

Abstract
Objective: We report a novel presentation of childhood cerebral X-linked adrenoleukodystrophy: status epilepticus followed by abrupt and catastrophic neurologic deterioration.
Methods: A description of the clinical presentation, laboratory evaluation, and imaging findings leading to a diagnosis of X-linked adrenoleukodystrophy.
Results: A 3-year-old male with prior history of autism presented with fever, diarrhea, and status epilepticus requiring a pentobarbital coma. Admission labs were notable only for a glucose level of 22 mg/dL, which stabilized after correction. The child never returned to his prior neurologic baseline, with complete loss of gross motor, fine motor, and speech skills. Serial brain magnetic resonance imaging (MRI)/magnetic resonance spectroscopy (MRS) was notable for progressive diffuse cortical signal changes with swelling, diffusion restriction, and ultimately laminar necrosis. Nine months after presentation, CSF (cerebrospinal fluid) protein and MRS lactate were persistently elevated, concerning for a neurodegenerative disorder. This led to testing for mitochondrial disease, followed by lysosomal and peroxisomal disorders. Very long-chain fatty acids were elevated. Identification of a pathogenic ABCD1 mutation confirmed the diagnosis of X-linked adrenoleukodystrophy.
Conclusions: Boys with childhood cerebral X-linked adrenoleukodystrophy typically present with gradual behavioral changes. Rare reports of boys presenting with transient altered mental status or status epilepticus describe a recovery to their pre-presentation baseline. To our knowledge this is the first X-ALD patient to present with status epilepticus with abrupt and catastrophic loss of neurologic function. X-linked adrenoleukodystrophy should be suspected in young males presenting with seizures, acute decline in neurologic function, with persistently elevated CSF protein and MRS lactate.
Introduction
X-linked adrenoleukodystrophy (X-ALD, OMIM 300100) is a disorder of impaired transmembrane transport of very long-chain fatty acids (VLCFA) into the peroxisome. As a result, VLCFAs accumulate in tissues of the body. ALD presents as childhood cerebral ALD, adrenomyeloneuropathy, or Addison’s disease. In ALD, patients usually present with several years of progressive cognitive decline and behavioral problems. Findings on neuroimaging usually include MRI FLAIR hyperintensities in the parietal-occipital regions (Engelen et al. 2012). Approximately 9% of patients with childhood cerebral ALD present acutely, usually at a younger age than ALD typically presents (around 5.5 versus 7.2 years old) (Stephenson et al. 2000). We report the case of a 3-year-old male, with a past medical history of autism, who presented with fever, vomiting, diarrhea, and status epilepticus. He had catastrophic loss of developmental milestones post-pentobarbital coma. After extensive testing and imaging, he was found to have X-ALD. This is the first report of a patient with childhood cerebral ALD presenting acutely with status epilepticus and no recovery of prior function.
Case Presentation
A 3-year-old Caucasian male presented to the emergency room with a first-time seizure. The child had been ill with fever, vomiting, and diarrhea for the preceding 12 h. His mother witnessed the onset of a generalized tonic-clonic seizure, which lasted 10 min. In the emergency room he had a seizure of bilateral extremity jerking with eye deviation to the left, which resolved with 0.1 mg/kg lorazepam intravenous. Labs at this time were notable for a glucose of 22 mg/dL, which was corrected with an intravenous dextrose bolus. A head CT was normal. Given the hypoglycemia, fever, and back-to-back seizures, the patient was admitted to the critical care unit for medical management. After admission he had a reassuring lumbar puncture (WBC 0 mm3, RBC 0 mm3, glucose 74 mg/dL, protein 21 mg/dL). Serum lactate and pyruvate were 2.3 mmol/L (normal range 1.00–2.40 mmol/L) and 0.11 mmol/L (normal range 0.06–0.17 mmol/L), respectively. His electrolytes, including sodium, and renal function were within normal limits after the initial correction of glucose.
His past medical history was notable for receptive and expressive speech delay. He was in speech therapy and had been diagnosed with autism. His speech had rapidly improved over the prior 6 months, and he was no longer displaying any repetitive behaviors. One week prior to this presentation, he had fallen on stairs and hit his head, with no loss of consciousness.
He was watched overnight in the critical care unit with continuous video electroencephalography (EEG) monitoring. During this time he was lethargic, but no seizures were observed. On the second hospital day, he had a generalized tonic-clonic seizure, after which his EEG showed the development of subclinical status epilepticus. His seizures stopped with initiation of a pentobarbital drip. He remained in burst suppression for 72 h, and then a pentobarbital infusion wean was attempted. He again had clinical and subclinical seizures, and pentobarbital was titrated to a burst suppression pattern for an additional 72 h. On hospital day 8 he was weaned off of pentobarbital and was seizure-free. At that time the patient was minimally responsive: his eyes would spontaneously open and rove, but he had no purposeful movements or speech.
A brain MRI/MRS with and without contrast obtained on hospital day 5 (FLAIR imaging shown in Fig. 1) was strikingly abnormal, with bilateral diffuse cortical swelling, hyperintensities, diffusion restriction, and subtle leptomeningeal enhancement. The MRS of the left frontal white matter showed a lactate peak. Though several of these findings can be seen as sequela of status epilepticus (Milligan et al. 2009), the overall MRI pattern was concerning for an underlying metabolic etiology. Extensive testing was performed with normal or nondiagnostic results: initial total and free carnitine were low (repeat testing after carnitine supplementation was normal); urine organic acids; repeat lumbar puncture on hospital day 8 (WBC 4 mm3, RBC 0 mm3, glucose 65 mg/dL, protein 46 mg/dL, lactate and pyruvate); neurotransmitter metabolites (pyridoxal 5-phosphate, 5-methyltetrahydrofolate, neopterin, tetrahydrobiopterin, 5-hydroxyindoleacetic acid, homovanillic acid, and 3-0-methyldopa); repeat serum lactate and pyruvate; viral polymerase chain reaction (PCR) testing (CSF, serum, and stool) including HSV, HIV, enterovirus, EBV, CMV, mycoplasma, human metapneumovirus, influenza, parainfluenza, RSV, rhinovirus, West Nile, California encephalitis, St. Louis encephalitis, Eastern equine encephalitis, Western equine encephalitis, JC virus, and rotavirus; serum and CSF paraneoplastic and anti-NMDA receptor antibody panels; serum and CSF amino acids; a third lumbar puncture on hospital day 12 which was notable for high protein (WBC 2 mm3, RBC 5,550 mm3, glucose 58 mg/dL, protein 96 mg/dL); inflammatory markers (C-reactive protein and sedimentation rate); and thyroid studies (thyroid-stimulating hormone, free thyroxine, and anti-thyroid autoantibodies). To evaluate his initial hypoglycemia, he was also evaluated by endocrinology with a reassuring insulin level of 0.8 mcIU/mL (normal fasting level is < 13 mcIU/mL) and reassuring morning cortisol level of 10.9 mcg/dL (normal for his age is 3–21 mcg/dL).
Fig. 1.
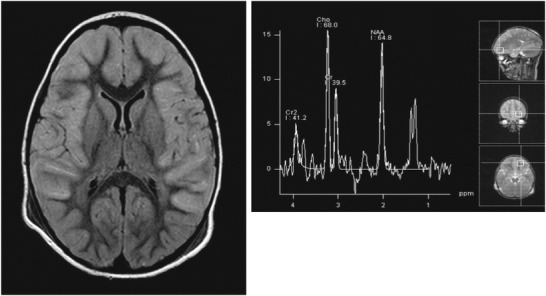
Brain MRI and MRS 5 days after initial presentation, showing diffuse cortical signal changes, edema, and a lactate peak
Repeat brain MRI with and without contrast 1 month later (flair imaging shown in Fig. 2) showed progressive and diffuse white matter changes, along with laminar necrosis. Findings were also notable for significant atrophy and contrast enhancement throughout the frontal, parietal, temporal, and left occipital regions. There was a persistent lactate peak on MRS of the frontal white matter in the absence of diffusion restriction.
Fig. 2.
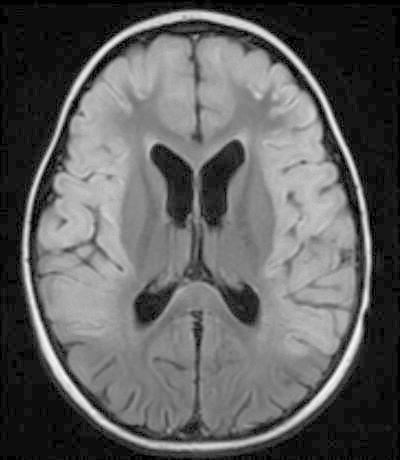
Brain MRI 30 days after onset showing progression of cortical changes with laminar necrosis
The following genetic testing was performed and normal: microarray; fragile X; whole mtDNA sequencing, duplication, and deletion; sequencing of POLG1, TYMP, SCN1A, SCN1B, and GABARG2; and TPP1 and PPT1 enzyme activity. Genetic testing for a primary carnitine deficiency syndrome as well as multiple fatty acid oxidation defects revealed a single variant of unknown significance in acyl-CoA dehydrogenase, C-4 to C-12 straight chain (ACADM), felt to be noncontributory. A third brain MRI/MRS with and without contrast was performed on hospital day 60 and was similar to the prior studies, demonstrating laminar necrosis, extensive T2 signal abnormality of white matter diffusely including corticospinal tracts, contrast enhancement, and a lactate peak. Throughout the hospital stay, the patient was seizure-free on a regimen of phenobarbital and lacosamide, but unfortunately his development and level of consciousness did not improve. The patient was discharged home with extensive support.
At 4 years of age, 9 months after presentation, the patient had a repeat lumbar puncture and brain MRI/MRS with and without contrast (flair imaging shown in Fig. 3). The CSF was notable for continued elevation of the protein (WBC 2 mm3, RBC 1 mm3, glucose 69 mg/dL, protein 70 mg/dL). The brain MRI/MRS showed progression of cerebral volume loss, signal abnormalities of both white matter and gray matter with extension of abnormalities into the bilateral cerebellar hemispheres, and the development of bilateral subdural fluid collections. Based on the MRI findings of a progressive chronic neurodegenerative disorder and continued protein elevation in the CSF, which though nonspecific is an indicator of a potential neurodegenerative disorder (commonly seen in Krabbe and metachromatic leukodystrophy), the patient had further testing including copper and ceruloplasmin levels (normal), leukocyte lysosomal enzyme-screening assay (normal), and VLCFAs to rule out peroxisomal disorders.
Fig. 3.
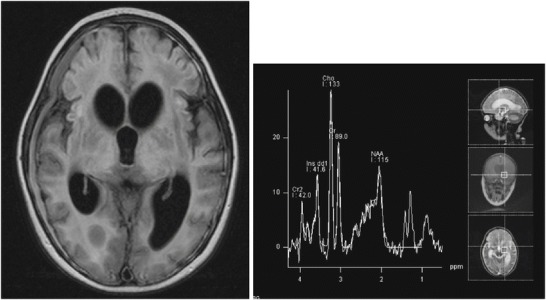
Brain MRI and MRS 9 months after onset showing extensive cortical signal changes, atrophy, and laminar necrosis, along with a persistent lactate peak on MRS
VLCFA profile was notable for elevation of C26:0 at 1.17 ug/mL (normal 0.23 ± 0.09), elevation of C26:1 at 0.900 ug/mL (normal 0.18 ± 0.09), elevation of C24/C22 at 1.793 ug/mL (normal 0.84 ± 0.10), and elevation of C26/C22 at 0.109 ug/mL (normal 0.01 ± 0.004). Analysis of the ABCD1 (ATP-binding cassette, subfamily D) gene revealed a novel deletion (c.136*>-AGCGAGCGCACAGA; p.X46X.), consistent with a diagnosis of X-linked adrenoleukodystrophy. This frameshift deletion resulted in a premature stop codon. Further testing of the patient with an adrenocorticotropin (ACTH) stimulation test showed that he had adrenal insufficiency (baseline cortisol level was 9.3 mcg/dL, 30 min after stimulation cortisol level was 8.6 mcg/dL, and 60 min after stimulation cortisol level was 9.7 mcg/dL; both renin and aldosterone levels were normal). Given the advanced stage of the patient’s disease, he was not a candidate for bone marrow or stem cell transplant, and management transitioned to palliative care. The patient’s mother was found to be a carrier of the same deletion. Due to insurance concerns the patient’s maternal grandmother was not tested for the mutation, but there were three maternal uncles who were potentially at risk for X-ALD, each of whom had a normal VLCFA profile, ruling out the diagnosis.
Discussion
Childhood cerebral adrenoleukodystrophy classically presents with neuropsychiatric symptoms, including attention-deficit/hyperactivity disorder, followed by slow behavioral decline and developmental regression. Seizures are known to affect patients with X-ALD, typically occurring 2–4 years after onset of other neurological symptoms (Wang et al. 2001). In a retrospective chart review of 485 boys, Stephenson et al. (2000) noted that 9% (n = 45) had an acute presentation of adrenal crisis, seizures, or encephalopathy.
Seizures occurred in approximately 44% (n = 20) of the children who presented acutely (Stephenson et al. 2000). Of the patients presenting with seizures, four presented in status epilepticus, six presented with focal seizures, and ten presented with generalized tonic-clonic seizures (Stephenson et al. 2000). In Stephenson’s study three of the patients had seizures within hours of sustaining a head injury, while our patient had a head injury 1 week prior to presentation. Many of the patients who presented with seizures did not have neuroimaging initially, and the diagnosis of X-ALD was not suspected. All of the patients who presented with seizures, including status epilepticus, had full recovery to prior baseline (Stephenson et al. 2000).
Adrenal crisis occurred in around 44% (n = 20) of the children with an acute presentation (Stephenson et al. 2000). Signs and symptoms included vomiting, lethargy, hyponatremia, hypoglycemia, and dehydration. Additionally, half of these patients had fever on presentation, and many were diagnosed with encephalopathy. Several of these boys had been hospitalized multiple times for dehydration prior to the diagnosis of X-ALD. Ravid et al. (2000) describe one child who presented with fever, vomiting, and subsequent coma found to have adrenal insufficiency (low baseline cortisol level, confirmed with inadequate response of cortisol levels to ACTH stimulation). This patient had minimal improvement in mental status after diagnosis and treatment with steroids (Ravid et al. 2000). Zammarchi et al. (1994) describe another child who presented with symptoms initially thought to be consistent with acute encephalitis (fever, vomiting, and altered mental status), but found on laboratory evaluation to have hyponatremia and adrenal insufficiency (inadequate response to ACTH stimulation). This child’s mental status rapidly returned to baseline after steroids were started (Zammarchi et al. 1994). Our patient’s initial lack of hyponatremia and normal cortisol level argued against adrenal insufficiency.
A minority of patients (n = 5 or 11%) in Stephenson’s review had an acute presentation of encephalopathy or coma with no seizures or adrenal dysfunction (Stephenson et al. 2000). Follow-up of these patients was limited. At least one child gradually returned to his neurologic baseline before decline and diagnosis of X-ALD.
Stephenson et al. (2000) provide a cohesive review of the common acute presentations of X-ALD. Of the 20 patients who presented with seizures including status epilepticus, all recovered to their prior level of function (Stephenson et al. 2000). Though there are many reports of children presenting with adrenal insufficiency and full recovery, there is one case report of a child presenting with adrenal insufficiency and no return to prior baseline (Zammarchi et al. 1994; Ravid et al. 2000; Stephenson et al. 2000). Several of the patients with acute presentations had the pathognomonic posterior white matter changes on neuroimaging. However, other patients had head CTs that were interpreted as normal at initial presentation. This highlights the difficulty of making an early diagnosis of ALD.
Our patient’s initial imaging findings were not pathognomonic for X-ALD. Status epilepticus can lead to MRI changes such as diffusion restriction in the hippocampus, gyri, and splenium (Milligan et al. 2009). This diffusion restriction may be secondary to hypoxemia or vasogenic and cytotoxic edema. These findings would be expected to improve on repeat imaging after the seizures are controlled (Milligan et al. 2009). Laminar necrosis has been reported in pediatric patients in the setting of status epilepticus with persistent hypoglycemia (Christiaens et al. 2003; Huang et al. 2009). Although our patient was initially hypoglycemic, subsequent serum and CSF glucoses were normal. The progressive nature of our patient’s imaging findings, along with the persistence of a lactate peak on MRS, was more consistent with a neurodegenerative disease than with the sequela of status epilepticus.
X-ALD rarely presents acutely. To our knowledge, our patient is the first with an acute status epilepticus presentation and no recovery to prior baseline. The previously reported patients with acute presentations of seizures and coma all returned, some gradually and others quickly, to their baseline level of function. Our patient, on the other hand, did not. He remained neurologically devastated, which correlated with the diffuse and extensive brain injury seen on serial MRIs. X-ALD is a devastating disorder and can be difficult to diagnose. Early diagnosis is vital for both the patient and family members who may be affected yet still asymptomatic.
Conclusion
Boys with childhood cerebral adrenoleukodystrophy typically present with gradual onset of learning and behavioral changes; however, 9% of boys with X-ALD have an acute presentation of altered mental status, adrenal insufficiency, or seizures (Stephenson et al. 2000). With the exception of one boy with adrenal insufficiency, prior reports describe a full recovery to the prior level of function (Ravid et al. 2000). To our knowledge this is the first patient to present with status epilepticus followed by an abrupt, catastrophic, and irreversible loss of neurologic function. X-linked adrenoleukodystrophy should be considered in young males presenting with seizures and acute neurologic deterioration, especially if there is supporting data, such as persistently elevated CSF protein or lactate peaks on serial MRS.
Synopsis
Practitioners should consider the diagnosis of X-linked adrenoleukodystrophy in males presenting with status epilepticus followed by neurologic deterioration, especially if the patient has elevated CSF protein, MRI white matter changes, or a lactate peak on MRS.
Compliance with Ethics Guidelines
Conflict of Interest
Marissa Vawter-Lee, Barbara Hallinan, T. Andrew Burrow, Christine Spaeth, and Todd Arthur declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the guardian of the patient included in this study. Proof that informed consent has been obtained is available upon request.
Author Contributorship Statement
Marissa M Vawter-Lee: conception and design, data acquisition, analysis, interpretation of data, writing of the manuscript, final approval of article
Barbara E Hallinan: conception and design, interpretation of data, writing of the manuscript, final approval of article
T. Andrew Burrows: interpretation of data and writing of the manuscript
Christine G Spaeth: interpretation of data and writing of the manuscript
Todd M Arthur: conception and design, interpretation of data, writing of the manuscript, final approval of article
Footnotes
Competing interests: None declared
Contributor Information
M. M. Vawter-Lee, Email: Marissa.Vawter@cchmc.org
Collaborators: Johannes Zschocke
References
- Christiaens FJ, Mewasingh LD, Christophe C, Goldman S, Dan B. Unilateral cortical necrosis following status epilepticus with hypoglycemia. Brain Dev. 2003;25:107–112. doi: 10.1016/S0387-7604(02)00164-X. [DOI] [PubMed] [Google Scholar]
- Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51. doi: 10.1186/1750-1172-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YC, Weng HH, Tsai YT, et al. Periictal magnetic resonance imaging in status epilepticus. Epilepsy Res. 2009;86:72–81. doi: 10.1016/j.eplepsyres.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Milligan TA, Zamani A, Bromfield E. Frequency and patterns of MRI abnormalities due to status epilepticus. Seizure. 2009;18:104–108. doi: 10.1016/j.seizure.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Ravid S, Diamond AS, Eviatar L. Coma as an acute presentation of adrenoleukodystrophy. Pediatr Neurol. 2000;22:237–239. doi: 10.1016/S0887-8994(99)00141-1. [DOI] [PubMed] [Google Scholar]
- Stephenson DJ, Bezman L, Raymond GV. Acute presentation of childhood adrenoleukodystrophy. Neuropediatrics. 2000;31:293–297. doi: 10.1055/s-2000-12952. [DOI] [PubMed] [Google Scholar]
- Wang PJ, Hwu WL, Shen YZ. Epileptic seizures and electroencephalographic evolution in genetic leukodystrophies. J Clin Neurophysiol. 2001;18:25–32. doi: 10.1097/00004691-200101000-00006. [DOI] [PubMed] [Google Scholar]
- Zammarchi E, Donati MA, Tucci F, Fonda C, Fanelli F, Pazzaglia R. Acute onset of X-linked adrenoleukodystrophy mimicking encephalitis. Brain Dev. 1994;16:238–240. doi: 10.1016/0387-7604(94)90077-9. [DOI] [PubMed] [Google Scholar]