

Abstract
Purine nucleoside phosphorylase (PNP) is an enzyme active in the purine salvage pathway. PNP deficiency caused by autosomal recessive mutations in the PNP gene leads to severe combined immunodeficiency (SCID) and in two thirds of cases also to neurological effects such as developmental delay, ataxia, and motor impairment.
PNP deficiency has a poor outcome, and the only curative treatment is allogenic hematopoietic stem cell transplantation (HSCT). We present the first Swedish patient with PNP deficiency with novel mutations in the PNP gene and the immunological results of the HSCT and evaluate the impact of HSCT on the neurological symptoms. The patient presented early in life with neurological symptoms and suffered later from repeated serious respiratory tract infections. Biochemical tests showed severe reduction in PNP activity (1% residual activity). Genetic testing revealed two new mutations in the PNP gene: c.729C>G (p.Asn243Lys) and c.746A>C (p.Tyr249Cys). HSCT was performed with an unrelated donor, resulting in prompt and sustained engraftment and complete donor chimerism. There was no further aggravation of the patient’s neurological symptoms at 21 months post HSCT, and appropriate developmental milestones were achieved. HSCT is curative for the immunological defect caused by PNP deficiency, and our case strengthens earlier reports that HSCT is effective as a treatment even for neurological symptoms in PNP deficiency.
Keywords: Developmental delay, Hematopoietic stem cell transplantation, Immunodeficiency, Lymphocytes, Matched unrelated donor, Pediatric, Purine nucleoside phosphorylase
Introduction
Purine nucleoside phosphorylase (PNP) is an enzyme active in purine metabolism (Fig. 1), in which it degrades guanosine, deoxyguanosine, inosine, and deoxyinosine (Bzowska et al. 2000). PNP is found in most cells of the body, but different tissues have different levels of PNP activity, with the highest expression in lymphoid tissue. Deficiency in PNP enzymatic activity leads to the accumulation of its substrates, which in turn leads to accumulation of deoxyguanosine triphosphate (dGTP) intracellularly, especially in the thymus, where high cell turnover takes place. dGTP may interfere with DNA synthesis or repair directly or by inhibition of ribonucleotide reductase activity, thereby preventing the cellular proliferation required for an immune response.
Fig. 1.

Schematic presentation of the role of PNP in the degradation and salvage pathways of purine nucleosides. ADA adenosine deaminase, AMP adenosine monophosphate, dGTP deoxyguanosine triphosphate, GMP Guanine monophosphate, IMP inosine monophosphate, HGPRT hypoxanthine guanine phosphoribosyltransferase, PNP purine nucleoside phosphorylase, d deoxy
PNP deficiency is an autosomal recessive disorder and leads to a progressive form of severe combined immunodeficiency (SCID) with decreased numbers of T cells and lymphopenia (Giblett et al. 1975). Some studies have shown that B cell function can be disrupted as well (Markert 1991).
In two thirds of cases of PNP deficiency with the SCID phenotype, neurodevelopmental problems also arise (Markert 1991). Symptoms usually develop towards the end of the patient’s second year of life and include ataxia, mental retardation, developmental delay, and muscle spasticity. In addition, PNP deficiency is associated with an increased risk of autoimmune disorders like autoimmune hemolytic anemia, immune thrombocytopenia, neutropenia, thyroiditis, and lupus.
Low uric acid in plasma has been suggested as a marker for PNP deficiency, but a recent study found normal uric acid in five of seven patients, suggesting that this marker is not reliable (Walker et al. 2011). If the disease is suspected, PNP enzyme activity should be measured either in a red blood cell lysate, blood spots, or in white blood cells (van Kuilenburg et al. 2010).
To confirm the diagnosis, a genetic analysis can be done. The PNP gene containing six exons is located on the long arm of chromosome 14 (14q13.1). Exon skipping, missense, and frameshift mutations in these six exons have been found to cause PNP deficiency, and the most common mutation is a change from A to C or G nucleotides at cDNA position 58 or 234, respectively (Alangari et al. 2009; Walker et al. 2011).
PNP deficiency has a poor prognosis, and the only successful treatment known to date is hematopoietic stem cell transplantation (HSCT). The effectiveness of this treatment on the neurological and autoimmune problems is unclear because of the small number of transplanted patients and limited number of studies (Grunebaum et al. 2013).
We present a patient with PNP deficiency with two novel mutations in the PNP gene. The findings of new mutations are important for future diagnostics and treatment of children with this deficiency. We also address whether the neurological defects of PNP deficiency are treatable by HSCT in order to further evaluate the usefulness of this therapy.
Patient
Our patient is the only child of Kurdish parents who are first-degree cousins. He was healthy until 9 months of age, when he was referred to a pediatric department for developmental delay and obesity. He was unable to sit up without support and had general muscular hypotonia but could hold his head stable. He measured 14 kg and 79 cm (both ≥3 standard deviations for age). The investigations at that age were inconclusive.
At 2 years of age, he was admitted to the local hospital with his first pneumonia. He was noted to have persistent cough but had no significant history of infections. Neurologically, he showed trunk ataxia, areflexia of the lower extremities, and general muscular hypotonia with inability to walk independently and bilateral pes planus. Unfortunately, a more extensive neurological assessment was not done at this time. His speech development was difficult to assess at that time because his native language is not Swedish. In the following 4 months, he suffered three more pneumonias.
Investigations
Standard blood investigations showed leukopenia (2.5 × 109/L) with a low lymphocyte count (0.3–0.8 × 109/L) and mild anemia (hemoglobin 98 g/L) with hypochromic and microcytic erythrocytes. Other laboratory tests, including electrolytes, ALT, AST, GT, pancreatic amylase, bilirubin, and thyroid hormone status, were normal.
Urine investigations showed increased purine metabolites and a low uric acid concentration at 12 μmol/L (ref: 230–480) pointing towards PNP deficiency.
Immunological investigation revealed low B and T cell counts (Table 1). Immunoglobulin levels and antibody response to tetanus vaccine were normal, while specific antibody levels to pneumococci were very low. The lymphocyte stimulation test showed almost no proliferation with T cell mitogens and low activity with T cell-dependent B cell mitogen.
Table 1.
Lymphocyte subset levels before and 21 months after HSCT
| Cell concentration (×109/L) | At diagnosis | 21 months after HSCT | Ref interval |
|---|---|---|---|
| Lymphocytes | 0.6 | 2.4 | 2.30–5.40 |
| T cells (CD3+) | 0.39 | 1.89 | 1.40–3.70 |
| T helper cells (CD4+) | 0.13 | 1.09 | 0.70–2.20 |
| T cytotoxic cells (CD8+) | 0.15 | 0.71 | 0.49–1.30 |
| B cells (CD19+) | 0.07 | 0.36 | 0.39–1.40 |
| NK cells (CD16 + CD56+) | 0.10 | 0.16 | 0.13–0.72 |
T cell receptor excision circles (TRECs), a byproduct of the maturation of T cells that reflects the number of recently formed T cells in peripheral blood, have been suggested as a screening test for early detection of SCID (Chan and Puck 2005). For identification of B cell deficiencies in patients, the measurement of κ-deleting recombinant excision circles (KRECs) is used (Nakagawa et al. 2011). The retrospective analysis of KRECs on the patient’s Guthrie card taken at birth was low at 4.2/μL (ref: >8), whereas TRECs were normal at 13.5/μL (ref: >10).
Genetic and Enzymatic Analysis
The analysis of the PNP enzyme activity in blood spots and the analysis of the PNP gene were performed by PCR amplification of all six coding exons, and flanking intronic regions of the PNP gene was carried out using intronic primer sets. Sequence analysis of genomic fragments amplified by PCR was carried out on an Applied Biosystems (Carlsbad, CA, USA) model 3730 automated DNA sequencer using the dye-terminator method (van Kuilenburg et al. 2010). The genetic analysis showed that the patient was compound heterozygous for two novel mutations in exon 6 of the PNP gene (14q13.1) c.729C>G (p.Asn243Lys) and c.746A>C (p.Tyr249Cys). The parents were carriers for the mutations.
PNP enzyme activity was almost absent at 16 mol/h/mg (normal 823–2387), thus establishing the diagnosis of PNP deficiency.
Transplantation
Considering the low numbers of lymphocytes, the poor results of the lymphocyte functional tests, and the recurrent infections, the decision was made to perform HSCT. The parents signed a written informed consent.
Because the patient had no siblings, he underwent a 9/10 HLA loci identical female unrelated donor bone marrow transplantation (DPB1 mismatch). There was a major blood type mismatch; the patient was type 0 and the donor type B. The patient was CMV IgG positive, while the donor was negative.
According to guidelines (ESID/EBMT 2011) regarding HSCT for primary immunodeficiencies, the patient was conditioned with myeloablative regimen. This consisted of exposure-targeted i.v. busulfan given in 8 doses every 12 h, from day −6 to −3 (first day 4.9 mg/kg and then adjusted aiming for AUC day 0–4 90 ± 5 mg h/L), and fludarabine (40 mg/m2/day, from day −6 to −3). Serotherapy with s.c. alemtuzumab (0.2 mg/kg) was administered for 5 days (day −12 to −8).
He received 5 × 106 CD34+ cells/kg body weight. The posttransplant graft vs. host disease (GvHD) prophylaxis consisted of four doses of methotrexate, cyclosporine, and steroids.
Engraftment and Immunological Reconstitution
Complete hematological recovery was achieved at day +24. Despite profound lymphocytopenia, chimerism analysis showed mixed chimerism in B cells (23% recipient origin) and T cells (35% recipient origin) and with 100% donor chimerism in all other cell lines. Two months after HSCT together with the increasing total lymphocyte count, increasing number of autologous cells were detected, 65% autologous T lymphocytes at day +70. Due to the risk of graft rejection and the persistent CMV viremia immunosuppression was discontinued. Four weeks later, the patient achieved stable 100% donor chimerism in all cell lines. IVIG supplementation was discontinued 23 weeks post HSCT. At 21 months after HSCT, the patient had a normal lymphocyte count with normal lymphocyte subsets (Table 1 and Fig. 2), and neither new nor exacerbations of preexisting infections were observed.
Fig. 2.
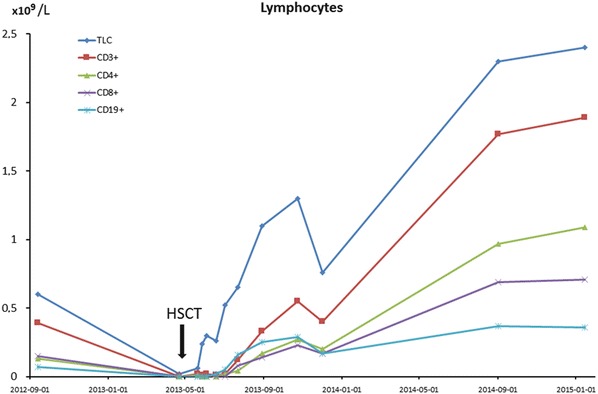
Total lymphocyte count (TLC) and concentration of different lymphocyte subsets as indicated by CD positivity over time in relation to hematopoietic stem cell transplantation (HSCT). The arrow indicates time of HSCT. TLC total lymphocyte count
The earlier high levels of purine metabolites in the urine, especially inosine, were normalized as a sign of functioning PNP enzyme.
Posttransplant Morbidity
On day +10 after HSCT, he suffered from mucositis grades II–III, metapneumovirus pneumonia, and CMV reactivation with CMV enteritis. On day +15, he developed engraftment syndrome with sudden weight gain, fever, and fluid retention and was treated successfully with steroids.
Eight weeks after the discontinuation of the immunosuppressive drugs, the patient developed acute cutaneous GvHD (stage III, overall grade II) but responded to three treatments with steroids without sequelae. No chronic GvHD was observed.
Neurological Development
His neurologic development showed no further deterioration. At 55 months of age, his gross and fine motor skills corresponded to what is expected for a 3- to 4-year-old. His cognitive and social/emotional skills were close to that of a 4-year-old, while his language skills were similar to that of a 3-year-old child. He did not have spasticity but has a persisting hyporeflexia in the lower extremities. The assessment at that age concluded that he had clearly reached new developmental milestones.
Literature Review
To evaluate the neurological outcome of HSCT in our patient, we completed a literature review. We found ten articles describing 14 cases of HSCT-treated PNP deficiency (Markert 1991; Broome et al. 1996; Carpenter et al. 1996; Pannicke et al. 1996; Classen et al. 2001; Baguette et al. 2002; Myers et al. 2004; Delicou et al. 2007; Aytekin et al. 2008; Singh 2012). Of these, two articles describing six cases had no description of the neurological outcome (Markert 1991; Broome et al. 1996). In one case, the patient died 1 month following HSCT (Pannicke et al. 1996). The seven remaining cases and the case presented here were evaluated for this review.
Discussion
Only approximately 4% of all cases of SCIDs are caused by PNP deficiency. The first case was presented in 1975, and up to the end of 2014, fewer than 80 cases have been reported. A total of 24 mutations causing PNP deficiency have been described (Giblett et al. 1975; Markert 1991; Walker et al. 2011). The two new mutations in our patient, c.729C>G (p.Asn243Lys) and c.746A>C (p.Tyr249Cys), both in exon 6, resulted in a near-complete absence of the PNP activity and caused both SCID and neurological defects such as developmental delay and muscular hypotonia, but no autoimmunity. Other mutations and a nonsense and frameshift mutation causing deficiency have been identified in exon 6 of the PNP gene (Walker et al. 2011). There is marked heterogeneity in the phenotype and onset of symptoms in PNP deficiency. Thus, no certain conclusion can be drawn regarding our patient’s mutations and manifestation of specific symptoms.
Typically, PNP deficiency presents with severe T cell defects, but the effects on B cells have been variable (Markert 1991). In our patient, both T and B cell counts were low when he was age 2.5 years. There was a poor response on mitogen-stimulation tests for both B and T cells. Even though immunoglobulin levels were normal, the patient’s immunodeficiency clearly affected both his B and T cells.
The late onset of the immunodeficiency in PNP deficiency may allow for normal TREC and KREC levels with screening in infancy. In line with this possibility, our patient had only abnormal levels of KRECs, whereas TRECs were normal at birth. As described elsewhere (la Marca et al. 2014), we suggest tandem mass spectrometry on dried blood spots to identify PNP metabolites as a more accurate screening method.
Our patient was successfully treated with HSCT which is the only curative treatment available for SCID caused by PNP deficiency. No patient with PNP deficiency has survived longer than the second decade of life without HSCT. When treating SCID caused by deficiency in adenosine deaminase, another enzyme in the purine salvage pathway, the transplant-related mortality is 20% (Grunebaum et al. 2013). The mortality for treatment of PNP deficiency is not known because of the limited number of patients.
After HSCT, no further neurological deterioration was reported in any of the PNP-deficient patients. In most cases, neurological evaluation was done approximately 1 year after transplantation. Marked improvement was reported in two patients, while in one patient, only slight improvement was reported. For the rest of the patients, appropriate developmental milestones were reached, but some of the neurological impairment remained. Persistent spasticity or muscular hypotonia was reported in five patients.
One patient was evaluated after 24 months and another after 32 months, and in both cases, persistent neurological deficits were reported. Our patient, evaluated 21 months after HSCT, clearly belongs to the group showing obvious neurodevelopmental improvements.
The cause of the neurological impairments is not yet known, but the symptoms are similar to those of Lesch-Nyhan syndrome, a deficiency in hypoxanthine guanine phosphoribosyltransferase (HGPRT), which is the next enzyme in the purine salvage pathway (Fig. 1) (Cohen et al. 2000). Because HGPRT deficiency does not present with immunodeficiency, it has been suggested that the mechanism causing the neurological effects of PNP deficiency differs from that causing the immunological defects. Low levels of guanosine triphosphate (GTP) could be the reason for the neurological effects rather than toxic levels of dGTP. The de novo synthesis of GTP is limited in the brain, and because the GTP salvage pathway is disturbed in PNP deficiency, the result is low levels of GTP in the brain and disruption of metabolic processes.
A recent study showed smaller than average cerebellum, corpus callosum, and thalamus in PNP-deficient mice (Mansouri et al. 2012). There were also a reduced number of Purkinje cells in the cerebellum, and the existing Purkinje cells had an abnormal shape. The two patients in our review who had undergone MRI had normal results, but some reported cases had abnormal MRIs (Ozkinay et al. 2007; Madkaikar et al. 2011; Somech et al. 2013). One patient with substantial neurological impairment had an MRI late in the progress of the illness, showing atrophy of the cerebellum and the cerebral cortex with loss of white matter myelination (Ozkinay et al. 2007). Two other cases had mild to significant brain atrophy (Madkaikar et al. 2011; Somech et al. 2013). All three patients died in early childhood. These results indicate that the brain damage visible on MRI can develop late in the course of the disease.
Early treatment with PNP replacement in the studied mice prevented cerebellar damage, indicating that the time of detection and start of treatment are important for reversing the neurological damage (Mansouri et al. 2012).
In the reported cases, only one other patient in addition to our patient had detectable levels of PNP activity, and the neurological symptoms before and after HSCT did not remarkably differ from those with no detectable PNP activity. The age when HSCT was performed varied, and in this very limited number of patients, there was no correlation between improvement and age at transplantation.
In none of the cases was further neurological deterioration after HSCT reported, and improvement was even reported in some. One reason for the reduction in further neurological damage could be that after HSCT, the stem cells can differentiate into blood monocytes that can migrate over the BBB and become microglia cells (Singh 2012). These cells can then produce the missing enzyme, halting the progress of the disease.
The improvement after HSCT is hard to evaluate. Because new developmental milestones are normally reached over time and the patients are relatively young when transplanted, it is difficult to know what the neurological progress would have been if they had not been transplanted. The oldest reported untreated patient lived to age 13 years and died after a stroke (Tam and Leshner 1995). Two more patients have been reported to have seriously deteriorated neurologically before death, one because of a viral brain infection and one because of cerebral edema following intracranial hemorrhage (Dror et al. 2004; Parvaneh et al. 2007). Most patients with PNP deficiency die from infections during the first decade of life (Markert 1991). Given that the course of the neurological decline is not known, it is hard to assess the effect of HSCT. Both in our case and the cases reported, there was no further neurological deterioration after the treatment, and some patients even showed improvement, indicating that HSCT is partly effective as a treatment for these symptoms.
Conclusion
We present a case with two novel mutations in the PNP gene, resulting in severe PNP deficiency with almost absent PNP activity. The patient had a SCID-like phenotype and showed neurological impairment. He was successfully treated with HSCT from an unrelated donor and was infection free after 21 months. He did not show worsening of the neurological symptoms and reached new developmental milestones. This case and reports in the literature on neurological improvement after HSCT indicate that this is an effective treatment even for the neurological symptoms.
Synopsis
Hematopoietic stem cell transplant is an effective treatment immunodeficiency caused by purine nucleoside phosphorylase deficiency and ameliorates neurological symptoms of the disorder.
Compliance with Ethics Guidelines
Informed Consent
All procedures followed were in accordance with the ethical standards of the Helsinki Declaration of 1975, as revised in 2000.
Upon contact with the Regional Ethical Review Board in Lund, we were advised that signed consent from the patient’s parents is sufficient for publication of this case report and the consent was obtained.
Author Contributions
Nicholas Brodszki: conception, planning, collection and compiling of the clinical and laboratory data and writing the manuscript.
Maria Svensson: collection and compiling of the clinical and laboratory data and drafting the manuscript.
André B.P. van Kuilenburg: supervising enzymatic and genetic analysis, cowriting of the manuscript.
Judith Meijer: experimental analysis, cowriting of the manuscript.
Lida Zoetekouw: experimental analysis.
Lennart Truedsson and Jacek Toporski: interpretation of the results and supervising the writing of the manuscript.
All authors read and approved the final manuscript.
Conflict of Interest
Nicholas Brodszki, Maria Svensson, André B.P. van Kuilenburg, Judith Meijer, Lida Zoetekouw, Lennart Truedsson, and Jacek Toporski declare that they have no conflict of interest.
Footnotes
Competing interests: None declared
Contributor Information
Nicholas Brodszki, Email: nicholas.brodszki@skane.se.
Collaborators: Johannes Zschocke
References
- Alangari A, Al-Harbi A, Al-Ghonaium A, Santisteban I, Hershfield M. Purine nucleoside phosphorylase deficiency in two unrelated Saudi patients. Ann Saudi Med. 2009;29:309–312. doi: 10.4103/0256-4947.55320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytekin C, Yuksek M, Dogu F, et al. An unconditioned bone marrow transplantation in a child with purine nucleoside phosphorylase deficiency and its unique complication. Pediatr Transplant. 2008;12:479–482. doi: 10.1111/j.1399-3046.2007.00890.x. [DOI] [PubMed] [Google Scholar]
- Baguette C, Vermylen C, Brichard B, et al. Persistent developmental delay despite successful bone marrow transplantation for purine nucleoside phosphorylase deficiency. J Pediatr Hematol Oncol. 2002;24:69–71. doi: 10.1097/00043426-200201000-00018. [DOI] [PubMed] [Google Scholar]
- Broome CB, Graham ML, Saulsbury FT, Hershfield MS, Buckley RH. Correction of purine nucleoside phosphorylase deficiency by transplantation of allogeneic bone marrow from a sibling. J Pediatr. 1996;128:373–376. doi: 10.1016/S0022-3476(96)70285-8. [DOI] [PubMed] [Google Scholar]
- Bzowska A, Kulikowska E, Shugar D. Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol Ther. 2000;88:349–425. doi: 10.1016/S0163-7258(00)00097-8. [DOI] [PubMed] [Google Scholar]
- Carpenter PA, Ziegler JB, Vowels MR. Late diagnosis and correction of purine nucleoside phosphorylase deficiency with allogeneic bone marrow transplantation. Bone Marrow Transplant. 1996;17:121–124. [PubMed] [Google Scholar]
- Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115:391–398. doi: 10.1016/j.jaci.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Classen CF, Schulz AS, Sigl-Kraetzig M, et al. Successful HLA-identical bone marrow transplantation in a patient with PNP deficiency using busulfan and fludarabine for conditioning. Bone Marrow Transplant. 2001;28:93–96. doi: 10.1038/sj.bmt.1703100. [DOI] [PubMed] [Google Scholar]
- Cohen A, Grunebaum E, Arpaia E, Roifman CM. Immunodeficiency caused by purine nucleoside phosphorylase deficiency. Immunol Allergy Clin North Am. 2000;20(1):143–159. doi: 10.1016/S0889-8561(05)70139-9. [DOI] [Google Scholar]
- Delicou S, Kitra-Roussou V, Peristeri J, et al. Successful HLA-identical hematopoietic stem cell transplantation in a patient with purine nucleoside phosphorylase deficiency. Pediatr Transplant. 2007;11:799–803. doi: 10.1111/j.1399-3046.2007.00772.x. [DOI] [PubMed] [Google Scholar]
- Dror Y, Grunebaum E, Hitzler J, et al. Purine nucleoside phosphorylase deficiency associated with a dysplastic marrow morphology. Pediatr Res. 2004;55:472–477. doi: 10.1203/01.PDR.0000111286.23110.F8. [DOI] [PubMed] [Google Scholar]
- ESID/EBMT (2011) EBMT/ESID guidelines for haematopoietic stem cell transplantation for primary immunodeficiencies
- Giblett ER, Ammann AJ, Wara DW, Sandman R, Diamond LK. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet. 1975;1:1010–1013. doi: 10.1016/S0140-6736(75)91950-9. [DOI] [PubMed] [Google Scholar]
- Grunebaum E, Cohen A, Roifman CM. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr Opin Allergy Clin Immunol. 2013;13:630–638. doi: 10.1097/ACI.0000000000000006. [DOI] [PubMed] [Google Scholar]
- la Marca G, Canessa C, Giocaliere E, et al. Diagnosis of immunodeficiency caused by a purine nucleoside phosphorylase defect by using tandem mass spectrometry on dried blood spots. J Allergy Clin Immunol. 2014;134:155–159. doi: 10.1016/j.jaci.2014.01.040. [DOI] [PubMed] [Google Scholar]
- Madkaikar MR, Kulkarni S, Utage P et al (2011) Purine nucleoside phosphorylase deficiency with a novel PNP gene mutation: a first case report from India. BMJ Case Rep 2011 [DOI] [PMC free article] [PubMed]
- Mansouri A, Min W, Cole CJ, et al. Cerebellar abnormalities in purine nucleoside phosphorylase deficient mice. Neurobiol Dis. 2012;47:201–209. doi: 10.1016/j.nbd.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Markert ML. Purine nucleoside phosphorylase deficiency. Immunodefic Rev. 1991;3:45–81. [PubMed] [Google Scholar]
- Myers LA, Hershfield MS, Neale WT, Escolar M, Kurtzberg J. Purine nucleoside phosphorylase deficiency (PNP-def) presenting with lymphopenia and developmental delay: successful correction with umbilical cord blood transplantation. J Pediatr. 2004;145:710–712. doi: 10.1016/j.jpeds.2004.06.075. [DOI] [PubMed] [Google Scholar]
- Nakagawa N, Imai K, Kanegane H, et al. Quantification of kappa-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J Allergy Clin Immunol. 2011;128(223–225) doi: 10.1016/j.jaci.2011.01.052. [DOI] [PubMed] [Google Scholar]
- Ozkinay F, Pehlivan S, Onay H, et al. Purine nucleoside phosphorylase deficiency in a patient with spastic paraplegia and recurrent infections. J Child Neurol. 2007;22:741–743. doi: 10.1177/0883073807302617. [DOI] [PubMed] [Google Scholar]
- Pannicke U, Tuchschmid P, Friedrich W, Bartram CR, Schwarz K. Two novel missense and frameshift mutations in exons 5 and 6 of the purine nucleoside phosphorylase (PNP) gene in a severe combined immunodeficiency (SCID) patient. Hum Genet. 1996;98:706–709. doi: 10.1007/s004390050290. [DOI] [PubMed] [Google Scholar]
- Parvaneh N, Ashrafi MR, Yeganeh M, Pouladi N, Sayarifar F, Parvaneh L. Progressive multifocal leukoencephalopathy in purine nucleoside phosphorylase deficiency. Brain Dev. 2007;29:124–126. doi: 10.1016/j.braindev.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Singh V. Cross correction following haemopoietic stem cell transplant for purine nucleoside phosphorylase deficiency: engrafted donor-derived white blood cells provide enzyme to residual enzyme-deficient recipient cells. JIMD Rep. 2012;6:39–42. doi: 10.1007/8904_2012_126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somech R, Lev A, Grisaru-Soen G, Shiran SI, Simon AJ, Grunebaum E. Purine nucleoside phosphorylase deficiency presenting as severe combined immune deficiency. Immunol Res. 2013;56:150–154. doi: 10.1007/s12026-012-8380-9. [DOI] [PubMed] [Google Scholar]
- Tam DA, Jr, Leshner RT. Stroke in purine nucleoside phosphorylase deficiency. Pediatr Neurol. 1995;12:146–148. doi: 10.1016/0887-8994(94)00118-L. [DOI] [PubMed] [Google Scholar]
- van Kuilenburg AB, Zoetekouw L, Meijer J, Kuijpers TW. Identification of purine nucleoside phosphorylase deficiency in dried blood spots by a non-radiochemical assay using reversed-phase high-performance liquid chromatography. Nucleosides Nucleotides Nucleic Acids. 2010;29:466–470. doi: 10.1080/15257771003741455. [DOI] [PubMed] [Google Scholar]
- Walker PL, Corrigan A, Arenas M, Escuredo E, Fairbanks L, Marinaki A. Purine nucleoside phosphorylase deficiency: a mutation update. Nucleosides Nucleotides Nucleic Acids. 2011;30:1243–1247. doi: 10.1080/15257770.2011.630852. [DOI] [PubMed] [Google Scholar]