

Abstract
The role of PI4K and PI3K-AKT in ERK1/2 activation by GnRH was examined. A relatively long preincubation (60 min) with wortmannin (10nM and 10μM), and LY294002 (10μM and 100μM) (doses known to inhibit PI3K and PI4K, respectively), were required to inhibit GnRH-and PMA-stimulated ERK1/2 activity in αT3-1 and LβT2 gonadotrope cells. A similar preincubation protocol was required to demonstrate inhibition of IGF-1-stimulated AKT activation lending support for the need of prolonged incubation (60 min) with wortmannin in contrast to other cellular systems. To rule out that the inhibitors acted upon PI(4,5)P2 levels, we followed the [Ca2+]i response to GnRH and found that wortmannin has no significant effect on GnRH-induced [Ca2+]i responses. Surprisingly, GnRH and PMA reduced, while IGF-1 increased AKT phosphorylation. We suggest that PI3K inhibits GnRH-stimulated αGSU activity, has no effect upon GnRH-stimulated LHβ activity and enhanced the GnRH-stimulated FSHβ transcription. Hence, PI4K and PI3K-AKT play a role in GnRH to ERK1/2 signaling, while PI3K may regulate also GnRH-induced gonadotropin gene expression.
Keywords: GnRH, PKC, PI3K, PI4K, ERK1/2, MAPK, Gonadotropes, LβT2 cells, αT3-1 cells
1 Introduction
The mechanism of action of Gonadotropin releasing hormone (GnRH) in pituitary gonadotropes is complex and includes activation of Gαq, phospholipase Cβ (PLCβ), phosphoinositide (PI) turnover, calcium ions (Ca2+) mobilization and influx, activation of protein kinase C isoforms (PKCs) and mitogen-activated protein kinases (MAPKs) (ERK1/2, JNK1/2 and p38MAPK) culminating in synthesis and secretion of luteinizing hormone (LH) and follicle stimulating hormone (FSH) (Caunt, Finch, Sedgley et al., 2006, Naor, 2009, Naor and Huhtaniemi, 2013). Common to the pituitary heterodimeric glycoprotein hormones LH and FSH is the α-subunit (α-GSU), which binds noncovalently to a specific β-subunit, LHβ and FSHβ (Pierce and Parsons, 1981).
Phosphoinositides (PIs) are derivatives of phosphatidylinositol (PI) in which one or more of the hydroxyl groups at the 3-, 4- and 5- position of the inositol ring are esterified with a phosphate group in all possible combinations (Balla, 2006, Balla, 2013, Balla and Balla, 2006). Several inositol lipids kinases and phosphatases which interconvert PIs have been identified including phosphatidylinositol 3-kinases (PI3Ks) and phosphatidylinositol 4-kinases (PI4Ks). PI3K isoforms include three classes based on structural features and lipid substrate preferences (Vanhaesebroeck, Guillermet-Guibert, Graupera et al., 2010). Class I PI3Ks are subdivided to class IA including catalytic subunits p110α, β, δ, which bind the p85 regulatory subunit, and class IB including p110γ, which binds one of two related regulatory subunits, p101 and p87 (ibid). Four distinct PI4Ks have been identified in mammalian cells: PI4KIIα and β (54 and 55 kDa), which are insensitive to wortmannin and LY294002, and PI4KIIIα and β (230 and 92 kDa), which have a high degree of conservation between their catalytic domain and PI3K, explaining their sensitivity to wortmannin (Balla and Balla, 2006).
Although PIs are a tiny fraction of cellular phospholipids they control almost all aspects of a cell’s life and death, PIs are involved in vesicular trafficking, lipid distribution and metabolism, ion channels, pumps, endocytic and exocytic processes (Balla, 2013). PIs recruit regulatory proteins to signaling complexes organized in cellular membranes (Balla, 2013, Balla and Balla, 2006). PI4K, the first committed step in inositol lipid synthesis provides PtdIns(4)P for PIPKs (PI to PIP). PIP is then converted to PIP2 by PI4P5K (ibid). Class I PI3K phosphorylates the D3 position of phosphatidylinositol-4,5- bisphoaphate (PI(4,5)P2) (PIP2) to generate the biologically active phosphatidylinositol-3,4,5-trisphoaphate (PI(3,4,5)P3) (PIP3) (Balla, 2013, Balla and Balla, 2006, LoPiccolo, Blumenthal, Bernstein et al., 2008), a process which is reversed by protein phosphatase and tension homologue (PTEN) (Howes, Chiang, Lang et al., 2007, Ihle and Powis, 2009) and SH2-domain-containing inositol polyphosphate 5-phosphatase (SHIP) (New, Wu, Kwok et al., 2007). Upon generation in the cell membrane, PI(3,4,5)P3 (PIP3) binds to pleckstrin homology (PH) domain of phosphoinositide-dependent kinase-1/2 (PDK-1/2) and the serine/threonine kinase AKT (also named as PKB) (Ihle and Powis, 2009), resulting in translocation of both proteins to the cell membrane where they are subsequently activated and can now initiate downstream signaling (LoPiccolo et al., 2008, Mendoza, Er and Blenis, 2011).
MAPK cascades consist of a core of several tiers of protein kinases (MAP3K, MAP2K and MAPK) (Plotnikov, Zehorai, Procaccia et al., 2011). At least four distinct MAPK cascades are known in mammals: ERK1/2 (p42 and p44), Jun N-terminal kinase (JNK1/2/3), p38 (α, β, λ, δ) and ERK5 (ibid). Although MAPKs act mainly via transcription factors after translocation to the nucleus, MAPKs act also in the cytosol and the cell membrane, as evident from their diverse list of substrates such as the cytosolic p90 ribosomal S6 kinase (RSK) (Chuderland and Seger, 2005). GnRH activates the MAPKs (ERK1/2, JNK1/2 and p38α), which are thought to participate in the transcriptional control of the gonadotropin subunits and GnRH receptor genes by GnRH.
Whereas the available literature has increased our understanding of the biochemistry of phospholipids and MAPKs in general, several important questions remain unanswered, including the crosstalk between them during GPCRs actions. Accordingly, the purpose of the current study was to address these questions for the GnRHR in studies in which gonadotrope cell lines were used. Here we demonstrate that PI4K and PI3K-AKT play a role in GnRH activation of ERK1/2 in αT3-1 and LβT2 gonadotrope cells. Still, GnRH inhibits AKT phosphorylation in αT3-1 cells. Since gonadotropins consist of a common αGSU- and specific LHβ- and FSHβ-subunits we also looked for the role of PI3K in their regulation by GnRH. We found that wortmannin stimulates GnRH-induced αGSU activity, has no effect upon GnRH-induced LHβ activity and reduced the GnRH-activation of FSHβ gene expression.
2 Materials and Methods
2.1 Cell cultures and activation
αT3-1 and LβT2 cells, were kindly provided by Dr. P. Mellon (University of California, San Diego, USA), were grown in monolayer cultured in DMEM (Biological Industries, Beit-Haemek, Israel) supplemented with 10% fetal calf serum (FCS) (Biological Industries, Beit-Haemek, Israel) and antibiotics in humidified incubator 5% CO2 at 37°C. Serum starvation was with 0.1% serum DMEM in the same medium for 16 h before the experiment. At the next morning, cells were pretreated with or without wortmannin (Calbiochem, Merck Millipore, Darmstadt, Germany), two different lots of Calbiochem wortmannin were used, LY294002 (Calbiochem, Merck Millipore, Darmstadt, Germany), PD098059 and U0126 (A.G. Scientific, Inc. San Diego, CA). Then cells were stimulated with or without GnRH (100nM), PMA (100nM), EGF (10ng/ml) (Sigma, Rehovot, Israel) and IGF-1 (20ng/ml) (PeproTech Asia, Rehovot, Israel) for various times.
2.2 Cell harvesting, SDS-PAGE and Western Blotting
Cells were grown in 12-wells plates, serum starved (FCS 0.1%) for 16 h and later stimulated as described above. After that, cells were washed twice with ice-cold PBS and incubated with 0.12 ml of lysis buffer (Lysis buffer contained 20mM Tris-HCl pH 7.5, 20mM NaCl, 5mM MgCl2, 1mM Na3VO4, 0.5% Triton x-100, 50mM β-glycerophosphate, 30% glycerol, 1mM benzamidine, 10μg/ml aprotinin, 10μg/ml leupeptin, 1mM PMSF) for 15 min, harvested and centrifuged (15,000xg, 15 min, 4°C). The supernatants were collected and sample buffer was added (1:3) to each aliquots. Then the samples were separated on 10% SDS-PAGE followed by Western blotting with specifics antibodies: anti-diphosphorylated ERK1/2 (Mouse monoclonal, 1:10000; M9692, Sigma-Aldrich), anti-Ser(P)298-MEK1/2 (Rabbit polyclonal, 1:1000; Abcam) and anti-Ser(P)473-AKT and anti-Thr(P)308-AKT (Rabbit polyclonal, 1:1000; Cell Signaling Technology). Results were normalized to the levels obtained with total ERK1/2 (Rabbit polyclonal, 1:40000; Sigma-Aldrich) and AKT (Rabbit polyclonal, 1:10000 Sigma-Aldrich), respectively, and the activation values were detected for each treatment vs. its control or GnRH alone. The blots were developed with horseradish peroxidase (HRP)-conjugated (goat anti-rabbit antibody, Jackson Immuno-research Laboratories, West-Grove, PA, USA) followed by enhanced chemiluminescence (ECL). The blots were auto radiographed on Fuji Super RX films and the phosphorylation was quantized by densitometry by ImageQuant (Pharmacia).
2.3 Cells transfection and reporter gene assays
αT3-1 and LβT2 cells were transfected with αGSU (h-846 αLuc) or 5′-deletion mutants of h-846αLuc, were kindly provided by Dr. J. L. Jameson (Northwestern University Medical School, Chicago, IL), or with LHβ-Luc and FSHβ-Luc promoter constructs as previously described (Larder, Karali, Nelson et al., 2006), respectively, and Luc activity was analyzed (Harris, Chuderland, Bonfil et al., 2003). LβT2 cells reporter gene assays were carried out as previously described (Naor, Jabbour, Naidich et al., 2007).
2.4 Intracellular Ca2+ concentration ([Ca2+]i) measurement
LβT2 cells were plated on poly-L-lysine coated coverslips and bathed in Krebs-Ringer-like medium containing 2.5 μM Fura 2 AM (Life Technologies, Grand Island, NY) for 1 h at room temperature. After that, the coverslips were washed in Krebs-Ringer-like medium and mounted on the stage of an inverted Observer-D1 microscope (Carl Zeiss, Oberkochen, Germany) with an attached ORCA-ER camera (Hamamatsu Photonics, Hamamatsu City, Japan) and a Lambda DG-4 wavelength switcher (Sutter, Novato, CA). Hardware control and image analysis was performed using Metafluor software (Molecular Devices, Downingtown, PA). Experiments were performed under a 40X oil-immersion objective during exposure to alternating 340 and 380 nm excitation beams, and the intensity of light emission at 520 nm was followed simultaneously in approximately 20 single cells. Changes in [Ca2+]i are presented by the ratio of fluorescence intensities F340/F380.
2.5 Statistical analysis
Results from two or more experiments were expressed as mean± SEM. Where appropriate, data were subjected to statistical analysis by Student’s t test. Values of P<0.05 were considered statistically significant.
3 Results
3.1 GnRH and PMA-induced ERK1/2 activation in αT3-1 and LβT2 cells
The activation of ERK1/2 by GnRH is shown in Fig. 1. GnRH treatment resulted in a rapid and robust activation of ERK1/2 with a peak 5 min after stimulation and decline after 180 min in both αT3-1 (Fig. 1A) and LβT2 cells (Fig. 1C). Moreover, treatment with PMA, which mimics the effects of binding of DAG, the natural activator of PKC also, activated ERK1/2 in a robust pattern in both cell lines (Fig. 1B and D). However, unlike GnRH, the duration of PMA-induced ERK1/2 activation in LβT2 cells was prolonged (Fig. 1D) in comparison to GnRH (Fig. 1C). Since the phosphorylation did not decline to the basal level after 180 min, but still remained relatively high, we suggest that while ERK1/2 activation is mediated through PKC in both αT3-1 and LβT2 cells as shown before (Naor, Benard and Seger, 2000), the ERK1/2 termination mechanism is apparently partially PKC-independent and another mechanism may be involved in ERK1/2 signal termination in particular in the more mature gonadotrope derived LβT2 cells.
Figure 1.

Time course of ERK1/2 phosphorylation by GnRH and PMA in αT3-1 and LβT2 cells. αT3-1 (panels A and B) and LβT2 (panels C and D) cells were serum-starved for 16 h before treatment with GnRH or PMA (100nM each) for up to 180 min. After treatment cell lysates were analyzed for ERK1/2 activity by Western blotting using an antibody for phospho-ERK1/2 (pERK1/2). Total ERK1/2 (gERK1/2) was detected with polyclonal antibody as a control for sample loading. A representative blot is shown and similar results were observed in three other experiments. In this figure results from four experiments are shown as mean±S.E.M of maximal phosphorylation. **p-val≤0.01; *p-val≤0.05 vs. control. In this and other figures the statistical test was conducted for the combined values of ERK1 and ERK2.
3.2 Lack of inhibition by wortmannin (10nM; 30 min) of GnRH-, EGF- and PMA- induced ERK1/2 activation
In order to elucidate the role of PI3K in GnRH-induced ERK1/2 activation we have used PI3K inhibitors: wortmannin, which is a fungal metabolite that was first isolated from penicillium wortmanni, and LY294002 (LY) that is known as the first synthetic inhibitor of PI3K. LY294002 inhibits the p85α regulatory subunit of PI3K activity and also targets the ATP binding site of the p110 catalytic subunit of PI3K. Wortmannin targets the p110 catalytic subunit of PI3K through an irreversible covalent interaction. We incubated αT3-1 and LβT2 cells for 30 min with wortmannin (10nM and 10μM), or LY294002 (10μM and 100μM), doses and time which are known to inhibit PI3K and PI4K, respectively (Balla and Balla, 2006, Nakanishi, Catt and Balla, 1995, Rao, Paik, Zheng et al., 1997), followed by stimulation with GnRH, PMA (both at 100nM) or EGF (10ng/ml) for 5 min to detect ERK1/2 activation. GnRH-, PMA- and EGF-induced a robust ERK1/2 activation within 5 min of stimulation in both αT3-1 (Fig. 2A and B) and LβT2 cells (Fig. 2D and E). Preincubation with wortmannin for 30 min inhibited ERK1/2 activation by GnRH in both cell lines only at the high dose (10μM), while no inhibition was observed at 10nM (Fig. 2A, B, D and E). The same high dose, 10μM, inhibited ERK1/2 activation by EGF in αT3-1 cells (Fig. 2A), but not in LβT2 cells (Fig. 2D). PMA-induced ERK1/2 activation was not inhibited by wortmannin at the two doses examined in both cell lines (Fig. 2B and E). Thus, the role of PI4K in GnRH-activation of ERK1/2 is more easily detected and is cell specific.
Figure 2.

Effect of wortmannin and LY294002 (30 min) on ERK1/2 activation by GnRH, PMA and EGF in αT3-1 and LβT2 cells. αT3-1 (panels A–C) and LβT2 (panels D–F) cells were serum-starved for 16 h before treatment with wortmannin (wort), 10nM or 10μM, or LY294002 (LY), 10μM or 100μM, for 30 min before stimulation with GnRH or PMA (100nM each) or EGF (10ng/ml) for 5 min. After treatment cell lysates were analyzed for ERK1/2 activity as in Fig. 1. A representative blot is shown and similar results were observed in three other experiments. The results are shown as mean±S.E.M of maximal phosphorylation. **p-val≤0.01; *p-val≤0.05 vs. GnRH, PMA or EGF alone.
3.3 Effect of LY294002 (30 min preincubation) on GnRH- and PMA-induced ERK1/2 activation in αT3-1 and LβT2 cells
We next examined the effect of LY294002 (30 min preincubation) on GnRH- and PMA-induced ERK1/2 activation in αT3-1 and LβT2 cells (Fig. 2C and F, respectively). LY294002 inhibited GnRH-induced ERK1/2 activation in αT3-1 cells in both concentrations (10μM and 100μM) (Fig. 2C), while inhibition occurred only at 100μM in LβT2 cells (Fig. 2F). Moreover, only the high dose (100μM) also inhibited PMA-induced ERK1/2 activation in both cell lines (Fig. 2C and F). Again, as above, the role of PI4K in GnRH to ERK1/2 signaling is already detected after 30 min of incubation. According to the data so far, preincubation for 30 min with wortmannin at 10nM did not inhibit GnRH-, EGF- and PMA-induced ERK1/2 activation in both αT3-1 and LβT2 cells, whereas wortmannin at 10μM inhibited GnRH-, EGF- but not PMA-induced ERK1/2 activation in αT3-1 cells and GnRH- but not EGF- and PMA-induced ERK1/2 activation in LβT2 cells. Preincubation for 30 min with LY294002 at 10μM inhibited only GnRH-induced ERK1/2 activation in αT3-1 cells, whereas the high dose, 100μM, inhibited both GnRH- and PMA-induced ERK1/2 activation in both cell lines. Therefore, although LY294002 provided more information as to the role of PI3K in GnRH to ERK1/2 signaling, we further examined the responses of the cells to wortmannin.
3.4 Incubation with wortmannin or LY294002 for 1 h inhibits GnRH- and PMA-induced ERK1/2 activation in αT3-1 and LβT2 cells
Since we observed that preincubation for 30 min with wortmannin 10nM did not inhibit GnRH-, EGF- and PMA-induced ERK1/2 activation in both αT3-1 and LβT2 cells (Fig. 2), we decided to check whether prolonged preincubation will induce inhibition. Hence, we checked the effect of wortmannin and LY294002 after preincubation of 1 h at the two doses examined above and followed the inhibition upon GnRH or PMA on ERK1/2 activity (Figs. 3 and 4). Wortmannin (10nM and 10μM) reduced GnRH-induced ERK1/2 activation by 25% in αT3-1 cells and by 50% in LβT2 cells (Fig. 3A and Fig. 4A). Also, wortmannin (10nM and 10μM) reduced PMA-induced ERK1/2 activation by 40% in αT3-1 cells and by 35% in LβT2 cells (Fig. 3C and Fig. 4C, respectively). Similar results were obtained with two different lots of Calbiochem wortmannin. LY294002 at 10μM gave 35% inhibition on GnRH-induced ERK1/2 activation in αT3-1 and LβT2 cells, while the higher dose, 100μM, gave 85% inhibition in αT3-1 and 35% in LβT2 cells (Fig. 3B and Fig. 4B, respectively). Inhibition by LY294002 was observed also in PMA-induced ERK1/2 activation by 25% and 70% for 10μM and 100μM, respectively, in αT3-1 cells and by 35% and 55% in LβT2 cells (Fig. 3D and Fig. 4D, respectively). Preincubation for 1 h with wortmannin (10nM and 10μM), inhibited GnRH-induced MEK1/2 (an upstream effector of ERK1/2) phosphorylation at Ser298, by 30% in LβT2 cells (Fig. 4E). Thus, PI3K acts upstream to MEK1/2 during ERK1/2 activation by GnRH.
Figure 3.

Effect of wortmannin and LY294002 (1 h) on ERK1/2 activation by GnRH and PMA in αT3-1 cells. αT3-1 cells were serum-starved for 16 h before treatment with wortmannin (wort), 10nM or 10μM, or LY294002 (LY), 10μM or 100μM, for 1 h before stimulation with GnRH or PMA (100nM each) for 5 min. After treatment cell lysates were analyzed for ERK1/2 activity by Western blotting using an antibody for phospho-ERK1/2 (pERK1/2). Total ERK1/2 (gERK1/2) was detected with polyclonal antibody as a control for sample loading. A representative blot is shown and similar results were observed in five other experiments. The results are shown as mean±S.E.M of maximal phosphorylation. **p-val≤0.01; *p-val≤0.05 vs. GnRH or PMA alone.
Figure 4.

Effect of wortmannin and LY294002 (1 h) on ERK1/2, or MEK activation by GnRH and PMA in LβT2 cells. LβT2 cells were serum-starved for 16 h before treatment with wortmannin (wort), 10nM or 10μM, or LY294002 (LY), 10μM or 100μM, for 1 h before stimulation with GnRH or PMA (100nM each) for 5 min. After treatment cell lysates were analyzed for ERK1/2 or MEK1/2 activity by Western blotting using an antibody for phospho-ERK1/2 (pERK1/2) or phospho-MEKser298 (pMEK298). Total ERK1/2 (gERK1/2) was detected with polyclonal antibody as a control for sample loading. A representative blot is shown and similar results were observed in five other experiments. The results are shown as mean±S.E.M of maximal phosphorylation. **p-val≤0.01; *p-val≤0.05 vs. GnRH or PMA alone.
3.5 Lack of inhibition by wortmannin (10nM) on IGF-1-induced AKT activation after 30 min
The observation that the inhibitory effect by wortmannin on GnRH- and PMA-induced ERK1/2 activation in both αT3-1 and LβT2 cells was dependent on a long preincubation (1 h) raised the possibility that the effect of wortmannin was exerted via inhibition of another signaling pathway and not via inhibition of PI3K cascades. Therefore, we examined the effect of wortmannin (10nM and 10μM) and LY294002 (10μM and 100μM) on a known activator of PI3K, namely Insulin-like growth factor-1 (IGF-1) (Rose, Froment, Perrot et al., 2004, Mutiara, Kanasaki, Harada et al., 2008), and examined AKT phosphorylation as a measure for PI3K activation. αT3-1 and LβT2 cells were incubated for 30 min with wortmannin or LY294002 at the two doses examined previously followed by stimulation with IGF-1 (20ng/ml for 30 min) to detect AKT phosphorylation on Ser473. As shown in Figure 5, wortmannin at both concentrations has no effect on the basal AKT activation. Wortmannin at 10nM had no effect on IGF-1-induced AKT activation, whereas a strong inhibition was observed only at the high dose (10μM) in both cell types (Fig. 5A, D). Unlike wortmannin, LY294002 alone inhibited the basal high AKT phosphorylation and also inhibited IGF-1-induced AKT activation at both concentrations and in both cell types (Fig. 5B, E). The data supports the notion that short preincubation (30 min) with wortmannin (10nM) is not sufficient in our cell models to exert inhibition of PI3K. Prolonged incubation with wortmannin, 10nM and 10μM, for 1 h inhibited IGF-1-induced AKT Ser473 phosphorylation at both concentrations in αT3-1 and LβT2 cells (Fig. 5C and F, respectively). Therefore, this data suggests that prolong incubation with wortmannin is required to demonstrate the inhibitory effect on GnRH-induced ERK1/2 activation via PI3K.
Figure 5.

Effect of wortmannin and LY294002 (30–60 min) on AKT activation by IGF-1 in αT3-1 and LβT2 cells. αT3-1 (panels A–C) and LβT2 (panels D–F) cells were serum-starved for 16 h before treatment with wortmannin (wort) 10nM or 10μM, 30 min (A,D) or 1 h (C, F), or LY294002 (LY), 10μM or 100μM, 30 min (B, E) before stimulation with IGF-1 (20ng/ml) for 30 min. After treatment cell lysates were analyzed for AKT activity by Western blotting using an antibody for phospho-AKT Ser473 (pAKTser473). Total AKT was detected with polyclonal antibody as a control for sample loading. A representative blot is shown and similar results were observed in three other experiments. The results are shown as mean±S.E.M of maximal phosphorylation. **p-val≤0.01; *p-val≤0.05 vs. IGF-1 alone.
3.6 Wortmannin and LY294002 do not alter [Ca2+]i elevation by GnRH in LβT2 cells
It could be argued that the prolonged incubation with wortmannin affected another signaling cascade, which in his turn affected ERK1/2 activation. A case in point is the possibility that the inhibition of wortmannin and LY294002 was exerted at the level of PI(4,5)P2. In this case reduction in PI(4,5)P2 will lead to reduction in Ins(1,4,5)P3 and diacylglycerol (DAG) which can lead to reduction of [Ca2+] release and PKC activation, both known to participate in ERK1/2 activation in general (Naor et al., 2000) and in the GnRH response in particular (Bonfil, Chuderland, Kraus et al., 2004, Mulvaney, Zhang, Fewtrell et al., 1999, Mulvaney and Roberson, 2000). Thus, we next examined the effect of wortmannin and LY294002 on GnRH-induced Ca2+ signaling. Figure 6 illustrates typical patterns of GnRH-induced Ca2+ signaling in LβT2 cells bathed in Ca2+-deficient medium (Fig. 6A) and in 2mM Ca2+-containing medium (Fig. 6B). As shown in both experimental conditions, GnRH-induced a dose-dependent rapid spike response. In Ca2+-deficient medium, the spike rise in [Ca2+]i was followed by a decrease to basal level within 2–3 min whereas in Ca2+-containing medium the spike response was followed by fluctuations in [Ca2+]i (Fig. 6A and B, respectively). Treatment with LY294002 (10μM) alone increased [Ca2+]i (Fig. 6C), but it kept the spike response and the fluctuations in [Ca2+]i that are observed with GnRH 100nM treatment. Pre- treatment with wortmannin, 10nM and 10μM, for 10 or 30 min, had no obvious effect on GnRH (100nM)-induced response (Fig. 6D). Furthermore, the same observation occurred with pre-treatment for 15, 30 and 60 min before the GnRH (10nM)-induced response (Fig. 6E). These data indicate that wortmannin (10nM and 10μM) has no significant effect on GnRH-induced [Ca2+]i response in either short or prolonged preincubation in LβT2 cells.
Figure 6.
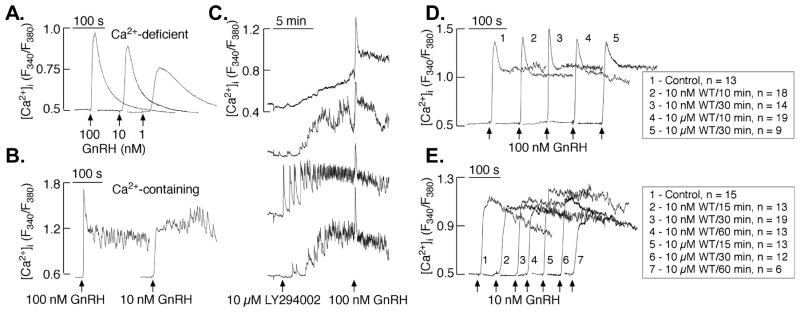
Effect of wortmannin and LY294002 on GnRH-induced calcium signaling in LβT2 cells. LβT2 cells bathed in Ca2+-deficient medium were treated with 1, 10 or 100nM GnRH to measure [Ca2+]i level (A). Typical fluctuations in [Ca2+]i levels in LβT2 cells bathed in 2mM Ca2+-containing medium in response to 100 or 10nM GnRH treatment (B). Examples of LY294002-induced [Ca2+]i increase and consequent response to 100 nM GnRH. (C). Average traces of 100nM GnRH-induced [Ca2+]i responses in Ca2+-containing controls and cells treated by 10nM or 10μM Wortmannin 10 or 30 min each before treatment with GnRH (D). Average traces of 10nM GnRH-induced [Ca2+]i responses in Ca2+-containing controls and cells treated by 10nM or 10μM Wortmannin 15, 30 or 60 min each before treatment with GnRH (E).
3.7 GnRH- and PMA-induced AKT dephosphorylation
Based on our previous study in αT3-1 cells (Ben-Ami, Yao, Naor et al., 2011), we evaluated the effect of GnRH and PMA upon AKT phosphorylation (Fig. 7). We treated the cells with GnRH (100nM) for up to 240 min, and followed AKT phosphorylation at both Thr308 and Ser473, which are crucial for AKT activity. Our results show that the basal phosphorylation of AKT in both sites was markedly high and reduced rapidly within 5 min of stimulation by GnRH, while the lowest phosphorylation was observed around 60 min in αT3-1 cells (Fig. 7A and B). There is a complex cross-talk between the Ras-ERK1/2 and PI3K-mTOR pathways (Mendoza et al., 2011). We therefore pretreated the αT3-1 cells with two selective MEK1/2 inhibitors: U0126 and PD98059, 30 min prior to GnRH addition for 5, 30 and 60 min. Both PD98059 and U0126 did not affect basal and GnRH-induced AKT dephosphorylation (data not shown). Therefore, GnRH-induced AKT dephosphorylation is independent of ERK1/2 activation. Similar treatment with PMA (100nM) for 90 min resulted in inhibition of pSer473-AKT in αT3-1 cells (Fig. 7C). An initial rise (5 min) was followed by prolonged inhibition at least for 90 min. Since PI3K is intimately involved in IGF-1 signaling (Rose et al., 2004), we studied its effect upon AKT. Treatments with IGF-1 (20ng/ml) induced a rapid and sustained pSer473-AKT phosphorylation in αT3-1 cells (Fig. 7D). Thus, IGF-1 to AKT signaling serves here as a positive control for GnRH and PMA.
Figure 7.

Time course of AKT activity by GnRH, PMA and IGF-1 in αT3-1 cells. αT3-1 cells were serum-starved for 16 h, followed by stimulation with GnRH, PMA (100nM each) or IGF-1 (20ng/ml) for up to 240 min. After treatment cell lysates were analyzed for AKT activity by Western blotting using an antibody for phospho-AKT- Ser473 (pAKTser473) or phospho-AKT-Thr308 (pAKTthr308). Total AKT was detected with polyclonal antibody as a control for sample loading. A representative blot is shown and similar results were observed in three other experiments. The results from three experiments are shown as mean±S.E.M of control phosphorylation.**p-val≤0.01; *p-val≤0.05 vs. control.
3.8 Effect of wortmannin on basal and GnRH-stimulated αGSU, LHβ and FSHβ transcription
As a key regulator of reproduction, GnRH stimulates gonadotropin (LH and FSH) synthesis and release. Gonadotropins share a common αGSU subunit and selective β subunits (LHβ and FSHβ). We therefore examined the role of PI3K in basal and GnRH-stimulated αGSU, LHβ and FSHβ transcription. The promoter’s transfected cells were pretreated for 1 h with wortmannin, followed by incubation with or without GnRH and promoter activity was determined (Fig. 8A–C). GnRH increased αGSU-, LHβ- and FSHβ-Luc activity by 2-, 4- and 4-fold, respectively. Wortmannin alone had no significant effect on promoter’s activity. However, wortmannin affected the GnRH response. Wortmannin enhanced the αGSU response to 6-fold, had no effect on the LHβ response (4-fold) and reduced the FSHβ response to 3-fold. As wortmannin is the inhibitor of PI3K, we suggest that PI3K inhibits GnRH-stimulated αGSU-, has no effect upon GnRH-stimulated LHβ- and enhanced the GnRH-stimulated FSHβ-gene expression.
Figure 8.

Effect of wortmannin on GnRH-iduced gonadotropin subunit promoter activities and PI3K-responsive regions in αGSU-promoter. αT3-1 cells were transiently transfected with αGSU-Luc (A) and LβT2 cells were transiently transfected with LHβ-Luc (B) and FSHβ-Luc (C) promoter constructs before being pretreated for 1 h with wortmannin (10nM) followed by addition of GnRH (100nM) for 6 h. Cells were harvested and assayed for an increase in reporter gene expression expressed as relative light units (RLU), after normalization of transfection efficiency with an internal control. One-way ANOVA determined that ***p-val<0.001; **p-val<0.01 and *p-val<0.05 were significantly different between treatment groups. D. Subconfluent αT3-1 cells were transfected with 5′-deletion mutants of h-846αGSU-Luc for 8 h. Cells were washed several times and incubated for 36 h. Cells were preincubated with or without wortmannin (10nM) followed by GnRH treatment (100nM) for 6 h. αGSU-Luc activity was determined as described in Materials and Methods. PGBE, pituitary glycoprotein basal element; αBE, α basal elements; GSE, glycoprotein specific element; URE, upstream response element; CRE, cAMP response element; JRE, junctional response element; TSS, transcription start site.
We then used 5′-deletion analysis to map the GnRH-responsive regions of αGSU promoter which are sensitive to PI3K inhibition (Fig. 8D). αT3-1 cells were transfected with the various αGSU-promoter deletion mutants linked to Luc and later preincubated with wortmannin (10nM) followed by GnRH-A (100nM). We have previously shown that basal h-846αGSU-Luc was increased by 3-fold by GnRH-A (Harris et al., 2003). When the GnRH response is taken as 100%, the presence of wortmannin increased it to 280% (Fig. 8D). The same response was still observed with the first deletion −846/ −420. However, the next deletion to −420/−346 had no effect on the GnRH response, but eliminated the stimulatory effect of wortmannin completely. Hence the inhibitory effect of PI3K upon GnRH-stimulated αGSU activity seems to be mediated by the −420/−346 region. Further deletion to −346-/280 reduced the GnRH-A response without effect on wortmannin in relation to GnRH. Interestingly, we have previously shown that ERK1/2 involvement in GnRH-stimulated αGSU activity is also mediated by the −346/−280 region (ibid). The data suggest that a region involved in GnRH-ERK1/2 stimulation of αGSU activity acts in a PI3K-independent manner. Indeed, this region includes the pituitary glycoprotein hormone basal element (PGBE) and αGSU basal element (αBE1/2) (Kay and Jameson, 1992, Heckert, Schultz and Nilson, 1995, Schoderbek, Roberson and Maurer, 1993).
4 Discussion
Here we investigate the role of PI4K and PI3K-AKT in GnRH to ERK1/2 signaling in the immortalized αT3-1 and LβT2 gonadotrope cells. The study is a continuation of our efforts to characterize the role of PKC isoforms in MAPK activation (Naor et al., 2000, Dobkin-Bekman, Rahamin-Ben Navi, Shterntal et al., 2010, Reiss, Llevi, Shacham et al., 1997, Benard, Naor and Seger, 2001). Combining the two efforts we also aimed to identify the site of PKC vs. PI3K during ERK1/2 activation by GnRH. ERK1/2 activation by GnRH is robust, rapid and protracted in both αT3-1 and LβT2 cells and is regarded as one of the strongest responses among GPCRs, implicating a major role for ERK1/2 in GnRH actions (Naor et al., 2000).
To investigate the role of PI4K and PI3K-AKT in GnRH-stimulated ERK1/2 activation, we incubated the αT3-1 and LβT2 cells with wortmannin (10nM and 10μM), or LY294002 (10μM and 100μM) for 30 min, followed by GnRH, PMA or EGF and measured ERK1/2 activation. The idea was that under these conditions (30 min preincubation), PI3K and PI4K will be inhibited by the low and high doses of both inhibitors, respectively (Balla, 2006, Nakanishi et al., 1995, Rao et al., 1997). Only the high dose of wortmannin (10μM) and LY294002 (100μM) gave consistent inhibition of ERK1/2 activation by GnRH at the 30 min preincubation time suggesting a potential role for PI4K in GnRH-stimulated ERK1/2 activation. The calcium-sensing receptor stimulates PI4K by Rho-dependent and Gαq-and Gαi-independent pathways (Huang, Handlogten and Miller, 2002). Indeed, Rho is involved in GnRHR signaling. Also, PI4K is the first step in inositol lipid biosynthesis converting PI to PI4-P (Balla, 2006). Since GnRH signaling is tightly coupled to phosphoinositides metabolism (Naor and Huhtaniemi, 2013) (e.g. IP3-Ca2+ and DAG-PKC), the requirement for a role for PI4K seems important due to the central role of PKC in ERK1/2 activation (ibid) (Fig. 9).
Figure 9.

A proposed model for PI4K and PI3K-AKT involvement in GnRH to ERK1/2 signaling and gonadotropins gene expression in αT3-1 and LβT2 gonadotrope cells. Binding of GnRH to its receptor exert activation of Gαq-PLCβ-PKC to mediate ERK1/2 activation, which results in its translocation to the nucleus to phosphorylate transcription factors involved in gonadotropin subunits gene expression. PI4K, known to be activated by Rho and Pyk2 converts PI to PIP. PI3K, known to be activated by Gβγ converts PIP2 to PIP3. PIP3 translocates PDK1 and mTORC2 to the membrane. PLCβ, known to be activated by Gαq converts PIP2 to IP3 and DAG, which are involved in Ca2+ mobilization and PKC activation, respectively. PDK1 and mTORC2 phosphorylate PKC and AKT during their activation. Still PKC inhibits AKT activation. Reduction in AKT during GnRHR activation alleviates Raf inhibition by AKT, thus supporting ERK activation. PKC acts upstream and downstream to PI3K, while some PKC isoforms are activated by PIP3. PI3K inhibits GnRH-stimulated αGSU transcription, has no effect on LHβ and stimulates GnRH-activation of FSHβ transcription.
The observation that wortmannin (10nM for 30 min) did not inhibit GnRH- and PMA-induced ERK1/2 activation in both αT3-1 and LβT2 cells, raised the possibility that a longer preincubation with wortmannin is required to exert inhibition on PI3K. This idea contradicted other reports that the inhibitory effect of wortmannin can be observed already within 10–20 min in rat adipocytes (Okada, Kawano, Sakakibara et al., 1994), cultured adrenal glomerulosa cells (Nakanishi et al., 1995) and RBL-2H3 cells (Yano, Nakanishi, Kimura et al., 1993). Still we decided to examine the effect of the two drugs after 1 h of preincubation. Indeed, preincubation with wortmannin (10nM and 10μM), or LY294002 (10μM and 100μM) for 1 h inhibits GnRH- and PMA-induced ERK1/2 activation in both αT3-1 and LβT2 cells, even at the low doses. Furthermore, wortmannin (10nM and 10μM), inhibited also phosphorylation of MEK1/2 (pSer298MEK) by GnRH in LβT2 cells. Thus, PI4K and PI3K-AKT act upstream to MEK1/2 during ERK1/2 activation by GnRH. Furthermore, the observation that wortmannin inhibited PMA-induced ERK1/2 activation reveals that PI3K is acting downstream of PKC. Still, we can estimate that the contribution of PI4K and PI3K-AKT to GnRH-evoked ERK1/2 activation is at the range of 40–60% in both cells and other players are acting also.
Our results suggest that 1 h preincubation is required for wortmannin to be effective in the two cell models used here. Once the concentration is reached wortmannin and LY294002 will participate in the inhibitory effect on GnRH- and PMA-induced ERK1/2 activation in both αT3-1 and LβT2 cells. This will explain why we missed the inhibitory effect of wortmannin on GnRH to ERK1/2 in a previous study, in which we used a short incubation only (Benard et al., 2001).
Previous findings revealed that wortmannin attenuated GnRH (100nM)-induced LH release in cultured rat pituitary cells in a concentration-dependent manner, with IC50 of 2μM (Rao et al., 1997). Indeed, in pituitary gonadotropes higher concentrations of wortmannin were found to inhibit secretion as compared to other cells (Balla, 2006, Rao et al., 1997). The authors concluded that the inhibition exerted by wortmannin was not mediated by PI3K, since nM of wortmannin is sufficient for PI3K inhibition (Rao et al., 1997).
To better demonstrate the critical role of the long preincubation with wortmannin we needed a positive control and decided to use IGF-1 a known activator of PI3K/AKT (Rose et al., 2004, Mutiara et al., 2008). IGF-1 activation of AKT phosphorylation was a good measure for PI3K activation. As with GnRH, a relatively long preincubation (60 but not 30 min) with wortmannin was required to observe inhibition of IGF-1-induced AKT activation. Thus, the IGF-1 data further supported a role for PI3K in ERK1/2 activation by GnRH. Nevertheless, the actual measurements of PI3K activity during GnRH action will be described elsewhere (Nadel et al., in preparation).
The possibility that the inhibitory effect of wortmannin during the prolonged preincubation was exerted via reduction in PI(4,5)P2 and therefore in Ins(1,4,5)P3 and DAG, which can lead to reduction of [Ca2+]i and PKC was investigated. We found no inhibition of [Ca2+]i responses to GnRH by wortmannin at both 30 and 60 min of preincubation and at the two doses examined above (10nM and 10μM).
PI3K are classically activated via growth factor receptors (RTKs) (Otsu, Hiles, Gout et al., 1991). Active RTKs elicit authophosphorylation of the receptor and binding of SH2 domain expressing proteins to the phosphorylated tyrosine residues in the receptor. Among them is the p85 regulatory subunit of PI3K, which binds to the RTKs, is phosphorylated on tyrosine residues, activated and can now recruit the catalytic subunits of PI3Ks to the membrane. Since GnRH does not transactivate the EGFR pathway in pituitary gonadotropes (Kraus, Benard, Naor et al., 2003), this pathway is not relevant in our case. GPCRs can also activate the PI3K pathway via Gβγ or directly through Ras-GTP (Vanhaesebroeck et al., 2010, New et al., 2007, Mendoza et al., 2011). It was previously reported that the muscarinic (m2) receptor activates MAPK via Gβγ-dependent activation of class IB PI3K (p110γ) (Lopez-Ilasaca, Crespo, Pellici et al., 1997). Also, PI3Kγ is thought to be activated by the βγ subunits of Gi/Go directly or via association with p101 or p84 regulatory subunits in hematopoietic cells (Stephens, Smrcka, Cooke et al., 1994). In another study it was shown that PI3K activity is required in the Gβγ-dependent activation of MAPK at a point upstream to Sos and Ras activation (Hawes, Luttrell, van Biesen et al., 1996). Much less is known about the activation of the other isoforms of class I PI3K (α,β,δ), the factors involved in their activation, the p85/p55 splice form associated with them and how they integrate with GPCR signaling (Balla, 2006, Balla, 2013). Class IA PI3K (p110α, β and δ) can be activated by GPCRs via Gβγ for p110β, or through RTKs transactivation for p110α and p110δ (Vanhaesebroeck et al., 2010, New et al., 2007). Indeed, the dominant negative mutant of p110β, but not p110γ, inhibits MAPK stimulation in response to lysophosphatidic acid (LPA) (Yart, Roche, Wetzker et al., 2002). P110β acts upstream of Ras via its lipid kinase activity and overexpression of the lipid phosphatases PTEN inhibited Ras stimulation by LPA, therefore p110β is the target of Gβγ and provides a novel link between βγ and Ras during LPA signaling (ibid).
Exploring the signaling interaction between GnRH receptor (GnRHR) and IGF-1R in αT3-1 cells Rose et al. 2004 observed that GnRH stimulates MAPK and PI3K activity but does not affect AKT phosphorylation (Rose et al., 2004). Co-treatment with both GnRH and IGF-1 reveals that GnRH inhibits IGF-1-indcuced AKT activation via PI3K-independent pathway but rather in a PKCα-dependent mechanism, and therefore inhibits the anti-apoptotic activity of IGF-1 in αT3-1 cells (ibid). Indeed, we have demonstrated that a GnRH agonist induced apoptotic effect in αT3-1 cells through Gαq-PKC-c-Src-JNK leading to apoptosis, while PKC inhibits AKT, alleviating its inhibitory effect on MLK3 and adding a support to the c-Src-JNK cascade (Ben-Ami et al., 2011). It was therefore of some surprise when we found that GnRH actually inhibited AKT activity via PKC (ibid). In the present results we confirmed the inhibitory effect of GnRH at both pThr308AKT and pSer473AKT in αT3-1 cells. Similar treatment with PMA resulted in inhibition of pSer473-AKT in αT3-1 cells. AKT negatively regulates ERK1/2 activation by phosphorylating inhibitory sites in Raf N-termimnus (Mendoza et al., 2011). Therefore the decrease of AKT observed here may alleviate its inhibition of Raf, enhancing ERK1/2 activity and implicating PI3K-AKT in the GnRH to ERK1/2 signaling (Fig. 9). Unlike the GnRH-PKC inhibition of AKT, IGF-1 stimulates Ser473-AKT phosphorylation in αT3-1 cells.
In our study PI4K most likely provides PtdIns 4,5-P2 (PIP2), which is the substrate for PI3K. The activated PI3K generates phosphatidylinositol 3,4,5-triphosphate (PIP3) in the membrane, inducing the recruitment of PH domain-containing proteins such as AKT, and PDK1 to the membrane. AKT is phosphorylated on Thr308 in the activation loop by PDK1 and on Ser473 in the hydrophobic motif by mTORC2, an activation process similar to PKC priming (Mendoza et al., 2011, Newton, 2003). PKC activation will follow the priming once PIP2 will be cleaved by PLCβ to generate the IP3-Ca2+ and DAG needed for relocation of PKC to the membrane, where it is activated. PKC can then participate in ERK1/2 activation at the Raf level. Still, wortmannin inhibited also GnRH-induced MEK1/2 phosphorylation suggesting that PI3K is acting above MEK1/2 in the GnRH signaling to ERK1/2. The findings here that PMA activation of ERK1/2 is sensitive to wortmannin and the activation of PKC by Ca2+ and DAG implicates PKC involvement upstream and downstream to PI3K (Fig. 9).
The relevance of the biochemical studies to the physiology of GnRH action was investigated by exploring the role of PI3K in GnRH-stimulated gonadotropin subunit gene expression. To that end the effect of wortmannin upon GnRH-stimulated αGSU-, LHβ- and FSHβ-Luc was examined. The data revealed that PI3K inhibits GnRH-stimulated αGSU activity, has no effect upon GnRH-stimulated LHβ activity and enhanced the GnRH-stimulated FSHβ gene expression. Our results are in partial agreement with Mutiara et al., 2008 who found that PI3K enhanced GnRH-stimulated αGSU activity has no effect upon GnRH-stimulated LHβ activity and enhanced the GnRH-stimulated FSHβ transcription (Mutiara et al., 2008). The only difference is in regard to αGSU and may arise from the fact that we have used αT3-1 cells while they used LβT2 cells. We have previously reported the involvement of ERK1/2 in GnRH-stimulated αGSU-, LHβ- and FSHβ gene expression (Harris et al., 2003, Bonfil et al., 2004, Harris, Bonfil, Chuderland et al., 2002). Hence the involvement of PI3K in GnRH action differs from that of ERK1/2. Thus, PI3K-ERK1/2 may participate in the activation of the FSHβ gene, while the involvement of ERK1/2 in the activation of the αGSU- and LHβ-gene is independent of PI3K. The most likely explanation is that PI3K and ERK1/2 may each affect different regions on the gonadotropin subunits promoters. To demonstrate this point we have used 5′-deletion analysis aimed to map the GnRH-responsive regions of αGSU promoter which are sensitive to PI3K inhibition. The data revealed the −420/−346 region, as the site of PI3K inhibition, while GnRH/ ERK1/2 activates the αGSU promoter via the −346/−280 region. Interestingly, all the seven known response elements in the full promoter −846 αGSU were still present at the −420/−346 region, suggesting that PI3K exerts its inhibition at a novel site. Thus, PI4K and PI3K-AKT are involved in ERK1/2 activation by GnRH but the PI3K and ERK1/2 signaling pathways can also operate separately during GnRH activation of the gonadotropin genes (Fig. 9).
Highlights.
Wortmannin and LY294002 reduced GnRH to ERK1/2 signaling at doses known to inhibit PI3K and PI4K.
PI4K and PI3K-AKT play a role in GnRH to ERK1/2 signaling, while PI3K regulates also GnRH-induced gonadotropins gene expression.
Understanding GnRH action will affect reproduction and cancer.
Acknowledgments
We thank Dr. P. Mellon for the gonadotropes cell lines, Dr. L. Jameson for the −846 αGSU and 5′-deletion mutants and Dr. R. P. Millar for the interest in this study. We thank Fiorenza Przedecki, Liat Rahamim-Ben Navi, Shany Mugami, Inna Ladin and Yathreb Easa for the interest in this study.
The studies were supported by The Israel Science Foundation (Grants No. 221/05 and 1932/15).
Footnotes
Disclosure summary: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Caunt CJ, Finch AR, Sedgley KR, McArdle CA. GnRH receptor signalling to ERK: kinetics and compartmentalization. Trends Endocrinol Metab. 2006;17:308–13. doi: 10.1016/j.tem.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Naor Z. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol. 2009;30:10–29. doi: 10.1016/j.yfrne.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Naor Z, Huhtaniemi I. Interactions of the GnRH receptor with heterotrimeric G proteins. Frontiers in neuroendocrinology. 2013;34:88–94. doi: 10.1016/j.yfrne.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annu Rev Biochem. 1981;50:465–95. doi: 10.1146/annurev.bi.50.070181.002341. [DOI] [PubMed] [Google Scholar]
- Balla T. Phosphoinositide-derived messengers in endocrine signaling. The Journal of endocrinology. 2006;188:135–53. doi: 10.1677/joe.1.06595. [DOI] [PubMed] [Google Scholar]
- Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiological reviews. 2013;93:1019–137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A, Balla T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 2006;16:351–61. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nature reviews. Molecular cell biology. 2010;11:329–41. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes AL, Chiang GG, Lang ES, Ho CB, Powis G, Vuori K, Abraham RT. The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Mol Cancer Ther. 2007;6:2505–14. doi: 10.1158/1535-7163.MCT-06-0698. [DOI] [PubMed] [Google Scholar]
- Ihle NT, Powis G. Take your PIK: phosphatidylinositol 3-kinase inhibitors race through the clinic and toward cancer therapy. Mol Cancer Ther. 2009;8:1–9. doi: 10.1158/1535-7163.MCT-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New DC, Wu K, Kwok AWS, Wong YH. G protein-coupled receptor-induced Akt activity in cellular proliferation and apoptosis. The FEBS journal. 2007;274:6025–36. doi: 10.1111/j.1742-4658.2007.06116.x. [DOI] [PubMed] [Google Scholar]
- Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends in biochemical sciences. 2011;36:320–8. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochimica et biophysica acta. 2011;1813:1619–33. doi: 10.1016/j.bbamcr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Chuderland D, Seger R. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol Biotechnol. 2005;29:57–74. doi: 10.1385/MB:29:1:57. [DOI] [PubMed] [Google Scholar]
- Larder R, Karali D, Nelson N, Brown P. Fanconi anemia A is a nucleocytoplasmic shuttling molecule required for gonadotropin-releasing hormone (GnRH) transduction of the GnRH receptor. Endocrinology. 2006;147:5676–89. doi: 10.1210/en.2006-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D, Chuderland D, Bonfil D, Kraus S, Seger R, Naor Z. Extracellular signal-regulated kinase and c-Src, but not Jun N-terminal kinase, are involved in basal and gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone alpha-subunit promoter. Endocrinology. 2003;144:612–22. doi: 10.1210/en.2002-220690. [DOI] [PubMed] [Google Scholar]
- Naor Z, Jabbour HN, Naidich M, Pawson AJ, Morgan K, Battersby S, Millar MR, Brown P, Millar RP. Reciprocal Cross Talk between Gonadotropin-Releasing Hormone (GnRH) and Prostaglandin Receptors Regulates GnRH Receptor Expression and Differential Gonadotropin Secretion. Mol Endocrinol. 2007;21:524–37. doi: 10.1210/me.2006-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab. 2000;11:91–9. doi: 10.1016/s1043-2760(99)00232-5. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Catt KJ, Balla T. A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5317–21. doi: 10.1073/pnas.92.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao K, Paik WY, Zheng L, Jobin RM, Tomic M, Jiang H, Nakanishi S, Stojilkovic SS. Wortmannin-sensitive and -insensitive steps in calcium-controlled exocytosis in pituitary gonadotrophs: evidence that myosin light chain kinase mediates calcium-dependent and wortmannin-sensitive gonadotropin secretion. Endocrinology. 1997;138:1440–9. doi: 10.1210/endo.138.4.5078. [DOI] [PubMed] [Google Scholar]
- Rose A, Froment P, Perrot V, Quon MJ, LeRoith D, Dupont J. The luteinizing hormone-releasing hormone inhibits the anti-apoptotic activity of insulin-like growth factor-1 in pituitary alphaT3 cells by protein kinase Calpha-mediated negative regulation of Akt. The Journal of biological chemistry. 2004;279:52500–16. doi: 10.1074/jbc.M404571200. [DOI] [PubMed] [Google Scholar]
- Mutiara S, Kanasaki H, Harada T, Oride A, Miyazaki K. The involvement of phosphatidylinositol 3-kinase in gonadotropin-releasing hormone-induced gonadotropin alpha- and FSHbeta-subunit genes expression in clonal gonadotroph LbetaT2 cells. Mol Cell Endocrinol. 2008;283:1–11. doi: 10.1016/j.mce.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone beta-subunit promoter. Endocrinology. 2004;145:2228–44. doi: 10.1210/en.2003-1418. [DOI] [PubMed] [Google Scholar]
- Mulvaney JM, Zhang T, Fewtrell C, Roberson MS. Calcium influx through L-type channels is required for selective activation of extracellular signal-regulated kinase by gonadotropin-releasing hormone. The Journal of biological chemistry. 1999;274:29796–804. doi: 10.1074/jbc.274.42.29796. [DOI] [PubMed] [Google Scholar]
- Mulvaney JM, Roberson MS. Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. The Journal of biological chemistry. 2000;275:14182–9. doi: 10.1074/jbc.275.19.14182. [DOI] [PubMed] [Google Scholar]
- Ben-Ami I, Yao Z, Naor Z, Seger R. Gq protein-induced apoptosis is mediated by AKT kinase inhibition that leads to protein kinase C-induced c-Jun N-terminal kinase activation. The Journal of biological chemistry. 2011;286:31022–31. doi: 10.1074/jbc.M111.247726. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kay TW, Jameson JL. Identification of a gonadotropin-releasing hormone-responsive region in the glycoprotein hormone alpha-subunit promoter. Mol Endocrinol. 1992;6:1767–73. doi: 10.1210/mend.6.11.1282668. [DOI] [PubMed] [Google Scholar]
- Heckert LL, Schultz K, Nilson JH. Different composite regulatory elements direct expression of the human alpha subunit gene to pituitary and placenta. The Journal of biological chemistry. 1995;270:26497–504. doi: 10.1074/jbc.270.44.26497. [DOI] [PubMed] [Google Scholar]
- Schoderbek WE, Roberson MS, Maurer RA. Two different DNA elements mediate gonadotropin releasing hormone effects on expression of the glycoprotein hormone alpha-subunit gene. The Journal of biological chemistry. 1993;268:3903–10. [PubMed] [Google Scholar]
- Dobkin-Bekman M, Rahamin-Ben Navi L, Shterntal B, Sviridonov L, Przedecki F, Naidich-Exler M, Brodie C, Seger R, Naor Z. Differential role of PKC isoforms in GnRH and phorbol 12-myristate 13-acetate activation of extracellular signal-regulated kinase and Jun N-terminal kinase. Endocrinology. 2010;151:4894–907. doi: 10.1210/en.2010-0114. [DOI] [PubMed] [Google Scholar]
- Reiss N, Llevi LN, Shacham S, Harris D, Seger R, Naor Z. Mechanism of mitogen-activated protein kinase activation by gonadotropin-releasing hormone in the pituitary of alphaT3-1 cell line: differential roles of calcium and protein kinase C. Endocrinology. 1997;138:1673–82. doi: 10.1210/endo.138.4.5057. [DOI] [PubMed] [Google Scholar]
- Benard O, Naor Z, Seger R. Role of dynamin, Src, and Ras in the protein kinase C-mediated activation of ERK by gonadotropin-releasing hormone. The Journal of biological chemistry. 2001;276:4554–63. doi: 10.1074/jbc.M006995200. [DOI] [PubMed] [Google Scholar]
- Huang C, Handlogten ME, Miller RT. Parallel activation of phosphatidylinositol 4-kinase and phospholipase C by the extracellular calcium-sensing receptor. The Journal of biological chemistry. 2002;277:20293–300. doi: 10.1074/jbc.M200831200. [DOI] [PubMed] [Google Scholar]
- Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. The Journal of biological chemistry. 1994;269:3568–73. [PubMed] [Google Scholar]
- Yano H, Nakanishi S, Kimura K, Hanai N, Saitoh Y, Fukui Y, Nonomura Y, Matsuda Y. Inhibition of histamine secretion by wortmannin through the blockade of phosphatidylinositol 3-kinase in RBL-2H3 cells. The Journal of biological chemistry. 1993;268:25846–56. [PubMed] [Google Scholar]
- Otsu M, Hiles I, Gout I, Fry MJ, Ruiz-Larrea F, Panayotou G, Thompson A, Dhand R, Hsuan J, Totty N. Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell. 1991;65:91–104. doi: 10.1016/0092-8674(91)90411-q. [DOI] [PubMed] [Google Scholar]
- Kraus S, Benard O, Naor Z, Seger R. c-Src is activated by the epidermal growth factor receptor in a pathway that mediates JNK and ERK activation by gonadotropin-releasing hormone in COS7 cells. The Journal of biological chemistry. 2003;278:32618–30. doi: 10.1074/jbc.M303886200. [DOI] [PubMed] [Google Scholar]
- Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science. 1997;275:394–7. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- Hawes BE, Luttrell LM, van Biesen T, Lefkowitz RJ. Phosphatidylinositol 3-kinase is an early intermediate in the G beta gamma-mediated mitogen-activated protein kinase signaling pathway. The Journal of biological chemistry. 1996;271:12133–6. doi: 10.1074/jbc.271.21.12133. [DOI] [PubMed] [Google Scholar]
- Yart A, Roche S, Wetzker R, Laffargue M, Tonks N, Mayeux P, Chap H, Raynal P. A function for phosphoinositide 3-kinase beta lipid products in coupling beta gamma to Ras activation in response to lysophosphatidic acid. The Journal of biological chemistry. 2002;277:21167–78. doi: 10.1074/jbc.M110411200. [DOI] [PubMed] [Google Scholar]
- Newton ACa. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–71. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHbeta-subunit promoter. Endocrinology. 2002;143:1018–25. doi: 10.1210/endo.143.3.8675. [DOI] [PubMed] [Google Scholar]