

Abstract
Alkaptonuria (AKU) is an ultrarare autosomal recessive disorder resulting from a deficiency of homogentisate 1,2 dioxygenase (HGD), an enzyme involved in the catabolism of phenylalanine and tyrosine. Loss of HGD function prevents metabolism of homogentisic acid (HGA), leading to increased levels of plasma HGA and urinary excretion. Excess HGA becomes deposited in collagenous tissues and subsequently undergoes polymerisation, principally in the cartilages of loaded joints, in a process known as ochronosis. This results in an early-onset, devastating osteoarthropathy for which there is currently no effective treatment. We recently described the natural history of ochronosis in a murine model of AKU, demonstrating that deposition of ochronotic pigment begins very early in life and accumulates with age. Using this model, we were able to show that lifetime treatment with nitisinone, a potential therapy for AKU, was able to completely prevent deposition of ochronotic pigment. However, although nitisinone has been shown to inhibit ochronotic deposition, whether it can also facilitate removal of existing pigment has not yet been examined. We describe here that midlife administration of nitisinone to AKU mice arrests further deposition of ochronotic pigment in the tibiofemoral joint, but does not result in the clearance of existing pigment. We also demonstrate the dose-dependent response of plasma HGA to nitisinone, highlighting its efficacy for personalised medicine, where dosage can be tailored to the individual AKU patient.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2015_437) contains supplementary material, which is available to authorized users.
Introduction
Alkaptonuria (AKU) has a unique place in the history of metabolic disease as the first disorder to be described as an “inborn error of metabolism” by distinguished English physician Sir Archibald Garrod (Garrod 1902). AKU is an ultrarare autosomal recessive disorder with a worldwide incidence of between 1 in 250,000 and 1,000,000 live births (Phornphutkul et al. 2002). AKU results from mutations in homogentisate 1,2-dioxygenase (HGD) (EC 1.13.11.5), the enzyme involved in the catabolism of phenylalanine and tyrosine (La Du et al. 1958) (Fig. 1a). Loss of HGD function results in both increased plasma levels of homogentisic acid (HGA) and HGA excretion. Urinary HGA darkens on exposure to air and is typically observed as the first symptom in patients who present with AKU. Elevated levels of plasma HGA ultimately lead to the pigmentation of cartilaginous tissues, following the deposition and subsequent polymerisation of HGA in a process known as ochronosis. This results in an early-onset, devastating osteoarthropathy for which there is currently no effective treatment.
Fig. 1.

(a) Diagram of the phenylalanine/tyrosine metabolic pathway showing the enzyme deficiency which results in AKU. The site of action of nitisinone is also highlighted. (b) The effect of midlife (34 weeks) dietary supplementation with 4 mg/L nitisinone on the plasma HGA concentration in AKU mice
Nitisinone (2-(2-nitro-4-(trifluoromethyl)benzoyl) cyclohexane-1,3-dione) is a reversible inhibitor of 4-hydroxyphenylpyruvate dioxygenase (HPPD) (EC 1.13.11.27), the enzyme responsible for producing HGA (Fig. 1a). Originally developed as a herbicide (Schulz et al. 1993), it is routinely used for the treatment of hereditary tyrosinaemia type 1 (McKiernan 2006). Nitisinone is viewed as a potential treatment for AKU as it prevents accumulation of HGA in plasma. Ochronosis has recently been described in two murine models of AKU (Taylor et al. 2012; Preston et al. 2014). In the latter of the two models, we have described the efficacy of nitisinone in treating ochronosis in a murine model of AKU and demonstrated that lifetime administration of nitisinone reduced plasma HGA by 88% and prevented ochronotic pigment deposition in the tibiofemoral joint (Preston et al. 2014). This was the first time that inhibition of ochronosis by nitisinone had been demonstrated and highlighted the efficacy of nitisinone as a treatment for AKU.
As a large proportion of AKU patients already suffer from osteoarthropathy, it is important to determine if nitisinone’s efficacy is purely prophylactic or whether it can facilitate repair and regeneration of damaged cartilage during natural metabolic turnover. Although pigmentation in AKU mice can be prevented throughout their lifetime by administration of nitisinone, there is no data on whether ochronosis is reversible. Here we describe that the midlife administration of nitisinone to AKU mice successfully arrests further deposition of ochronotic pigment in the tibiofemoral joint, but does not result in the reduction of existing pigment. We also demonstrate that the response of plasma HGA to nitisinone treatment is dose dependent, which should facilitate tailored treatment of AKU patients presenting with differing degrees of severity.
Materials and Methods
Mice
Hgd−/− (AKU) mice on a BALB/c or C57BL/6 background were used for all experiments. All mice were housed and maintained within the University of Liverpool’s Biological Services Unit (BSU) in accordance with Home Office UK guidelines.
Sample Preparation
Tail bleed samples were collected into Microvettes (Sarstedt, CB 300) and stored at 4°C prior to processing within 2 h, using an adaptation of the Bory method (Bory et al. 1990). Briefly, whole blood was centrifuged at 1500×g for 10 min at 4°C and the plasma deproteinised by adding 5.8 M perchloric acid (Sigma, UK) equivalent to 10% of the plasma and containing 0.1 mM 4-amino-2-chlorobenzoic acid (Sigma, UK) as the internal standard. Acidified supernatant was stored at −20°C. A 150 μL tail bleed volume yielded approximately 25 μL of deproteinised plasma.
Chromatographic Conditions
Plasma HGA concentration was determined via HPLC as described previously (Preston et al. 2014), on a Phenomenex Kinetex XB-C18 column, 2.6 μM (4.6 × 100 mm). Briefly, the initial mobile phase was 100% buffer A (12 mM orthophosphoric acid, Sigma, UK), before increasing buffer B (100% methanol, Sigma, UK) from 0 to 80% over 10 min. Detection was by UV at 290 nm.
Midlife Nitisinone Treatment
A cohort of eight BALB/c Hgd−/− mice (four males, four females) were provided with filtered water from 8 to 34 weeks of age. They were then provided with an ad libitum supply of water containing 4 mg/L of nitisinone (Shanghai Elittes Organics, China) from 34 to 81 weeks of age. The control group of 8 BALB/c Hgd−/− mice (four males, four females) was untreated over the same time period. Plasma was taken at 35 weeks and then sampled regularly by tail bleed over the mouse’s lifetime. Tibiofemoral joints were taken for histological analysis at the end of study. Analysis of joint pigmentation was also performed at different ages, in either BALB/c Hgd−/− or C57BL/6 Hgd−/− mice to build up a disease progression timeline.
Nitisinone Dose–Response
Six cohorts of four age-matched C57BL/6 Hgd−/− mice (two males, two females) had their plasma sampled at 54 weeks and then were immediately treated with an ad libitum supply of water containing either 4, 1, 0.5, 0.25, 0.125 or 0 mg/L of nitisinone for 13 days. Plasma was sampled again seven and 19 days posttreatment, and its HGA concentration was determined by HPLC.
Histological Analysis
Mice were euthanised with Pentoject (sodium pentobarbitone 20% w/v) and their tibiofemoral joints harvested and stored in 10% phosphate-buffered formalin solution, pH 7.4, for a minimum of 24 h. Tissues were washed in phosphate-buffered saline before decalcification in 12% EDTA for seven days. Tibiofemoral joints were dissected free of excess muscle then paraffin embedded in the coronal plane to enable simultaneous evaluation of both the medial and lateral compartments of the joint, as recommended by the Osteoarthritis Research Society International (OARSI) histopathology initiative (Glasson et al. 2010). The first section that encompassed both the tibial plateau and femoral condyles was selected as representative of each mouse. Sections were mounted on glass slides, rehydrated and stained with H&E or Schmorl’s stain, previously shown to be a sensitive method for the detection of ochronotic pigment (Tinti et al. 2011). Sections were dehydrated through graded alcohols and mounted with DPX resin (VWR International, UK) for examination by light microscopy.
Quantification of Pigmented Chondrons
The first whole section that encompassed the entire tibiofemoral joint (MTP, MFC, LTP and LFC) was selected as representative of each mouse for quantification analysis. From these sections, the total number of pigmented chondrons present in the articular cartilage and entheses of the femoral condyles and articular cartilage of the tibial plateau was assessed. Figure 2e illustrates the area quantified in a section of each tibiofemoral joint. Quantitation of each section required counting at least 36 fields. All sections were counted by a second blinded observer, and the correlation between observers was greater than 0.93.
Fig. 2.
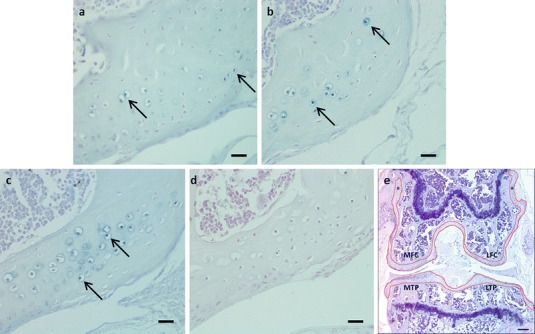
(a) Photomicrograph displaying pigmented chondrons (arrows) in a 35-week-old BALB/c Hgd−/− mouse, prior to treatment with nitisinone. (b) Administration of nitisinone (4 mg/L) at 35 weeks prevented large-scale pigmentation of chondrons in the tibiofemoral joint. The number of pigmented chondrons (arrows) observed in the nitisinone-treated BALB/c Hgd−/− mice at 79–81 weeks was comparable to those seen in untreated BALB/c Hgd−/− mice at 35 weeks (Figs. 1 and 2a). (c) Photomicrograph of an 80-week-old untreated BALB/c Hgd−/− mouse. Large numbers of pigmented chondrons (arrows) were present throughout the tibiofemoral joint, highlighting the effectiveness of nitisinone when given midlife (Fig. 2b). (d) Lifetime treatment with nitisinone (4 mg/L) prevented any deposition of ochronotic pigment in the tibiofemoral joint. (e) H&E photomicrograph displaying the a section of the entire tibiofemoral joint. The red lines on the image highlight the boundaries of the areas where pigmented chondrons where quantified (* = entheses). Images (a–d) were all taken from the lateral femoral condyle of BALB/c Hgd−/− mice. Bar = 20 μm (a–d), 50 μm (e)
Statistical Analyses
Comparisons in pigmentation rates between treated and untreated groups were performed using an independent sample t-test. Descriptive statistics are reported as mean and standard deviation (SD) as normality was achieved, while results were considered as statistically significant at the 5% level. Data analysis was undertaken using Stata 13 (StataCorp. 2013. Stata Statistical Software: Release 13. College Station, TX: StataCorp LP.).
Results
Midlife treatment with 4 mg/L nitisinone from 34 to 81 weeks of age suppressed plasma HGA concentration by approximately 15-fold, in agreement with previous work (Preston et al. 2014) (Fig. 1b). The reduction in plasma HGA following 44–46 weeks of nitisinone treatment translated into a statistically significant difference (t = 4.645, p = 0.001) in the mean number of pigmented chondrons visible in the nitisinone-treated mice at 79–81 weeks, equal to 88 (SD = 26.5) (Fig. 2b), relative to aged-matched untreated AKU mice, equal to 201.2 (SD = 48.4) (Fig. 3). The degree of pigmentation observed in nitisinone-treated BALB/c Hgd−/− mice at 79–81 weeks was considered equivalent to that observed in untreated 34-week-old BALB/c Hgd−/− mice (Fig. 2a), correspondent with the time at which treatment began. This demonstrated that midlife treatment with nitisinone arrested further deposition of ochronotic pigment but did not clear previously laid-down pigment, resulting in higher observable chondron pigmentation than AKU mice treated from birth (Fig. 2d). The number of pigmented chondrons observed in the untreated mice at 80 weeks (Fig. 2c) was consistent with levels previously reported in the natural history study of BALB/c Hgd−/− mice (Preston et al. 2014).
Fig. 3.

Quantification of pigmented chondrons in the tibiofemoral joint of AKU mice, depicting ochronotic pigment deposition over time and the effect of treatment with 4 mg/L nitisinone when administered midlife. Treatment at 34 weeks prevented further deposition of ochronotic pigment by week 79, but did not reverse the effects of previously laid-down pigment. Quantification of pigmented chondrons was performed on a single section from each mouse and does not represent the total cell number in each mouse
Quantification of pigmented chondrons over time (Fig. 3) confirmed that although the degree of deposition between AKU mice was highly variable (even within highly inbred mouse strains), progressive accumulation of ochronotic pigment was consistent with ageing. Nevertheless, midlife treatment with nitisinone effectively inhibited further deposition of ochronotic pigment.
Plasma HGA concentration was also highly variable in AKU mice, but was observed to respond in a dose-dependent fashion to nitisinone treatment, when plotted as a percentage of its pretreatment value (Fig. 4). Higher concentrations of nitisinone resulted in greater suppression of plasma HGA variability within cohorts, while removal of nitisinone after 13 days resulted in a rebound of plasma HGA levels.
Fig. 4.
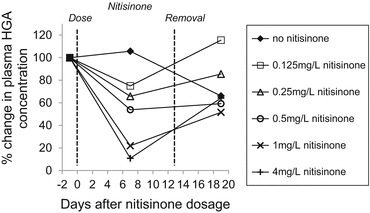
The dose–response to nitisinone (as percentage change of plasma HGA concentration) in AKU mice and recovery
Discussion
We have previously shown that nitisinone treatment from birth can prevent ochronosis in the adult AKU mouse (Preston et al. 2014). Here we demonstrate that beginning nitisinone treatment midway through life (34 weeks) is sufficient to arrest further disease progression. Remarkably, 44–46 weeks after treatment, the mean number of pigmented chondrons observed within the knee joint was no greater than that typical of untreated 34-week-old AKU mice, according to our disease progression timeline (Fig. 3). Unlike in mice treated with nitisinone from birth however (Fig. 2d), pigmentation could still be observed and quantified. It is evident therefore that while nitisinone can prevent further deposition of ochronotic pigment, it does not reduce pre-existing pigmentation by enabling turnover/replacement of damaged cartilage. This strongly implies that in order to minimise the irreparable joint damage typical of AKU disease progression in humans, treatment with nitisinone should begin as early as possible. Although there was no evidence that midlife treatment with nitisinone could facilitate removal of existing pigmentation, it did arrest any further deposition of ochronotic pigment which may lead to the prevention or slowing down of disease progression in patients with established ochronosis.
Establishing a minimum effective nitisinone dose is fundamental in reducing the cost of lifetime treatment and minimising potential side effects such as corneal keratopathy (Introne et al. 2011). As plasma HGA concentration in AKU patients is highly variable, we therefore examined dose–response sensitivity to nitisinone to determine the practicality of tailoring nitisinone treatment dose to the patient. A clear dose–response effect was observed between nitisinone and plasma HGA levels, which decreased consistently following increased doses of nitisinone. Treatment with 4 mg/L nitisinone reduced plasma HGA by 90% over a 13-day period when compared with baseline controls. Ranganath and colleagues recently showed a similar dose–response effect of nitisinone when analysing urinary HGA excretion over a 24 h period in AKU patients. Furthermore, they found that nitisinone was well tolerated within the studied dose range (Ranganath et al. 2014), which is consistent with our observations in mice. Both our data and that of Ranganath et al. highlight the efficacy of nitisinone in reducing the levels of circulating HGA. As excellent dose–response sensitivity was observed between differing nitisinone concentrations and plasma HGA levels, it may be possible to tailor individual treatment plans for AKU patients.
In summary, we have shown that nitisinone can effectively inhibit ochronotic deposition in an alkaptonuric mouse model and if introduced in midlife can arrest any further disease progression. Nitisinone treatment does not result in the removal of existing ochronotic pigmentation and cannot therefore be used to reverse existing joint damage. Plasma HGA concentrations display excellent sensitivity to treatment dose, facilitating the tailoring of therapy to patient disease severity.
Electronic Supplementary Material
Supplementary Fig. 1. (a) xyz scatter graph showing the location of pigmented chondrons across the tibial articular cartilage of a BALB/c Hgd−/− mouse knee, through 23 serial coronal sections. (b) xyz scatter graph showing the surface of the tibial articular cartilage from a BALB/c Hgd−/− mouse knee through 23 serial sections, with the most anterior and most posterior sections showing the area of entire tibial articular cartilage. Both graphs are 3D representations throughout the anterolateral plane
Synopsis
Nitisinone arrests further deposition of ochronotic pigment when administered midlife, it does not reduce existing pigmentation, and it reduces the plasma HGA levels in a dose-dependent manner.
Compliance with Ethics Guidelines
Conflicts of Interest
Craig M Keenan, Andrew J Preston, Hazel Sutherland, Peter J Wilson, Eftychia E Psarelli, Trevor F Cox, Lakshminarayan R Ranganath, Jonathan C Jarvis and James A Gallagher declare they have no conflict of interest.
Animal Rights
All institutional and national guidelines for the care and use of laboratory animals were followed.
Author Contributions
CMK and AJP contributed equally and were involved in data acquisition, analysis and reporting of the work.
HS and PJW were involved in data acquisition.
EEP and TFC were involved in data analysis.
LRR, JCJ and JAG were involved in the planning of the work.
JCJ and JAG contributed equally.
Footnotes
Competing interests: None declared
Craig M Keenan and Andrew J Preston contributed equally.
Jonathan C Jarvis and James A Gallagher contributed equally.
Contributor Information
James A Gallagher, Email: jag1@liverpool.ac.uk.
Collaborators: Johannes Zschocke
References
- Bory C, Boulieu R, Chantin C, Mathieu M. Diagnosis of alcaptonuria: rapid analysis of homogentisic acid by HPLC. Clin Chim Acta. 1990;189(1):7–11. doi: 10.1016/0009-8981(90)90228-K. [DOI] [PubMed] [Google Scholar]
- Garrod AE. The incidence of alkaptonuria: a study in chemical individuality. Lancet. 1902;ii:1616–1620. doi: 10.1016/S0140-6736(01)41972-6. [DOI] [Google Scholar]
- Glasson SS, Chambers MG, Van Den Berg WB, et al. The OARSI histopathology initiative – recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr Cartil. 2010;18(S3):S17–S23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307–314. doi: 10.1016/j.ymgme.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Du BN, Zannoni VG, Laster L, Seegmiller JE. The nature of the defect in tyrosine metabolism in alcaptonuria. J Biol Chem. 1958;230(1):251–260. [PubMed] [Google Scholar]
- McKiernan PJ. Nitisinone in the treatment of hereditary tyrosinaemia type 1. Drugs. 2006;66(6):743–750. doi: 10.2165/00003495-200666060-00002. [DOI] [PubMed] [Google Scholar]
- Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111–2121. doi: 10.1056/NEJMoa021736. [DOI] [PubMed] [Google Scholar]
- Preston AJ, Keenan CM, Sutherland H, et al. Ochronotic osteoarthropathy in a mouse model of alkaptonuria, and its inhibition by nitisinone. Ann Rheum Dis. 2014;73(1):284–289. doi: 10.1136/annrheumdis-2012-202878. [DOI] [PubMed] [Google Scholar]
- Ranganath LR, Milan AM, Hughes AT et al (2014) Suitability of nitisinone in alkaptonuria 1 (SONIA 1): an international, multicentre, randomised, open-label, no-treatment controlled, parallel-group, dose–response study to investigate the effect of once daily nitisinone on 24-h urinary homogentisic acid excretion in patients with alkaptonuria after 4 weeks of treatment. Ann Rheum Dis [DOI] [PubMed]
- Schulz A, Ort O, Beyer P, Kleinig H. SC-0051, a 2-benzoyl-cyclohexane-1,3-dione bleaching herbicide, is a potent inhibitor of the enzyme p-hydroxyphenylpyruvate dioxygenase. FEBS Lett. 1993;318(2):162–166. doi: 10.1016/0014-5793(93)80013-K. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Preston AJ, Paulk NK, et al. Ochronosis in a murine model of alkaptonuria is synonymous to that in the human condition. Osteoarthritis Cartilage. 2012;20(8):880–886. doi: 10.1016/j.joca.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinti L, Taylor AM, Santucci A, et al. Development of an in vitro model to investigate joint ochronosis in alkaptonuria. Rheumatology (Oxford) 2011;50(2):271–277. doi: 10.1093/rheumatology/keq246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. (a) xyz scatter graph showing the location of pigmented chondrons across the tibial articular cartilage of a BALB/c Hgd−/− mouse knee, through 23 serial coronal sections. (b) xyz scatter graph showing the surface of the tibial articular cartilage from a BALB/c Hgd−/− mouse knee through 23 serial sections, with the most anterior and most posterior sections showing the area of entire tibial articular cartilage. Both graphs are 3D representations throughout the anterolateral plane