

Abstract
Alkaptonuria (AKU) is a rare autosomal recessive disorder with incidence ranging from 1:100,000 to 1:250,000. The disorder is caused by a deficiency of the enzyme homogentisate 1,2-dioxygenase (HGD), which results from defects in the HGD gene. This enzyme converts homogentisic acid to maleylacetoacetate and has a major role in the catabolism of phenylalanine and tyrosine. To elucidate the mutation spectrum of the HGD gene in patients with alkaptonuria from 42 patients attending the National Alkaptonuria Centre, 14 exons of the HGD gene and the intron–exon boundaries were analysed by PCR-based sequencing. A total of 34 sequence variants was observed, confirming the genetic heterogeneity of AKU. Of these mutations, 26 were missense substitutions and four splice site mutations. There were two deletions and one duplication giving rise to frame shifts and one substitution abolishing the translation termination codon (no stop). Nine of the mutations were previously unreported novel variants. Using computational approaches based on the 3D structure, these novel mutations are predicted to affect the activity of the protein complex through destabilisation of the individual protomer structure or through disruption of protomer–protomer interactions.
Electronic supplementary material: The online version of this chapter (doi:10.1007/8904_2014_380) contains supplementary material, which is available to authorized users.
Keywords: Alkaptonuria, Homogentisic acid, Novel mutation, Rare genetic disorder, Sequencing
Introduction
Alkaptonuria (AKU) [OMIM #203500] was the first inborn error of metabolism described by Garrod, in 1902 (Garrod 1908). It is a rare autosomal recessive disorder, which results from mutations in the homogentisate 1,2-dioxygenase (HGD) gene. These mutations disrupt HGD [E.C.1.13.11.5] enzyme activity (La Du et al. 1958). The resulting deficiency of HGD activity leads to accumulation of homogentisic acid (HGA), a product of the tyrosine degradation pathway. The incidence of AKU is 1:100,000 to 1:250,000 with higher incidence in Slovakia and the Dominican Republic (1:19,000) (Phornphutkul et al. 2002; Srsen et al. 2002). Oxidation of HGA results in the formation of benzoquinones, which polymerise and bind to cartilage and connective tissue proteins leading to ochronotic pigmentation (Helliwell et al. 2008; Keller et al. 2005; Ranganath et al. 2013; Taylor et al. 2012). This deposition leads to degenerative premature arthritis and cardiac valve deterioration, leading AKU patients to experience considerable pain, incapacity and disability. The same reaction causes the urine of patients to darken upon standing, usually the first visible symptom of the disease (Fernandez-Canon et al. 1996).
The HGD gene has been mapped to chromosome 3q21-q23 (Pollak et al. 1993) with potential disruptive mutations identified in AKU patients and their families. The HGD genes carrying the AKU-associated mutations were then expressed and assayed demonstrating the loss of enzyme activity (Fernandez-Canon et al. 1996). The enzymatic defect in HGD is autosomal recessive, due to homozygosity or compound heterozygosity for mutations within the HGD gene. While AKU is not very common, a wide variety of causative mutations and polymorphisms have been reported (http://hgddatabase.cvtisr.sk).
The X-ray crystal structure of the wild-type protein revealed that the active enzyme consists of six protomers that form a dimer of trimers. Each protomer contains an N-terminal domain of 280 residues with a central β-sandwich and a C-terminal domain of 140 residues that sits aside a straddle formed by the N-terminal domain. The active site of the enzyme binds iron that is coordinated by the side chains of His335, Glu341 and His371 (Titus et al. 2000). Many non-covalent intra- and intermolecular interactions are essential to maintain the intricate structures of the protomer, trimer and hexamer. The complex structure of HGD, and hence its enzyme activity, can be easily disrupted by mutations.
In 2012, the NHS National Special Services Commissioning Group established the Robert Gregory National Alkaptonuria Centre (NAC) at the Royal Liverpool University Hospital in 2012. To date, 42 patients with confirmed AKU have attended for clinical assessments, with a national register establishing 75 patients in the UK with AKU, thus far. These patients have received trial therapy with nitisinone with subsequent metabolic investigations to monitor the safety and efficacy of the treatment. This study details the genetic analysis undertaken on all 42 patients who have been attending the NAC. Each patient has had genetic screening for small-scale mutations within the 14 exons of the HGD gene and in the intron–exon boundaries.
Material and Methods
Subjects
The 42 patients with AKU were diagnosed by clinical evaluation of their symptoms and by measurement primarily of urine homogentisic acid by liquid chromatography tandem mass spectrometry (Hughes et al. 2014). Classically, AKU patients excrete HGA in mmol/L quantities (normal reference range <0.29 mmol/mol creatinine) (Davison et al. 2014). The patient population included 34 non-related probands, three pairs of affected siblings and a father and son who were both affected. There were 24 males and 18 females who ranged from 20 to 74 years of age. The cohort included seven patients of non-Caucasian origin.
Mutation Analysis of the HGD Gene
Genomic DNA was extracted from whole blood using the QIAamp DNA blood mini kit (Qiagen Ltd, Crawley, West Sussex). Sequence variation was studied by amplification of the HGD coding regions and intron–exon boundaries by PCR and direct sequencing of the PCR products. Sequence variation was detected by comparison of the sample sequences with the genomic reference sequence of the HGD gene (NG_011957). The position of mutations was described with reference to the mRNA sequence (NM_000187.3) with the first base of the Met codon counted as position +1. The Mutalyzer 2.0β-8 https://mutalyzer.nl/name-generator name checker was used to check that the sequence variants were described according to the Human Genome Variation Society (HGVS) nomenclature.
Although the sequencing was performed in order to identify mutations affecting HGD enzyme function, the regions sequenced also covered the sites of some polymorphic variations. Nine single-nucleotide polymorphisms were analysed by sequencing: IVS2+35 (c.87+35A/T), H80Q (c.240T/A), IVS5+25 (c.342+25T/C), IVS6+16 (c.434+16C/T), IVS6+46 (c.434+46C/A), IVS7+24 (c.469+24C/A), IVS9+31 (c.549+31G/A), IVS9+39 (c.549+39T/G) and IVS11+18 (c.879+18A/G) (Supplementary Table 1)
The effects of missense mutations identified in the coding regions were analysed and predicted using the bioinformatics programs PolyPhen2 http://genetics.bwh.harvard.edu/pph2/ and SIFT http://sift.jcvi.org/ (Ramensky et al. 2002; Ng and Henikoff 2003). The PolyPhen server uses the UniProt entry, which is annotated for function, to check the site of the substitution and to identify homologous proteins. The resulting multiple alignment is used to calculate a profile matrix and a PSIC (position-specific independent counts) score. High values of this difference may indicate that the studied substitution is rarely or never observed in the protein family and could be potentially deleterious. The missense variations were also submitted to the SIFT (Sorting Intolerant from Tolerant) program, PROVEAN (Protein Variation Effect Analyzer) http://provean.jcvi.org/seq_submit.php (Choi et al. 2012) and SNAP (screening for non-acceptable polymorphisms) https://rostlab.org/services/snap/submit (Bromberg and Rost 2007). These are able to predict the consequences of the changes to the protein structure and function.
We also used mCSM (Pires et al. 2014a) and DUET (Pires et al. 2014b) in order to predict the effects of the novel missense mutations on a structural basis. These approaches are novel machine-learning algorithms that use the three-dimensional structure in order to predict quantitatively the effects of point mutations on protein stability and protein–protein and protein–nucleic acid affinities. Two crystal structures of human HGD have been published and were used in this analysis (PDB code 1EY2 and 1EYB) (Titus et al. 2000). The effects of the mutations were assessed in the context of the molecular interactions of the wild-type residue and using mCSM (Pires et al. 2014a) and DUET (Pires et al. 2014b) to predict the effects of the mutations on protomer and hexamer thermal stability and mCSM-PPI (Pires et al. 2014a) to predict the effects of the mutations on the affinity of the protomers to interact with each other.
Computer programs are also available for the analysis of more complex splice site, deletion and no-stop mutations which are all likely to be pathogenic. For splice site mutations, the Berkley Drosophila Genome Project (BDGP) fruit fly splice site predictor http://www.fruitfly.org/seq_tools/splice.html and the Net2Gene splice site predictor were used http://www.cbs.dtu.dk/services/NetGene2/ (Reese et al. 1997; Brunak et al. 1991) in order to predict the location of alternative splice sites. Both programs use artificial neural networks to predict splice sites with high levels of confidence. Most splicing mutations are found to eliminate the GT or AG dinucleotides that define the 5′ and 3′ ends of introns (Kralovicova and Vorechovsky 2007).
Splice site mutations cause either exon skipping of the respective exon or activation of pre-existing splice sites; the CRYP-SKIP utility found at http://www.dbass.org.uk/cryp-skip was developed to distinguish between these two outcomes and uses a multivariate discrimination procedure to predict which is the most likely (Divina et al. 2009).
The protein effect of either cryptic splice site activation or exon skipping can be described by the Mutalyzer 2.0 β-8 program which was primarily designed to assist in checking the nomenclature of variants using the Human Genome Sequence Variation Society (HGVS) standard human sequence variant guidelines and using annotated genomic reference sequences (Wildeman et al. 2008).
Once the position and nature of a sequence change has been established, the sequence change is analysed by a set of tools for the prediction of the variant effect on transcription and RNA processing such as splicing and translation.
Results and Discussion
A total of 34 mutations were identified in the 42 UK patients with AKU, including nine novel and 25 previously reported mutations (Table 1). The mutations from five patients included in this study have been previously reported (Zatkova et al. 2011). There were eleven recurrent mutations that have been identified in the HGD mutation database (Zatkova et al. 2011). The HGD mutation database currently lists 620 variants, from which almost eighty percent are missense substitutions. The remainder are deletions, duplications, insertions, indels or unknown. In their report of 72 AKU patients and review of previously reported mutations, Vilboux et al. (2009) described 91 variants, 62 missense, 10 frameshift, 13 splice site, 5 nonsense and one no-stop mutation. The results from the UK cohort mirror these findings; 26 of the 34 mutations identified were missense with the remaining eight mutations falling into the other categories. Fifteen of the subjects were homozygous for eleven different variants, six of these were common European variants, two were previously reported variants and three were novel.
Table 1.
The HGD gene mutations identified in the 42 AKU patients
| Patient ID | Variant 1 | Protein | Variant 2 | Protein | Other remarks |
|---|---|---|---|---|---|
| 001 sib of 022 | c.359G>T | C120F | c.589A>G | R197G | Compound heterozygous |
| 002 | c.481G>A | G161R | c.674G>A | R225H | Compound heterozygous |
| 003 | c.342+1G>T | L95_S114del | c.470-479del25G>T | V157fs | Compound heterozygous |
| 004 | c.343G>C | G115R | c.1282_1292 del GAGCCACTCAA | K431fs | Compound heterozygous |
| 005 | c.125A>C | E42A | c.125A>C | E42A | Homozygous |
| 006 | Unknown | Unknown | c.899T>G | V300G | Unknown |
| 007 | c.688C>T | P230S | c.688C>T | P230S | Homozygous |
| 008 | c.359G>T | C120F | c.359G>T | E42A | Compound heterozygous |
| 009 sib of 013 | c.125A>C | E42A | c.1102A>G | M368V | Compound heterozygous |
| 010 | c.125A>C | E42A | c.125A>C | E42A | Homozygous |
| 011 | c.481G>A | G161R | c.481G>A | G161R | Homozygous |
| 012 | c.808G>A | G270R | c.1120G>C | D374H | Compound heterozygous |
| 013 sib of 009 | c.125A>C | E42A | c.1102A>G | M368V | Compound heterozygous |
| 014 | Unknown | Unknown | c.367G>A | G123R | Unknown |
| 015 | c.16-1G>A | ivs1-1G>A | c.1102A>G | M368V | Compound heterozygous |
| 016 | c.828G>C | K276N | c.1081G>A | G361R | Compound heterozygous |
| 017 | c.828G>C | K276N | c.828G>C | K276N | Homozygous |
| 018 | c.469+2T>C | ivs7+2T>C | c.507T>G | F169L | Compound heterozygous |
| 019 | c.359G>T | C120F | c.1079G>C | G360R | Compound heterozygous |
| 020 | c.158G>A | R53Q | c.515T>C | M172T | Compound heterozygous |
| 021 | c.656A>G | N219S | c.656A>G | N219S | Homozygous |
| 022 sib of 001 | c.359G>T | C120F | c.589A>G | R197G | Compound heterozygous |
| 023 | c.367G>A | G123R | c.1075C>T | P359L | Compound heterozygous |
| 024 | c.175delA | S59fs | c.175delA | S59fs | Homozygous |
| 025 sib of 034 | c.158G>A | R53Q | c.158G>A | R53Q | Homozygous |
| 026 | c.175delA | S59fs | c.175delA | S59fs | Homozygous |
| 027 | c.175delA | S59fs | c.899T>G | V300G | Compound heterozygous |
| 028 | c.688C>T | P230S | c.899T>G | V300G | Compound heterozygous |
| 029 father of 036 | c.674G>C | R225P | c.674G>C | R225P | Homozygous |
| 030 | c.52G>A | D18N | c.175delA | S59fs | Compound heterozygous |
| 031 | c.359G>T | C120F | c.359G>T | C120F | Homozygous |
| 032 | Unknown | Unknown | c.664_674dupGCCAATCCTCG | D226PfsX7 | Unknown |
| 033 | c.37G>A | E13K | c.733G>T | V245F | Compound heterozygous |
| 034 sib of 025 | c.158G>A | R53Q | c.158G>A | R53Q | Homozygous |
| 035 | c.125A>C | E42A | c.481G>A | G161R | Compound heterozygous |
| 036 son of 029 | c.674G>C | R225P | c.674G>C | R225P | Homozygous |
| 037 | c.343G>C | G115R | c.1102A>G | M368V | Compound heterozygous |
| 038 | Unknown | Unknown | c.125A>C | E42A | Unknown |
| 039 | c.1009A>G | N337D | c.1009A>G | N337D | Homozygous |
| 040 | c.125A>C | E42A | c.899T>G | V300G | Compound heterozygous |
| 041 | c.347T>C | L116P | c.674G>C | R225P | Compound heterozygous |
| 042 | c.513G>T | K171N | c.513G>T | K171N | Homozygous |
In four of the subjects, only one mutation was identified in the HGD gene, in these cases there may be mutations in the unsequenced regulatory regions of the gene, or there could be large-scale deletions that have gone undetected by the sequencing method used. There were 26 missense mutations, 24 of these were predicted as damaging by PolyPhen2 and 25 were predicted to affect protein function by SIFT and all 26 were predicted deleterious by PROVEAN. Twenty of the missense mutations were predicted as non-neutral and five as neutral by SNAP. The E13K and D18N mutations were predicted as damaging by PolyPhen2 and damaging by SIFT and PROVEAN but as neutral by SNAP. One mutation (F169L) was predicted “benign” by PolyPhen2 and “tolerated” by SIFT, deleterious by PROVEAN and neutral by SNAP (Table 2). One mutation (M172T) was predicted as benign by PolyPhen2, damaging by SIFT, deleterious by PROVEAN and non-neutral by SNAP.
Table 2.
The effects of the missense mutations in the NAC patients as predicted by PolyPhen2, SIFT, PROVEAN and SNAP
| Protein effect | Nucleotide change NM_000187.3 | PolyPhen2 prediction | SIFT score/prediction | PROVEAN score/prediction | SNAP |
|---|---|---|---|---|---|
| E13K | c.37G>A | 0.842 possibly damaging | 0.01 affect protein function | −2.959 deleterious | Neutral |
| D18N | c.52G>A | 1.00 probably damaging | 0.01 affect protein function | −2.758 deleterious | Neutral |
| E42A | c.125 A>C | 1.00 probably damaging | 0.0 affect protein function | −5.639 deleterious | Non-neutral |
| R53Q | c.158G>A | 1.00 probably damaging | 0.0 affect protein function | −3.724 deleterious | Non-neutral |
| H80Q | c.240T>A | 0.01 benign | 0.65 tolerated | 1.231 neutral | Neutral |
| G115R | c.343G>C | 0.998 probably damaging | 0.0 affect protein function | −7.013 deleterious | Non-neutral |
| C120F | c.359G>T | 0.984 probably damaging | 0.0 affect protein function | −8.373 deleterious | Non-neutral |
| G123R | c.367G>A | 1.00 probably damaging | 0.0 affect protein function | −7.547 deleterious | Non-neutral |
| G161R | c.481G>A | 1.00 probably damaging | 0.0 affect protein function | −7.957 deleterious | Non-neutral |
| F169L | c.507T>G | 0.097 benign | 0.80 tolerated | −4.260 deleterious | Neutral |
| K171N | c.513G>T | 0.744 possibly damaging | 0.01 affect protein function | −3.283 deleterious | Neutral |
| M172T | c.515T>C | 0.446 benign | 0.0 affect protein function | −4.492 deleterious | Non-neutral |
| R197G | c.589A>G | 1.00 probably damaging | 0.0 affect protein function | −6.496 deleterious | Non-neutral |
| N219S | c.656A>G | 0.992 probably damaging | 0.0 affect protein function | −4.973 deleterious | Non-neutral |
| R225H | c.674G>A | 0.983 probably damaging | 0.0 affect protein function | −4.573 deleterious | Non-neutral |
| R225P | c.674G>C | 0.993 probably damaging | 0.004 affect protein function | −6.229 deleterious | Non-neutral |
| P230S | c.688C>T | 1.00 probably damaging | 0.0 affect protein function | −7.957 deleterious | Non-neutral |
| V245F | c.733G>T | 0.900 possibly damaging | 0.004 affect protein function | −3.741 deleterious | Neutral |
| G270R | c.808G>A | 1.00 probably damaging | 0.0 affect protein function | −7.957 deleterious | Non-neutral |
| K276N | c.828G>C | 1.00 probably damaging | 0.0 affect protein function | −4.973 deleterious | Non-neutral |
| V300G | c.899T>G | 0.999 probably damaging | 0.0 affect protein function | −6.879 deleterious | Non-neutral |
| N337D | C.1009A>G | 1.00 probably damaging | 0.0 affect protein function | −4.891 deleterious | Non-neutral |
| P359L | c.1075C>T | 0.999 probably damaging | 0.0 affect protein function | −9.037 deleterious | Non-neutral |
| G360R | c.1078G>C | 1.00 probably damaging | 0.0 affect protein function | −7.572 deleterious | Non-neutral |
| G361R | c.1081G>A | 0.995 probably damaging | 0.0 affect protein function | −7.338 deleterious | Non-neutral |
| M368V | c.1102 A>G | 0.596 possibly damaging | 0.0 affect protein function | −3.652 deleterious | Non-neutral |
| D374H | c.1120G>C | 0.994 probably damaging | 0.0 affect protein function | −6.861 deleterious | Non-neutral |
DNA variant numbering is based on cDNA (NM_000187.3), +1 corresponding to the A of the AGT
Protein effect numbering is based on NP_000178.2. The Uniprot ID Q93099 was used for submission of queries to PolyPhen
PolyPhen PSIC profile scores are based on sequence annotation and then multiple alignment to compute the absolute values of the difference between profile scores of both allelic variants in the position of the substitution and then map it to the known 3D protein structure. Based on this data, it predicts one of the four outcomes, PSIC <0.5 benign, >1–1.5 either possibly damaging or probably damaging dependent on the substitutions structural properties and >2.0 probably damaging. Larger PSIC values indicate that the substitution is rarely or never observed in the protein family
The disparities between the predictions made by the prediction tools used may reflect the variations in performance between them. The maximum level of accuracy achieved is around 83% (SIFT), and the sensitivity and specificity vary considerably.
The commonly observed European mutation, M368V, was predicted as possibly damaging by PoyPhen2, deleterious by SIFT and PROVEAN and non-neutral by SNAP. The M368 residue is involved in the formation of the active site of the enzyme, and the effect of the M368V mutation is to disrupt interaction at the subunit interfaces (Titus et al. 2000).
The complex structure of the enzyme can be disrupted by mutations in many different ways. Some will affect the assembly of the hexamer, others will affect the stability of the protomer and some will interfere with the active site of the enzyme. Additionally, the majority of patients carry two different mutations, and the combined effects of these on enzyme structure and function are difficult to predict. The protomer interfaces are non-isologous, and heterozygous hexamers appear likely to be as or more disruptive than the homozygous complex. The novel mutations are spread throughout the structure of HGD: with four buried within the protomer structure (D18N, M172T, V245F, G361R) and four located at the interface between two protomers (E13K, R53Q, R225P, N337D). The effects of mutations on the HGD structure were quantitatively analysed from two perspectives: changes on affinity between protomer interfaces and the effects on protomer stability. To assess the effects of the mutations on the affinity between subunits, we used mCSM-PPI (Pires et al. 2014a), a machine-learning method that uses graph-based signatures to predict the effects of the mutations on protein–protein affinity. To evaluate the effects of the mutations on the stability of the monomeric units, we used DUET (Pires et al. 2014b), an approach that combines the graph-based signatures of mCSM (Pires et al. 2014a) with the statistical potential energy function of SDM (Worth et al. 2011) to estimate the change in Gibbs free energy of folding caused by the mutation. The structural analysis of the novel mutations revealed that they affect either protomer stability and/or hexamer formation (Fig. 1 and Table 3). The majority of the variants were predicted by mCSM-PPI to reduce the affinity of protomers to interact, destabilising the hexameric structure.
Fig. 1.
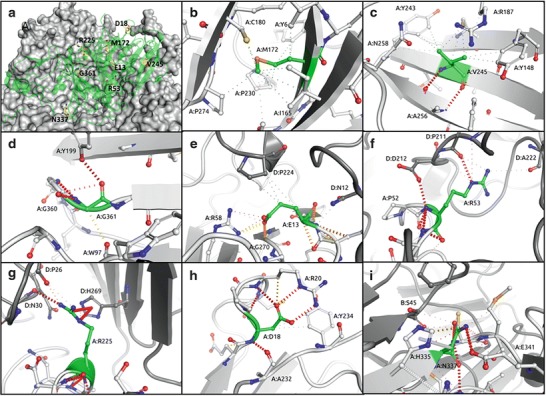
Analysis of wild-type interactions made by the novel AKU mutations. The novel mutations (yellow) are located throughout the protomer structure (green), with the other protomers shown as a grey surface (a). Mutation of M172 (b), V245 (c) and G361 (d) is expected to decrease the stability of the protomer through disruption of the intramolecular hydrophobic (grey dashes) and hydrogen bonds (red dashes). Mutation of E13 (e), R53 (f) or R225 (g) is expected to disrupt intermolecular interactions and decrease the stability of the hexamer. Mutation of D18 (h) or N337 (i) is predicted by mCSM to destabilise the structure of both the protomer and hexamer. The sites of the novel mutations are shown in green, while partner protomer chains are shown in dark grey
Table 3.
The analysis of the novel UK HGD gene mutations
| Patient ID | DNA | Protein | PolyPhen2 | SIFT | PROVEAN | DUET (ΔΔG Kcal/mol) | mCSM-PPI (ΔΔG Kcal/mol) | Proposed effect |
|---|---|---|---|---|---|---|---|---|
| 033 | c.37G>A | E13K | 0.842 possibly damaging | 0.01 affect protein function | −2.959 deleterious | 0.7 | −1.562 | Decreased protomer–protomer affinity |
| 030 | c.52G>A | D18N | 1.00 probably damaging | 0.01 affect protein function | −2.758 deleterious | −1.55 | −1.004 | Decreased protomer stability and protomer–protomer affinity |
| 034 | c.158G>A | R53Q | 1.00 probably damaging | 0.0 affect protein function | −3.724 deleterious | 0.245 | −3.348 | Decreased protomer–protomer affinity |
| 020 | c.515T>C | M172T | 0.446 benign | 0.0 affect protein function | −4.492 deleterious | −2.569 | −0.309 | Decreased protomer stability |
| 036 | c.674G>C | R225P | 0.993 probably damaging | 0.004 affect protein function | −6.229 deleterious | 0.289 | −2.216 | Decreased protomer–protomer affinity |
| 032 | c.664_674dupGCCAATCCTGC | D226PfsX7 | – | – | – | |||
| 033 | c.733G>T | V245F | 0.900 possibly damaging | 0.004 affect protein function | −3.741 deleterious | −1.797 | −0.431 | Decreased protomer stability |
| 039 | c.1009A>G | N337D | 1.00 probably damaging | 0.0 affect protein function | −4.891 deleterious | −1.205 | −1.772 | Decreased protomer stability and protomer–protomer affinity |
| 016 | c.1081G>A | G361R | 0.995 probably damaging | 0.0 affect protein function | −7.338 deleterious | −0.448 | −2.173 | Decreased protomer stability |
Mutations that affect the production of active HGD enzyme through destabilisation of the protomer structure typically perturb the local secondary structure through the introduction of energetically unfavourable mutations, disrupting the wild-type interactions. For example, M172 and V245 both make strong intramolecular hydrophobic interactions. While M172T results in the loss of these interactions, V245F disrupts the local secondary structure by introducing a steric clash. G361 is a buried residue that adopts a positive phi angle, which would usually be energetically unfavourable for non-glycine amino acids. Furthermore, mutation to arginine would introduce a steric clash, destabilising the protomer.
Mutations that disrupt the network of intermolecular interactions typically lower the stability of the 3–2 symmetrical homohexameric structure. Disruption of the intricate hexameric structure of HGD is also expected to affect enzyme activity. E13 is located along a protomer interaction interface, making intra- and intermolecular interactions. Mutation to lysine reverses the local charge and is predicted to destabilise the hexamer. R53 makes intra- and intermolecular polar interactions and side-chain to main-chain hydrogen bonds. Mutation to glutamine results in the loss of the intermolecular interactions, consistent with the prediction that it would destabilise the hexamer. R225 is also located on an interface between protomers, where it makes polar hydrogen bonds and nitrogen-pi and carbon-pi intermolecular interactions. In addition to altering the backbone geometries, mutation to proline will reduce the affinity of this interaction and is predicted to destabilise the hexamer.
A few mutations were predicted by mCSM-PPI and DUET to be highly disruptive to both the protomer (predicted to reduce the free energy of folding by a factor greater than 1.2 kcal/mol) and hexamer structures (predicted to reduce the free energy of association by a factor greater than 1 kcal/mol), highlighting the essential nature of the wild-type residues. D18 is a buried residue located near the interaction interface for a loop from a companion protomer that fits into a deep groove. D18 makes a network of strong intramolecular hydrogen bonds. Mutation to asparagine disrupts these interactions and is predicted to destabilise the protomer and disrupt hexamer formation. N337 forms intermolecular polar and carbon-pi interactions at the interface between two protomers. Mutation to aspartic acid will introduce a repulsive intramolecular force which is predicted to destabilise both the protomer and hexamer.
The splice site mutations can give rise either to exon skipping or to the activation of alternative splice sites; frameshift mutations either cause truncation or extension of the protein (Table 4).
Table 4.
A summary of the analysis of the splice site, frameshift and no-stop mutations
| HGD exon | Short name | Nucleotide change | Protein change | CrypSkip prediction | BDGP splice sites prediction | Net2Gene splice sites prediction | Mutalyzer protein effect prediction | Effect |
|---|---|---|---|---|---|---|---|---|
| 01i | IVS1-1G>A | c.16-1G>A | p.(Tyr6_Gln29del) | 0.16 exon skipping strongly favoured | 1 alternative acceptor site | No acceptable acceptor sites above threshold | ||
| 3 | S59fs | c.175delA | p.Ser59Alafsx52 | – | – | – | Severely truncated protein 52 novel amino acids |
Frameshift |
| 5 | IVS5+1G>T | c.342+1G>T | Uncertain (p.Leu95_Ser114del)* | 0.42 cryptic splice site activation | 2 alternative donor sites | 2 alternative donor sites | No change in protein (if exon is skipped deletion of 20 amino acids) | Aberrant splicing |
| 7 | IVS7+2T>C | c.469+2T>C | p.Arg154SerfsX22 | 0.01 exon skipping strongly favoured | No alternative donor splice sites | No alternative donor splice sites | No change in protein | Exon skipping aberrant splicing |
| 8 | IVS8-1G>T (V157fs) | c.470-1_494del25 | p.Val157Glufsx11 or if exon skipped p.Pro 158Thr fsx25 | 0.12 exon skipping strongly favoured | Acceptor splice site is abolished by the deletion | One alternative acceptor site | If skipped, a severely truncated protein with novel 10 residues + stop or a truncated protein with novel 24 residues + stop | Exon skip and frameshift |
| 10 | D226PfsX7 | c.664_674dupGCCAATCCTCG | p.(Asp226Profs*7) | – | – | – | Severely truncated protein 7 novel amino acids |
Frameshift |
| 14 | K431fs | c.1282-1292del11 | p.Lys431Hisfsx52 | – | – | – | Truncated protein with novel 10 residues + stop | Deletion causing frameshift |
| 14 | X446ext | c.1336G>C | p.X446ArgxtX24 | – | – | – | Extended protein with additional 24 residues + stop | Extension |
DNA variant numbering is based on cDNA (NM_000187.3), +1 corresponding to the A of the ATG. Protein effect numbering is based on NP_000178.2
The effects of the splice site and frameshift mutations were expected to cause large-scale alterations to the protein structure. It is likely that these changes would either result in nonsense-mediated decay and in a null allele or introduce unfavourable steric contacts and disrupt the assembly of the subunits which is critical to the formation of the active site of the enzyme.
Conclusion
The HGD gene analysis of the NAC patients found mutations in 12 of the 14 exons; none were observed in exon 1 or 4. The mutations were not evenly distributed; the largest numbers of variants were found in exons 3, 6, 8, 10 and 13 (Supplementary Table 2). Exon 13 carries the residues that bind HGA in the active site, and there were ten mutated alleles identified in this exon in the UK cohort.
The mutations most commonly found in this study were E42A, R53Q, S59fs, C120F, R225P, V300G and M368V. Five of these can be found in the HGD mutation database; the notable exceptions were R53Q and R225P which were both novel mutations. R53Q was found in three patients, two of these were siblings and they were homozygotes. The R225P mutation was seen in two patients who were siblings, and these were also homozygotes.
Nine single-nucleotide polymorphisms in the HGD gene were identified including the H80Q polymorphism in exon 4. The identification of the novel mutations and studying the distribution of AKU mutations in the United Kingdom demonstrate the genetic heterogeneity of this rare disease. The analysis of AKU mutations using mCSM and DUET provides further insight into their effects on the structure and function of the defective enzyme and provides a useful source of data for making genotype–phenotype correlations.
Electronic Supplementary Material
Supplementary Table 1. The single-nucleotide polymorphisms (SNPs) found in the NAC AKU patients
Supplementary Table 2. The exon distribution and frequency of HGD variants in the 42 NAC patients
Acknowledgements
DBA is supported by a CJ Martin Fellowship from the National Health and Medical Research Council (NHMRC; GNT1072476). DEVP is funded by the Brazilian agency Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). TLB receives funding from the University of Cambridge and the Wellcome Trust.
Compliance with Ethics Guidelines
Jeannette Usher declares that she has no conflict of interest.
Anna Milan declares that she has no conflict of interest.
Lakshminarayan Ranganath declares that he has no conflict of interest.
David Ascher declares that he has no conflict of interest.
Douglas Pires declares that he has no conflict of interest.
Tom Blundell declares that he has no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
This article does not contain studies with animal subjects performed by any of the authors.
Details of the Contributions of Individual Authors
JU performed the sequence analysis of the HGD gene mutations, the analysis of the mutations with the freely available bioinformatics programs and the initial writing of the manuscript.
DA and DP performed the computation modelling by MCSM and DUET and the interpretation of the results.
AMM and LRR assisted with data interpretation and writing of the manuscript.
TB reviewed the result interpretation.
All authors contributed to reviewing of the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Jeannette L. Usher, Email: jeanette.usher@rlbuht.nhs.uk
Collaborators: Johannes Zschocke
References
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35(11):3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220(1):49–65. doi: 10.1016/0022-2836(91)90380-O. [DOI] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effects of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison AS, Milan AM, Hughes AT, Dutton JJ, Ranganath LR (2014) Serum concentrations and urinary excretion of homogentisic acid and tyrosine in normal subjects. Clin Chem Lab Med. doi:10.1515/cclm-2014-0668 [DOI] [PubMed]
- Divina P, Kvitkovicova A, Buratti E, Vorechovsky I. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur J Hum Genet. 2009;17:759–765. doi: 10.1038/ejhg.2008.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:19–24. doi: 10.1038/ng0996-19. [DOI] [PubMed] [Google Scholar]
- Garrod AE. The Croonian lectures on inborn errors of metabolism. Lecture II. Alkaptonuria. Lancet. 1908;2:73–79. [Google Scholar]
- Helliwell TR, Gallagher JA, Ranganath L. Alkaptonuria - a review of surgical and autopsy pathology. Histopathology. 2008;53(5):503–512. doi: 10.1111/j.1365-2559.2008.03000.x. [DOI] [PubMed] [Google Scholar]
- Hughes AT, Milan AM, Christensen P, et al. Urine homogentisic acid and tyrosine: simultaneous analysis by liquid chromatography tandem mass spectrometry. J Chromatogr B. 2014;963:106–112. doi: 10.1016/j.jchromb.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Keller JM, Macaulay W, Ohannes A, et al. New developments in ochronosis: review of the literature. Rheumatol Int. 2005;25(2):81–85. doi: 10.1007/s00296-004-0498-1. [DOI] [PubMed] [Google Scholar]
- Kralovicova J, Vorechovsky I. Global control of aberrant splice-site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition. Nucleic Acids Res. 2007;5(19):6399–6413. doi: 10.1093/nar/gkm680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Du BN, Zannoni VG, Laster L, et al. The nature of the defect in tyrosine metabolism in alkaptonuria. J Biol Chem. 1958;230:251–260. [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347:2111–2121. doi: 10.1056/NEJMoa021736. [DOI] [PubMed] [Google Scholar]
- Pires DE, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30:335–342. doi: 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires DE, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42(W1):W314–W319. doi: 10.1093/nar/gku411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak MR, Chou YH, Cerda JJ, et al. Homozygosity mapping of the gene for alkaptonuria to chromosome 3q2. Nat Genet. 1993;5:201–204. doi: 10.1038/ng1093-201. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganath LR, Jarvis JC, Gallagher JA. Recent advances in management of alkaptonuria. J Clin Pathol. 2013;66:367–373. doi: 10.1136/jclinpath-2012-200877. [DOI] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comp Biol. 1997;4(3):311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Srsen S, Muller CR, Fregin A, et al. Alkaptonuria in Slovakia: thirty-two years of research on phenotype and genotype. Mol Genet Metab. 2002;75(4):353–359. doi: 10.1016/S1096-7192(02)00002-1. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Preston AJ, Paulk NK, et al. Ochronosis in a murine model of alkaptonuria is synonymous to that in the human condition. Osteoarthritis Cartilage. 2012;20(8):880–886. doi: 10.1016/j.joca.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus GP, Mueller CR, Burgner J, et al. Crystal structure of human homogentisate dioxygenase. Nat Struct Biol. 2000;7:542–546. doi: 10.1038/76756. [DOI] [PubMed] [Google Scholar]
- Vilboux T, Kayser M, Introne W, et al. Mutation spectrum of homogentisic acid oxidase (HGD)in alkaptonuria. Hum Mutat. 2009;30(12):1611–1619. doi: 10.1002/humu.21120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the MUTALYZER sequence variation nomenclature checker. Hum Mutat. 2008;29(1):6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
- Worth CL, Preissner R, Blundell TL. SDM - a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222. doi: 10.1093/nar/gkr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatkova A, Sedlackova T, Radvansky J, et al. Identification of 11 novel homogentisate 1,2 dioxygenase variants in alkaptonuria patients and establishment of a novel LOVD-based HGD mutation database. JIMD Rep. 2011;4:455–465. doi: 10.1007/8904_2011_68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. The single-nucleotide polymorphisms (SNPs) found in the NAC AKU patients
Supplementary Table 2. The exon distribution and frequency of HGD variants in the 42 NAC patients