m, 1H), 1.90 – 2.21 (m, 2H), 1.69 – 1.90 (m, 2H), 1.49 – 1.70 (m, 3H), 1.26 – 1.49 (m, 9H), 0.76 (dd, J = 6.88, 17.61 Hz, 6H); 13C NMR (75 MHz, CHLOROFORM-d) δ 171.4, 164.6, 161.3, 155.6, 149.0, 148.9, 129.8, 129.7, 129.3, 124.4, 122.5, 122.5, 114.9, 113.8, 113.5, 113.0, 112.7, 59.7, 54.5, 51.3, 44.8, 42.0, 33.3, 33.0, 30.3, 28.4, 19.1, 17.3.
[35S]GTPγS Assay
The [35S]GTPγS assays were conducted using the methods previously reported.[3, 4]
Calculated Pharmacokinetic Properties
The topological polar surface area (TPSA) and calculated lipophilicity (clogP) values were calculated using the ChemAxon Instant JChem package. Predictions of the logarithm of the in-vivo blood-brain ratio (logBB) were based on the Clark and Pickett model (equation 3).[13]
MDCK-mdr1 permeability assays
MDCK-mdr1 cells obtained from the Netherlands Cancer Institute were grown on Transwell type filters (Corning) for 4 days to confluence in DMEM/F12 media containing 10% fetal bovine serum and antibiotics as has been described previously.[14] Compounds were added to the apical side at a concentration of 10 μM in a transport buffer comprising of 1X Hank’s balanced salt solution, 25 mM D-glucose and buffered with HEPES to pH 7.4. Samples were incubated for 1 h at 37 °C and carefully collected from both the apical and basal side of the filters. Compounds selected for MDCK-mdr1 cell assays were infused on an Applied Biosystems API-4000 mass spectrometer to optimize for analysis using multiple reaction monitoring (MRM). Flow injection analysis was also conducted to optimize for mass spectrometer parameters. Samples from the apical and basolateral side of the MDCK cell assay were dried under nitrogen on a Turbovap LV. The chromatography was conducted with an Agilent 1100 binary pump with a flow rate of 0.5 mL/min. Mobile phase solvents were A, 0.1% formic acid in water, and B, 0.1% formic acid in methanol. The initial solvent conditions were 10% B for 1 minute, then a gradient was used by increasing to 95% B over 5 minutes, then returning to initial conditions. Data reported are average values from 2–3 measurements.
In vitro stability testing
Stability of compounds to plasma and S9 fraction was performed as previously described.[15] In vitro testing for metabolic stability was conducted in mixed gender pooled hepatic S9 fraction supplied by Xenotech, LLC, Lenexa, KS. Identity of the donors was unknown.
For the hepatic S9 metabolism studies, all samples were tested at 10μM final concentration in a 1 mL volume containing 1 mg/mL S9. Samples were incubated in a buffer containing 50 mM potassium phosphate, pH 7.4 with 3 mM MgCl2 and a NADPH regeneration system comprising of NADP (1 mM), glucose-6-phosphate (5 mM) and glucose-6-phosphate dehydrogenase (1 unit/mL). Triplicate samples were incubated for 60 min. Reactions were terminated by addition of 3 volumes of acetonitrile and processed as described for the MDCK-mdr1 assays, but standard curves were prepared in blank matrix for each compound for quantitative assessment. Data reported are average values from 3 measurements.
PAMPA
PAMPA was conducted using a 96-well plate based kit from BD Biosciences (RTP, NC) and manufacturer’s instructions were followed closely. Briefly, all samples were tested at 10μM final concentration by adding them to the apical (top) compartment in PBS at pH 5.5 or 7.4. Samples were incubated at room temperature for 4 hours. Apical to basal transport was evaluated by sampling from the top and bottom wells of the plate and measuring concentrations of compounds in each compartment using LC-MS. Processing and methods for analytical evaluation were similar to those described for MDCK-mdr1 transport assays. Data reported are average values from 3 measurements.
hERG Assay
Preparations of membranes overexpressing human hERG were purchased from Perkin Elmer. The binding assays were performed for 60 min using 4 μg hERG expressing membranes, ~3 nM [3H]Astemizole, and various concentrations of the test agent in a binding buffer (10 mM HEPES, pH 7.4, 130 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 1 mM NaEDTA, 10 mM glucose, 0.1% BSA). Binding was terminated by rapid filtration onto GF/B fiber filtermats, presoaked in 0.3% polyethyleneimine, followed by rapid washing 6 times (2 mL) with ice-cold solution containing 25 mM Tris-HCl, pH 7.4, 130 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 0.05 mM CaCl2, and 0.1% BSA using a Brandel harvester. Filters were dried and counted after addition of a scintillant. Data were analyzed using non-linear regression (GraphPad Prism) and Ki values were determined as described before.[16] All experiments were performed at least twice in duplicate and data reported are mean values.
Solubility Determination
For these experiments, 10 mM DMSO stocks of compounds were directly diluted into 10 mM phosphate buffer at pH 7.4 or 3 and shaken for 90 min at room temperature. The final concentration of DMSO was 1%. After the incubation, samples were filtered through a 0.4 micron filterplate (Millipore). Filtrates were carefully collected. Analysis of compounds was performed by LC/MS using previously available methods and concentrations determined. Data are reported as mean values from three determinations.
Supplementary Material
Acknowledgments
This research was supported by the National Institute on Drug Abuse Grant DA09045. We thank Tiffany Langston, Keith Warner, and Rodney Snyder for conducting the in vitro testing and in vitro preclinical studies. RM was supported by AA022235 and DK100414 from NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, Mascarella SW, Rothman RB, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. J Med Chem. 2003;46:3127. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- 2.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. J Med Chem. 2001;44:2687. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 3.Kormos CM, Cueva JP, Gichinga MG, Runyon SP, Thomas JB, Brieaddy LE, Mascarella SW, Gilmour BP, Navarro HA, Carroll FI. J Med Chem. 2014;57:3140. doi: 10.1021/jm500184j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kormos CM, Gichinga MG, Maitra R, Runyon SP, Thomas JB, Brieaddy LE, Mascarella SW, Navarro HA, Carroll FI. J Med Chem. 2014;57:7367. doi: 10.1021/jm5008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaveri NT, Journigan VB, Polgar WE. ACS Chem Neurosci. 2015;6:646. doi: 10.1021/cn500367b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werner JA, Cerbone LR, Frank SA, Ward JA, Labib P, Tharp-Taylor RW, Ryan CW. J Org Chem. 1996;61:587. doi: 10.1021/jo951403y. [DOI] [PubMed] [Google Scholar]
- 7.Skiles JW, Miao C, Sorcek R, Jacober S, Mui PW, Chow G, Weldon SM, Possanza G, Skoog M, Keirns J, et al. J Med Chem. 1992;35:4795. doi: 10.1021/jm00104a004. [DOI] [PubMed] [Google Scholar]
- 8.Nagai Y, Hino K, Uno H, Minami S. Chem Pharm Bull (Tokyo) 1980;28:1387. doi: 10.1248/cpb.28.2618. [DOI] [PubMed] [Google Scholar]
- 9.Kosterlitz HW, Lees GM, Wallis DI, Watt AJ. Br J Pharmacol. 1968;34:691P. [PMC free article] [PubMed] [Google Scholar]
- 10.Summerfeld SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. J Pharmacol Exp Ther. 2007;322:205. doi: 10.1124/jpet.107.121525. [DOI] [PubMed] [Google Scholar]
- 11.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. ACS Chem Neurosci. 2012;3:50. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark DE. J Pharm Sci. 1999;88:815. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 13.Clark DE, Pickett SD. Drug Discov Today. 2000;5:49. doi: 10.1016/s1359-6446(99)01451-8. [DOI] [PubMed] [Google Scholar]
- 14.Fulp A, Bortoff K, Zhang Y, Seltzman H, Snyder R, Maitra R. Bioorg Med Chem Lett. 2011;21:5711. doi: 10.1016/j.bmcl.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fulp A, Bortoff K, Seltzman H, Zhang Y, Mathews J, Snyder R, Fennell T, Maitra R. J Med Chem. 2012;55:2820. doi: 10.1021/jm201731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.