

Abstract
Deficiency of cyclin-dependent kinase 5 (Cdk5) has been linked to the death of postmitotic cortical neurons during brain development. We now report that, in mouse cortical neurons, Cdk5 is capable of phosphorylating the transcription factor FOXO1 at Ser249 in vitro and in vivo. Cellular stresses resulting from extracellular stimulation by H2O2 or β-amyloid promote hyperactivation of Cdk5, FOXO1 nuclear export and inhibition of its downstream transcriptional activity. In contrast, a loss of Cdk5 leads to FOXO1 translocation into the nucleus: a shift due to decreased AKT activity but independent of S249 phosphorylation. Nuclear FOXO1 upregulates transcription of the proapoptotic gene, BIM, leading to neuronal death, which can be rescued when endogenous FOXO1 was replaced by the cytoplasmically localized form of FOXO1, FOXO1-S249D. Cytoplasmic, but not nuclear, Cdk5 attenuates neuronal death by inhibiting FOXO1 transcriptional activity and BIM expression. Together, our findings suggest that Cdk5 plays a novel and unexpected role in the degeneration of postmitotic neurons through modulation of the cellular location of FOXO1, which constitutes an alternative pathway through which Cdk5 deficiency leads to neuronal death.
Keywords: Cdk5, FOXO1, neuronal death, phosphorylation
Introduction
Cyclin-dependent kinase 5 (Cdk5) is a unique member of the Cdk family. Although its cloning was based on its sequence homology to other Cdks (Hellmich et al., 1992), Cdk5 is not activated by traditional cyclins but by the cyclin paralogs p35 and p39 (Lew et al., 1994; Tsai et al., 1994; Dhavan and Tsai, 2001). Cdk5 is ubiquitously expressed, yet the high neuronal expression of p35 and p39 largely confines physiological Cdk5 enzyme activity to a limited number of tissues, most prominently the nervous system. Cdk5 is expressed early in embryo genesis and plays pivotal roles in neuronal development, including survival, migration, and maturation (Ohshima et al., 1996, 1999; Gilmore and Herrup, 2001; Cruz and Tsai, 2004; Hawasli and Bibb, 2007; Hisanaga and Endo, 2010).
In addition to its developmental functions, in the mature nervous system, Cdk5 is involved in synaptic function and neuronal survival. For example, hyperactive Cdk5 is reported to promote neuronal death and has been proposed to play a role in the pathogenesis of Alzheimer's disease (AD). In AD, the overactivation of Cdk5 is thought to induce tau phosphorylation and tangle formation, which promotes neuron loss. One mechanism by which the hyperactivation of Cdk5 might occur is through the proteolytic cleavage of p35 by the calcium-dependent protease calpain to create p25. This cleavage is prominent in the neuronal response to β-amyloid-induced stress (Patrick et al., 1999; Su and Tsai, 2011). Although too much Cdk5 activity might be bad, too little Cdk5 is also dangerous to the health and survival of adult neurons. For example, there is florid neuronal death in Cdk5−/− mouse cortex (Cicero and Herrup, 2005). Cdk5-deficient neurons are also more sensitive to extracellular stressors, such as UV irradiation (Li et al., 2002) and β-amyloid challenge. In forebrain, conditional Cdk5 knock-out leads to a progressive neuronal degeneration in adult brain (Takahashi et al., 2010). Restoring Cdk5 function in Cdk5−/− neurons reverses their chromatolytic morphology (Tanaka et al., 2001; Cheung and Ip, 2004). To date, however, the mechanism by which the loss of Cdk5 leads to neuronal death remains unclear. Although cell cycle regulation undoubtedly plays a part, there are suggestions that independent pathways are also at work.
The FOXO (forkhead) transcription factors have a broad impact on various cellular processes, including cell cycle regulation, apoptosis, DNA repair, and autophagy (Lehtinen et al., 2006; Yuan et al., 2009; Xie et al., 2012). Activation of FOXO promotes cell death by regulating the expression of genes involved in apoptosis, such as BIM, FAS ligand, and TRAIL (Modur et al., 2002; Essafi et al., 2005; Real et al., 2005). In the current study, we report that FOXO1 is a key link in a cell cycle-independent chain of events leading from Cdk5 deficiency to the death of CNS neurons. Our data demonstrate that, in Cdk5-deficient postmitotic neurons, FOXO1 phosphorylation at Ser249 is lost. This initiates the expression of proapoptotic genes and facilitates neuronal death.
Materials and Methods
Antibodies and chemical reagents.
Antibodies against FOXO1 and cleaved caspase-3 were from Cell Signaling Technology. Anti-Cdk5, GFP, p35, hnRNP A2/B1, 14-3-3β, and GAPDH antibodies were from Santa Cruz Biotechnology. Anti-α-tubulin and anti-Flag (M2) were from Sigma-Aldrich. Anti-BrdU and FOXO1 antibodies were from Abcam. Phospho-FOXO1-S319, phospho-AKT (Ser473), and Akt1 were from Sangon Biotech. The phospho-FOXO1-S249 antibody was described previously (Yuan et al., 2008). Secondary antibodies used for immunocytochemistry were as follows: goat anti-mouse Alexa-488 and -594 and goat anti-rabbit Alexa-488 and -594 (Invitrogen).
DAPI was used as a nuclear counterstain at 1 mg/ml; LY-294002 hydrochloride, BrdU, and roscovitine were purchased from Sigma-Aldrich. The dual-luciferase reporter assay kit was purchased from Promega.
Animals.
A colony of Cdk5+/− mice was maintained on a mixed C57BL/6J background. Homozygous mutant embryos of both sexes were produced by intercrossing heterozygous Cdk5+/− mice. Wild-type C57BL/6J mice were purchased from SLRC Laboratory. Timed pregnancies were established, and the embryos were taken at embryonic day 16.5 for primary cortical cultures or brain lysates. We assume each litter contained a nearly equal mixture of the two sexes, although no attempt was made to assess the sex of the harvested embryos. All animal procedures were performed in accordance with Xiamen University Institutional Animal Care and Use Committee standards.
Cell culture.
Neuro2A and 293T cells were cultured in DMEM medium containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were maintained at 37°C and 5% CO2. Primary cortical neurons were cultured from cortex of E16.5 d mice. For Cdk5-deficient cultures, all of the embryos from a Cdk5+/−xCdk5+/− mating were harvested and treated separately as described previously (Zhang et al., 2010a). Primary cortical neurons were cultured in Neurobasal medium supplemented with B27 and 2 mm glutamine and grown on coverslips or plastic culture plate coated with poly-l-lysine (25 μg/ml).
Plasmids and small interference RNA (siRNA).
The generosity of many laboratories allowed us to assemble a majority of the vectors used in this study. Pcmv-Flag-FOXO1-AAA and sh-foxo vectors were kindly provided by Dr. You Han (Xiamen University, Xiamen); Bim-luciferase and 3xIRS-luciferase reporter genes vectors were provided by Dr. KL Guan (University of California, San Diego). GST-FOXO1 was provided by Dr. Haojie Huang (Mayo Clinic, Rochester, MN). CMV-p35 was a gift from Dr. Li-Huei Tsai (Massachusetts Institute of Technology, Cambridge, MA). Flag-FOXO1 was purchased from Addgene. GFP-Cdk1 to GFP-Cdk6, GFP-cyclinA, GFP-cyclinB, GFP-cyclinC, and GFP-cyclinD were generated by inserting target genes into pEGFP-C3 vector (Clontech). GFP-FOXO1-S249A, GFP-FOXO1-S249D, GFP-FOXO1-AAA-S249A, GFP-FOXO1-AAA-S298A, GFP-FOXO1-AAA-S249D, and Flag-tagged FOXO1 mutations were generated by site-directed mutagenesis (Stratagene). CMV-mCherry-IRES-U6-scramble and CMV-mCherry-IRES-U6-shcdk5 were from GENECHEM. The Flag-14-3-3β plasmid was cloned by PCR into the pFLAG-CMV-2 expression vector. GFP, GFP-p25, GFP-p35, GFP-Cdk5, GFP-Cdk5-KD (D144N), GFP-NLS, GFP-Cdk5-NLS, GFP-NES, and GFP-Cdk5-NES vectors were described previously (Zhang et al., 2010a, 2012). Gene-specific siRNAs targeting Cdk5 and foxo1 were purchased from Ribobio.
Luciferase assay.
Cells were transfected with 1 μg of 3xIRS or Bim-luciferase reporter plasmid and 200 ng of the tk-renilla luciferase reporter plasmid, the latter serving as an internal control. At 72 h after transfection, cells were left untreated or treated with drugs. The cells were then washed with PBS and lysed in the buffer provided in the Promega luciferase system and subjected to luciferase assays. Relative luciferase activities were obtained by normalizing the firefly luciferase activity against renilla luciferase activity.
Western blotting and coimmunoprecipitation.
Immunoblotting and coimmunoprecipitation were performed as described previously (Zhang et al., 2010a). Briefly, harvested cells were homogenized in ice-cold NP-40 lysis buffer with protease inhibitor mix. The samples were centrifuged at 12,000 × g for 15 min at 4°C. The supernatant was collected, and total protein levels were measured by the micro BCA protein assay kit (Thermo Fisher Scientific). Fractionation of cells into cytoplasmic and nuclear components was accomplished with an NER-mammalian kit from Thermo Fisher Scientific according to the manufacturer's instructions. For Western blots, the lysates were separated with SDS-PAGE and electrophoretically transferred onto nitrocellulose membranes. The membranes were blocked with 5% nonfat milk in TBST and probed with primary antibodies, followed by treatment with HRP-linked secondary antibodies and ECL Western blotting detection reagents. The intensity of immunoreactive bands was quantified using National Institutes of Health ImageJ software. For immunoprecipitation, the cell lysates were incubated with immunoprecipitation antibody at 4°C for 4 h, followed by overnight incubation with protein G. The beads were washed five times with ice-cold NP-40 lysis buffer, and the bound proteins were analyzed by SDS-PAGE and immunoblotting analysis.
Immunofluorescence staining.
The neuronal cells were immunostained as described previously (Zhang et al., 2010a). In brief, the cells were fixed with 4% PFA for 20 min. After three washes with PBS, the cells were blocked with 10% goat serum in PBS containing 0.5% Tween 20 to reduce nonspecific antibody binding. Neurons were then incubated with the primary antibody at 4°C overnight. After washing with PBS four times, AlexaFluor-488- or 546-conjugated secondary antibody was used to detect the signal. Nuclear morphology was visualized using the DNA dye DAPI.
Statistical analysis.
All data were obtained from at least three different preparations. Statistical analysis of the data was performed with a two-tailed Student's t test or a one-way ANOVA using Graphpad Prism 5. Data are presented as the mean ± SEM. p < 0.05 was taken to represent statistical significance.
Results
Cdk5 associates with FOXO1 and phosphorylates serine 249
We coexpressed Cdk5 with p35 or its cleaved form p25 in 293T cells together with wild-type human FOXO1 or a nonphosphorylatable human FOXO1-S249A mutant. Using a previously generated antibody (Yuan et al., 2008) that specifically recognizes FOXO1 phosphorylated at Ser249, we immunoblotted gels of total cell lysates. We found that both Cdk5/p35 and Cdk5/p25 transfection can dramatically increase the amount of phosphor-S249-FOXO1 labeling. No labeling was detected when the mutant FOXO1-S249A was used, even in the presence of Cdk5/p35 or Cdk5/p25 cotransfection (Fig. 1A). The kinase dead-mutation of Cdk5 (Cdk5-KD) complex, such as Cdk5-KD/p35 or Cdk5-KD/p25, was unable to phosphorylate FOXO1 at Ser249 (Fig. 1A). Other members of the Cdk family, such as Cdk1 and Cdk2, were also capable of phosphorylating FOXO1 at Ser249 (Fig. 1B). By in vitro kinase assay, we found that the phosphor-S249-FOXO1 antibody recognized recombinant FOXO1 that was phosphorylated by Cdk5/p35, Cdk5/p25, Cdk1/cyclin B, or Cdk2/cyclin A in vitro but did not recognize unphosphorylated FOXO1-S249A mutant (Fig. 1C,D). This particular experiment demonstrates that activated Cdk5 is able to directly phosphorylate FOXO1. We next investigated the physical interactions among FOXO1, Cdk5, and p35. By coimmunoprecipitation in both directions, FOXO1 was found to associate with both Cdk5 and p35 either when all three proteins were coexpressed or when protein pairs (FOXO1/Cdk5 and FOXO1/p35) were coexpressed separately (Fig. 1E). These data suggested that both Cdk5 and p35 physically associate with FOXO1.
Figure 1.

Cdk5 phosphorylates FOXO1 at serine 249. A, B, Lysates of 293T cells transfected with Cdk5/p35, Cdk5/p25, Cdk5-KD/p35, Cdk5-KD/p25 (Cdk5 kinase-dead plasmid), Cdk1/Cyclin B, Cdk2/Cyclin A, or control vector, together with GFP-FOXO1 or the GFP-FOXO1–S249A mutant, were analyzed by immunoblotting with antibodies to pS249-FOXO1, FOXO1, Cdk5, p35/p25, GFP, or GAPGH as shown. C, D, In vitro kinase assays with immunoprecipitated (IP) Cdk5. Recombinant GST-FOXO1 or GST-FOXO1–S249A proteins were subjected to an in vitro kinase assay with IP GFP fusion proteins from 293T cells expressing the indicated constructs. Kinase reaction products were immunoblotted with the pS249-FOXO1 antibody. FOXO1 proteins levels were evaluated by Coomassie Blue staining. E, The interactions among FOXO1, Cdk5, and p35 were investigated in 293T cells. The 293T cells were cotransfected Flag-FOXO1 with GFP-Cdk5 and/or GFP-p35. The antibodies used for IP are indicated in the top of the gel. After immunoprecipitation, the gels were blotted with indicated antibodies. F, Primary cortical neurons were transfected with the GFP-FOXO1-WT or GFP-FOXO1–S249A plasmid together with p25/Cdk5 or p25/Cdk5-KD or control vector in combination with the 3xIRS luciferase reporter system and subjected to luciferase assay. Data are mean ± SEM; n = 5. *p < 0.05. G, Primary cortical neurons were transfected with different Cdk/cyclin pairs together with the 3xIRS-luciferase reporter gene and subjected to luciferase assays. Data are mean ± SEM; n = 5. *p < 0.05.
FOXO1 has been reported to be phosphorylated by Cdk1 or Cdk2 at the S249 site in cancer cells or cerebellar granule cells (Huang et al., 2006; Yuan et al., 2008); however, the potential role of this pathway in cortical neurons remains completely unknown. Considering that the active Cdk5 is specifically found in the nervous system especially in adult neurons, we therefore asked whether the phosphorylation of FOXO1 by Cdk5 altered its transcription activity in primary cortical neurons. In primary postmitotic cortical neurons, active Cdk5, but not kinase-dead Cdk5, dramatically reduced the 3xIRS luciferase expression (Fig. 1F). To confirm that the decreased FOXO1 transcriptional activity was mediated by Cdk5-directed phosphorylation, the FOXO1-S249A mutant was used to replace the wild-type. As shown in Figure 1F, Cdk5/p25 had no effect on the expression of the 3xIRS-luciferase reporter gene when the mutant FOXO1-S249A was used. Active Cdk2/cyclinA also lowered 3xIRS activity, but active Cdk1/cyclin B increased it. Other classic Cdk/cyclin complexes (Cdk3/cyclin C, Cdk4/cyclin D, or Cdk6/cyclin D) had no significant effect on FOXO1 transcriptional activity (Fig. 1G).
Activation of Cdk5 accelerates FOXO1 nuclear export
Inactivation of FOXO1 is commonly associated with its export from the nucleus. We have documented that Cdk5 can serve as a FOXO1 kinase, yet AKT can also phosphorylate FOXO1, promoting its interaction with 14-3-3β proteins, its cytoplasmic sequestration, and its inhibition as a transcription factor (Burgering and Kops, 2002; van der Heide et al., 2004). We found that introduction of Cdk5/p25 significantly increased the association between FOXO1 and 14-3-3β compared with control vector. However, Cdk5/p25 failed to increase the interaction between FOXO1-S249A and 14-3-3β proteins consistent with the hypothesis that phosphor-S249 is one key 14-3-3β interacting residue (Fig. 2A).
Figure 2.
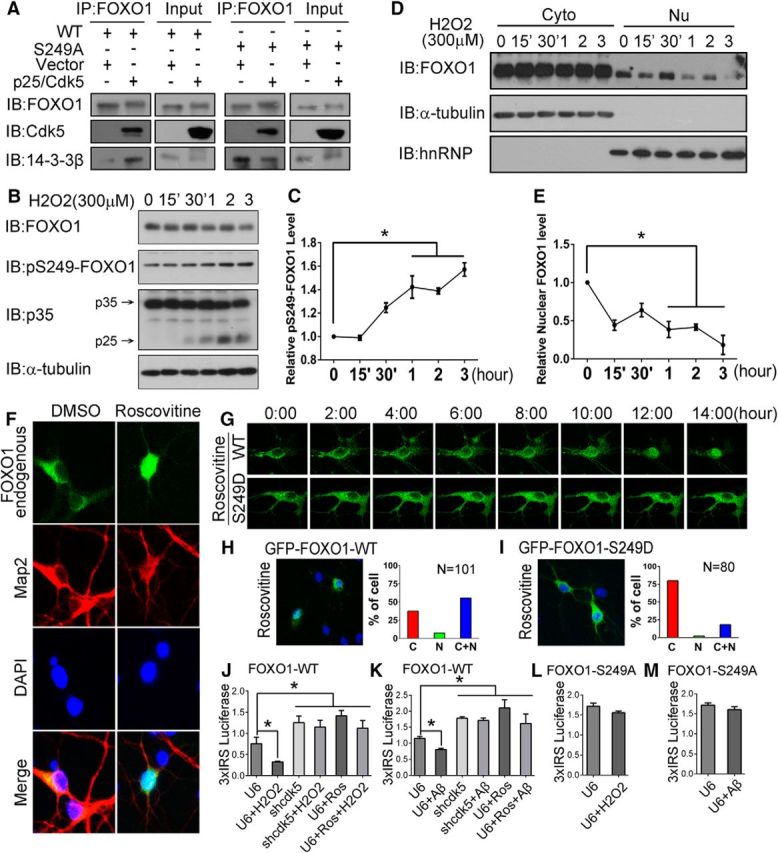
Oxidative stress induces FOXO1 nuclear export by active Cdk5. A, Lysates of 293T cells transfected with GFP-FOXO1 or GFP-FOXO1–S249A, together with Cdk5/p25 or the control vector, were immunoprecipitated with the FOXO1 antibody and immunoblotted with the FOXO1, Cdk5, and 14-3-3β antibodies. B, Primary cortical neurons were treated with 300 μm H2O2 for the indicated time. Whole-cell lysates were then analyzed by immunoblotting with the FOXO1, pS249-FOXO1, p35/p25, and α-tubulin antibodies. C, Quantifications of the relative pS249-FOXO1 intensities. Data are mean ± SEM; n = 3. *p < 0.05. D, E, Primary cortical neurons were treated with 300 μm H2O2 for the indicated time. The cytosolic and nuclear extracts were isolated and immunoblotted with the indicated antibodies. E, Quantifications of the relative nuclear FOXO1 intensities. Data are mean ± SEM; n = 3. *p < 0.05. F, Primary cortical neurons were treated with 50 μm roscovitine or vehicle control (DMSO). Endogenous FOXO1 and Map2 (Microtubule-associated protein 2) were immunostained to investigate the localization of FOXO1 in neurons (Map2-positive cells). G–I, GFP-FOXO1 or GFP-FOXO1–S249D was transfected in primary cortical neurons. G, After 50 μm roscovitine administration, the nuclear translocation time of FOXO1 proteins (GFP-tag) was monitored by live cell imaging. Representative images and quantifications of subcellular localization of GFP-FOXO1 and GFP-FOXO1–S249D are shown in H and I, respectively. J–M, Primary cortical neurons were transfected with control U6 or shCdk5 plasmid together with GFP-FOXO1-WT or GFP-FOXO1–S249A in combination with the 3xIRS luciferase reporter system. On DIV 7, the neurons were treated with 300 μm H2O2 for 3 h or 3 μm β-amyloid for 10 h, and then subjected to luciferase assay. Data are mean ± SEM; n = 5. *p < 0.05.
To determine the consequences of endogenous activation of Cdk5 on FOXO1, we exposed cultures of primary cortical neurons to H2O2, which is known to induce the cleavage of p35 to p25 and activate Cdk5 (Fig. 2B). H2O2 treatments significantly increased the phosphorylation level of S249-FOXO1 while having little effect on the total FOXO1 protein level (Fig. 2B,C). H2O2 administration also dramatically induced the nuclear export of FOXO1 (Fig. 2D,E). When Cdk5 activity was inhibited by roscovitine (a Cdk5 activity inhibitor), the endogenous FOXO1 (Fig. 2F) or the exogenous GFP-FOXO1 (Fig. 2G) relocated from cytoplasm to nucleus within 12 h. Compared with wild-type-FOXO1, roscovitine treatments failed to induce the nuclear relocation of phosphomimetic FOXO1-S249D mutant (Fig. 2H,I).
H2O2 treatment and the resulting shift in FOXO1 localization also significantly decreased the expression of the 3xIRS-luciferase reporter (Fig. 2J). Importantly, this effect of H2O2 was abolished either when Cdk5 was silenced by shRNA or the mutant FOXO1-S249A was used instead of wild-type FOXO1 (Fig. 2L). Similar results were obtained when β-amyloid was applied to the cultured neurons instead of H2O2 (Fig. 2K,M). These findings demonstrate that the Ser249 phosphorylation by Cdk5 is vital both for the cytoplasmic retention of FOXO1 and for suppressing its activity as a transcription cofactor. The fact that roscovitine treatment blocks the downregulation of the 3xIRS-luciferase reporter by H2O2 or β-amyloid (Fig. 2J,K) further implies that it is Cdk5 activity, not just the protein, which is the key to this effect.
Cdk5 phosphorylation of FOXO1 at S249 is sufficient for its cytoplasmic retention and reverses nuclear localization of FOXO1 resulting from AKT inhibition
Nuclear accumulation of FOXO1 can also be achieved by inhibiting the AKT pathway (Burgering and Kops, 2002; van der Heide et al., 2004), and we have replicated these findings in our system. Significantly, whereas the nuclear localizations of both wild-type and mutant (S249A) FOXO1 were enhanced in the presence of the AKT inhibitor LY294002 (Fig. 3A,B,D,E), we found that the phosphomimetic FOXO1-S249D mutant retained significant cytoplasmic localization, even in the presence of the AKT inhibitor (Fig. 3C,F). To address the same issue in reverse, we used an AKT-nonphosphorylatable FOXO1 mutant by converting the three AKT target residues to alanine (FOXO1-AAA). On its own, this mutant lacks the ability to associate with 14-3-3β and thus is imported to the nucleus (Fig. 3G). Blocking the Cdk5 sites in addition (FOXO1-AAA-S249A) had no additional effect on protein localization (Fig. 3H). However, when the Cdk5 site was altered to its phosphomimetic form (FOXO1-AAA-S249D), the protein lost its exclusively nuclear localization and could be found in both cytoplasm and nucleus (Fig. 3I). The levels of expression of the 3xIRS and BIM reporters were consistent with the localization data. FOXO1-AAA-S249D had a reduced effect on the reporter constructs compared with the AKT-nonphosphorylatable FOXO1-AAA mutant (Fig. 3J,K). To demonstrate the Cdk5 dependence of this phenomenon, we showed that LY294002-induced nuclear accumulation of wild-type FOXO1 was abolished by Cdk5/p35 or Cdk5/p25, but not by Cdk5-KD/p35 or Cdk5-KD/p25 (Fig. 4A,B). The same results were obtained when the AKT-unresponsive mutant FOXO1-AAA was used (Fig. 4C). Furthermore, when the Cdk5 phosphorylation site on FOXO1-AAA was removed (FOXO1-AAA-S249A), activation of Cdk5/p25 or Cdk5/p35 failed to translocate the nuclear FOXO1 into cytoplasm (Fig. 4D).
Figure 3.

Ser249-FOXO1 is less affected by AKT inhibition. A–F, GFP-FOXO1, GFP-FOXO1–S249A, or GFP-FOXO1–S249D was transfected in primary cortical neurons treated with DMSO or LY294002 for 16 h. Representative images and the quantification of subcellular localization of GFP-FOXO1, GFP-FOXO1–S249A, and GFP-FOXO1–S249D were shown in top and bottom panels, respectively. G–I, GFP-FOXO1-AAA, GFP-FOXO1-AAA-S249A, or GFP-FOXO1-AAA-S249D was transfected in primary cortical neurons. Representative images and the quantification of subcellular localization of GFP-FOXO1-AAA, GFP-FOXO1-AAA-S249A, and GFP-FOXO1-AAA-S249D were shown in top and bottom panels, respectively. J–K, Primary cortical neurons were transfected with GFP-FOXO1-AAA, GFP-FOXO1-AAA-S249A, or GFP-FOXO1-AAA-S249D plasmids together with the 3xIRS-luciferase (J) or Bim-luciferase (K) reporter gene and subjected to luciferase assays. Data are mean ± SEM; n = 5. *p < 0.05.
Figure 4.

Cdk5 promotes an AKT-independent nuclear export of FOXO1. A, B, Primary cortical neurons were transfected with Flag-FOXO1 together with GFP, GFP-p25/Cdk5, GFP-p35/Cdk5, GFP-p25/Cdk5-KD, or GFP-p35/Cdk5-KD on DIV 3. On DIV 7, the neurons were treated with vehicle control (DMSO) (A) or LY294002 (B) for 16 h. After treatments, the cells were fixed to measure the subcellular localization of FOXO1 by Flag-immunofluorescent staining. Representative images are shown on the left: green represents GFP; red represents Flag; blue represents DAPI. Quantification of subcellular localization of FOXO1 is shown on the right. C, D, Primary cortical neurons were transfected with Flag-FOXO1-AAA or Flag-FOXO1-AAA-S249A together with GFP, GFP-p25/Cdk5, GFP-p35/Cdk5, GFP-p25/Cdk5-KD, or GFP-p35/Cdk5-KD. On DIV 7, the cells were fixed to measure the subcellular localization of FOXO1 by Flag-immunofluorescent staining. Representative images are shown on the left: green represents GFP; red represents Flag; blue represents DAPI. Quantification of subcellular localization of FOXO1 is shown on the right.
Consistent with the localization data, the LY294002-induced upregulation of the FOXO1 transcriptional activity was not observed in the presence of Cdk5/p35 or Cdk5/p25 but still evident when p35 or p25 was coexpressed with the kinase-dead form of Cdk5 (Fig. 5A, red columns). However, when FOXO1 (FOXO1-S249A) failed to be phosphorylated by Cdk5, AKT inhibition by LY294002 remained capable of activating FOXO1's transcriptional activity, even in the presence of Cdk5/p35 or Cdk5/p25 (Fig. 5A, green columns). As predicted from the above results, AKT inhibition has relatively little effect on the Cdk5 phosphomimetic form of FOXO1 (FOXO1-S249D) (Fig. 5B). Overexpression of Cdk5/p35 or Cdk5/p25 decreased FOXO1's transcriptional activity on wild-type FOXO1-AAA but had no effect on the FOXO1-AAA-S249A mutant (Fig. 5C). These results are consistent with the decreased nuclear localization of the FOXO1-AAA due to the activation of Cdk5/p35 or Cdk5/p25 (Fig. 4C,D).
Figure 5.

Cdk5 inhibits FOXO1 transcriptional activity in an AKT-independent way. Primary cortical neurons were transfected with the GFP-FOXO1-WT, GFP-FOXO1–S249A, GFP-FOXO1-AAA, or GFP-FOXO1-AAA-S249A plasmid together with indicated vectors in A, B, C or control vector in combination with the 3xIRS luciferase reporter system. With or without drug treatments, the neurons were subjected to luciferase assay. Data are mean ± SEM; n = 5. *p < 0.05.
As mentioned above, Cdk1 and Cdk2 can both phosphorylate FOXO1 at S249, and both kinases can regulate FOXO1 transcriptional activity. In exploring the specificity of the Cdk5 effect, we found that, when either Cdk1 or Cdk2 was transfected with their activating cyclins, they were not able to downregulate the AKT-insensitive FOXO1-AAA mutant, even though the S249 residue was present (Fig. 5C).
Cell cycle, cell death, and the interplay of AKT, Cdk5, and FOXO1
We have previously shown that, in the Cdk5-deficient brain, there is a distinct loss of cell cycle suppression in maturing cortical neurons accompanied by massive cell death (Cicero and Herrup, 2005). We wondered whether abnormal activation of FOXO1 might be involved either of these processes. We first asked whether deficiency of Cdk5 affects any aspects of the FOXO1 phenotype. In neurons derived from the E16.5 brains of Cdk5+/+ or Cdk5+/− mice, FOXO1 immunoreactivity was found predominately in the cytoplasm. In the Cdk5-deficient mouse brain (Cdk5−/−), however, we observed strong FOXO1 staining in neuronal nuclei (Fig. 6A). Similar results were obtained in vitro: the endogenous FOXO1 or the GFP-tagged FOXO1 was found in the nucleus of cultured Cdk5−/− neurons (Fig. 6B,C). Furthermore, the endogenous FOXO1 protein was found predominately in the nucleus in cells of the Cdk5-deficient mice (Cdk5−/−) or when Cdk5 was downregulated by siRNAs in neuronal cultures derived from wild-type mice (Fig. 6D–F). FOXO1 localized in cytoplasm is normally inactive (Gan et al., 2005); after relocation to the nucleus, its transcription activity increases. This is equally true in our system. Depletion of endogenous Cdk5 in neurons increased the expression of the 3xIRS-luciferase reporter gene, which suggests that endogenous Cdk5 normally suppresses FOXO1-dependent transcription (Fig. 6G,H).
Figure 6.
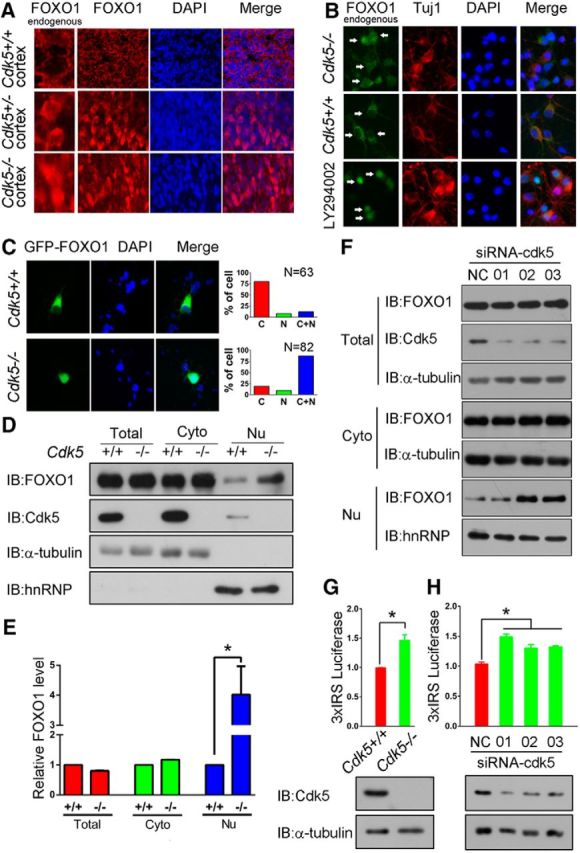
Nuclear accumulation of FOXO1 in Cdk5-deficient neurons leads to the activation of FOXO1. A, Representative images of FOXO1 immunoreactivity in cortical regions of E16.5 Cdk5+/+, Cdk5+/−, and Cdk5−/− mice. B, C, Cdk5+/+ and Cdk5−/− neurons were isolated from E16.5 embryos obtained from crosses of Cdk5+/−mice. B, Subcellular localization of endogenous FOXO1 in Cdk5+/+ and Cdk5−/− neurons was evaluated by FOXO1 immunofluorescence staining in Tuj1-positive neurons. LY294002 treatments induced nuclear accumulation of FOXO1. C, Subcellular localization of exogenous FOXO1 was evaluated following transfection of GFP-FOXO1 in Cdk5+/+ and Cdk5−/− neurons. Representative images are shown on the left, and quantification of subcellular localization of FOXO1 is shown on the right. D, E, The cytoplasmic and nuclear extracts were isolated from cortices of E16.5 Cdk5+/+ and Cdk5−/− mice. Lysates were immunoblotted with the indicated antibodies. E, The relative level of FOXO1 in total/cytoplasm/nucleus was quantified. Data are mean ± SEM; n = 3. *p < 0.05. F, Lysates from primary cortical neurons were transfected with Cdk5-specific siRNA (01, 02, 03) and control siRNA (NC) on DIV 3. On DIV 7, the cytosolic and nuclear extracts were isolated and immunoblotted with the indicated antibodies. G, Cdk5+/+ and Cdk5−/− neurons were transfected with the 3xIRS luciferase reporter system on DIV 3. On DIV 7, luciferase activities for 3xIRS were measured and analyzed. Data are mean ± SEM; n = 5. *p < 0.05. The protein level of Cdk5 in lysates was also provided. H, Primary cortical neurons (DIV 3) were transfected with plasmids or specific siRNA targeting Cdk5 as indicated. On DIV 7, luciferase activities for 3xIRS were measured and analyzed. Data are mean ± SEM; n = 5. *p < 0.05. The protein level of Cdk5 in lysates was also provided.
We next investigated whether the dephosphorylation of S249 on FOXO1 is involved in any of these events. It is worth noting that the phosphomimetic FOXO1-S249D mutant retained significant cytoplasmic localization in Cdk5-deficient neurons. When we transfected the Cdk5-inactive FOXO1 mutant (S249A) into Cdk5-deficient neurons, it localized to the nucleus as did the wild-type FOXO1 (Fig. 7A); and knockdown Cdk5 also reduced the interaction between FOXO1 with 14-3-3β (Fig. 7B). The activities of wild-type FOXO1 or FOXO1-S249A on the 3xIRS-luciferase reporter gene and a reporter gene controlled by the Bim promoter were both increased by Cdk5 knockdown (Fig. 7C,D). These effects may be due in part to the inactivation of AKT under conditions of Cdk5 deficiency. First, increasing Cdk5 activity (overexpression of Cdk5/p35 or Cdk5/p25) did not affect the activation of AKT as assessed by the phosphorylation of S473 and the phosphorylation of FOXO1 on S319 (one of the phosphorylation targets of AKT) (Fig. 7E,F). This suggests that active Cdk5 promotes the cytoplasmic translocation of FOXO1 through mechanisms independent of AKT regulation. However, when Cdk5 was silenced by shRNA, phosphorylation of FOXO1 at the AKT site (S319) was significantly decreased (Fig. 7G,H). Consistent with the in vitro observation, phosphorylation at S473 of AKT and S319 of FOXO1 was significantly decreased in Cdk5−/− brain lysates (Fig. 7I,J). As expected, the phosphorylation at S249 of FOXO1 was also decreased in Cdk5−/− brain lysates (Fig. 7I,J). Thus, it would appear that the nuclear accumulation of FOXO1 under conditions of Cdk5 deficiency is in part mediated by inhibition of AKT. These effects are summarized in Table 1. Inhibition of Cdk5, either genetically or pharmacologically, permits the nuclear accumulation of FOXO1. Activation of Cdk5 promotes FOXO1 cytoplasmic localization even if AKT is inhibited.
Figure 7.
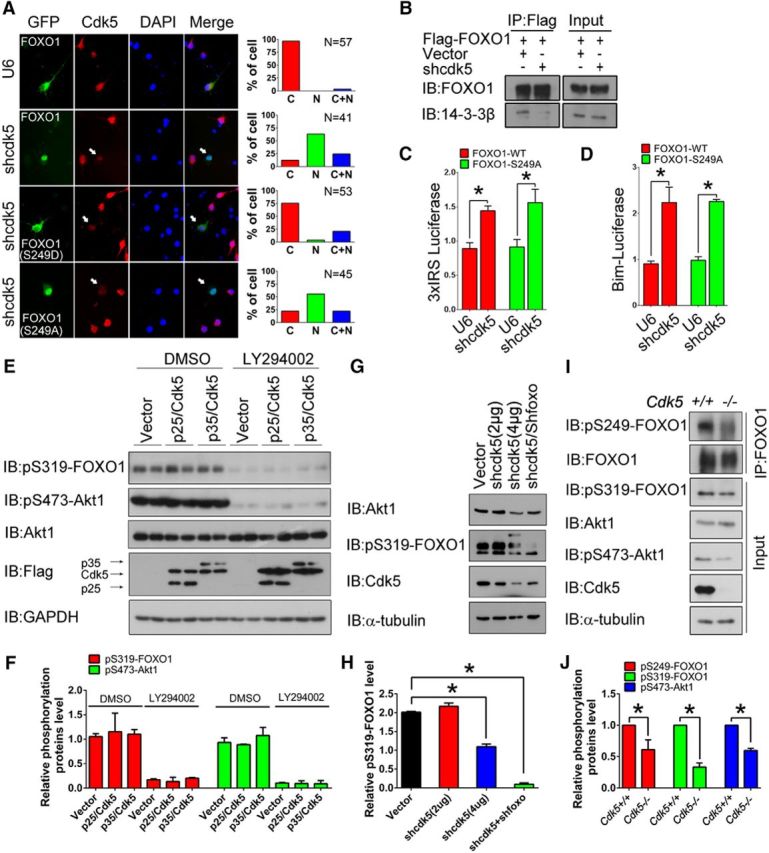
Cdk5 deficiency induces nuclear accumulation of FOXO1 by inactivating AKT. A, Primary cortical neurons were transfected with control U6 or shcdk5 plasmid together with GFP-FOXO1-WT, GFP-FOXO1–S249D, or GFP-FOXO1–S249A on DIV 3, then immunostained with Cdk5 antibody on DIV 7. The subcellular localizations of GFP-FOXO1-WT, GFP-FOXO1–S249D, or GFP-FOXO1–S249A were measured in neurons displaying low Cdk5 expression; arrows indicate shcdk5 effective neurons. Representative images are shown on the left: green represents GFP; red represents Cdk5; blue represents DAPI. Quantification is shown on the right. B, Lysates of 293T cells transfected with Flag-FOXO1 together with control U6 or shcdk5 plasmid were immunoprecipitated with the Flag antibody and immunoblotted with the FOXO1 and 14-3-3β antibodies. C, D, Primary cortical neurons transfected with the GFP-FOXO1-WT or GFP-FOXO1–S249A plasmid together with the control U6 or shcdk5 plasmid plus either the 3xIRS reporter (C) or Bim reporter gene (D). Neurons were subjected to luciferase assays. Data are mean ± SEM; n = 5. *p < 0.05. E, F, N2a cells were transfected with Cdk5/p25, Cdk5/p35, or control Flag vectors. Forty-eight hours after transfection, the cells were treated with DMSO or 50 μm LY294002 for another 12 h. The lysates were immunoblotted with the indicated antibodies. F, The relative levels of pS319-FOXO1 and pS473-Akt1 were quantified. Data are mean ± SEM; n = 3. G, H, N2a cells were transfected with shRNA targeted to either Cdk5 or foxo1. Scrambled shRNA was used as a control. Forty-eight hours after transfection, the lysates were immunoblotted with the indicated antibodies. H, The relative level of pS319-FOXO1 was quantified. Data are mean ± SEM; n = 5. *p < 0.05. I, J, Brain lysates from the cortices of E16.5 Cdk5+/+ and Cdk5−/− mice were prepared, and the levels of pS249-FOXO1 were detected following immunoprecipitation with FOXO1 antibody. The other proteins were measured by directly immunoblotting with the indicated antibodies. J, The relative levels of pS249-FOXO1, pS319-FOXO1, and pS473-Akt1 were quantified. Data are mean ± SEM; n = 3. *p < 0.05.
Table 1.
Summary of FOXO1 localization in primary cortical neuronsa
| FOXO1 | 249A | 249D | AAA | AAA-249A | AAA-249D | |
|---|---|---|---|---|---|---|
| Cdk5+/+ | C | C | C | N | N | C + N |
| Cdk5−/− | C + N | C + N | C | N | N | C + N |
| Roscovitine | C + N | C | C | N | N | C + N |
| Cdk5/p35 or Cdk5/p25 | C | C | C | C + N | N | C + N |
| Cdk5-KD/p25 or Cdk5-KD/p35 | C | C | C | N | N | C + N |
| LY294002 | N | N | C + N | N | N | C + N |
| Cdk5/p35 or Cdk5/p25 + LY294002 | C + N | N | C + N | |||
| Cdk5-KD/p35 or Cdk5-KD/p25 + LY294002 | N |
aThe indicated plasmids were transfected into Cdk5+/+ or Cdk5−/− neurons on DIV 3; drug treatment was administered if desired on DIV 5. The localizations of FOXO1 were detected on DIV 7.
With respect to cell cycle regulation, we have previously shown that Cdk5 is vital for keeping postmitotic neurons from reentering the cell cycle (Zhang and Herrup, 2008, 2011; Zhang et al., 2010b, 2012). To pursue these findings further, we cultured cortical neurons from Cdk5−/− mice for 7 DIV. As expected, spontaneous neuronal death and cell cycle reactivation were both detected. As shown in Figure 8A, Cdk5-deficient postmitotic neurons (TuJ1-positive) exhibit two distinct phenotypes: neuronal cell cycle reactivation (BrdU-positive) and neuronal death (activated caspase-3). We previously reported that Cdk5 negatively regulates E2F1, blocking neuronal cell cycle reentry; however, little is known regarding the molecular mechanism underlying neuronal death phenotype in Cdk5−/− neurons. To address this, we silenced foxo1 and Cdk5 in primary neurons and examined effects on neuronal viability and BIM expression. We found that silencing of FOXO1 markedly diminished neuronal cell death under Cdk5 deficiency conditions by decreasing BIM expression (Fig. 8B,D,E). To further interrogate this pathway, we performed rescuing experiments by forced expression of various silencing-resistant forms of FOXO1. We found that Cdk5 deficiency-induced apoptosis was restored by expression of silencing-resistant FOXO1 proteins WT-Res or S249A-Res, but not cytoplasmic localized S249D-Res (Fig. 8C,D). Together, these results indicate that neuronal apoptosis induced by the loss of Cdk5 is indeed mediated by the nuclear localization of FOXO1.
Figure 8.
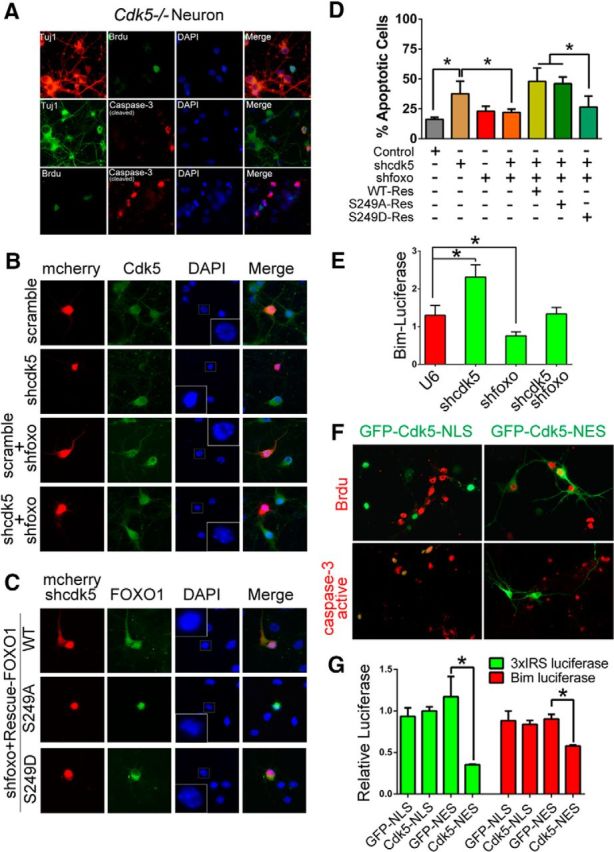
Interaction of Cdk5-dependent cell cycle and cell death activities. A, Cleaved-caspase-3 and BrdU incorporation was double stained on DIV 7 Cdk5−/− neurons. TuJ1 staining serves as neuronal cell marker. B–D, Effects of expressing mutant or wild-type FOXO1 on cortical neuronal apoptosis. B, Primary cortical neurons (DIV 3) were transfected with CMV-mCherry-IRES-U6-shcdk5 or control CMV-mCherry-IRES-U6-scramble vector either alone or together with U6-shfoxo plasmid. Cells were then immunostained with Cdk5 antibody at DIV 7. DAPI was used to counterstain nuclei to measure the neuronal death by nuclei condensation. C, Primary cortical neurons (DIV 3) were transfected with CMV-mCherry-IRES-U6-shcdk5 and shfoxo1 vector together with GFP-FOXO1-WT-Res, GFP-FOXO1–S249A-Res, or GFP-FOXO1–S249D-Res plasmid. The cells were fixed on DIV 7 and stained with DAPI to evaluate neuronal death by nuclei condensation. D, The quantification of neuronal apoptotic rate in B, C is presented in D. Data are mean ± SEM; n = 3. *p < 0.05. F, Primary cortical Cdk5−/− neurons were transfected with GFP-Cdk5-NLS or GFP-Cdk5-NES. Cleaved-caspase-3 and BrdU incorporation was detected by immunostaining. E, G, Primary cortical neurons were transfected with the indicated plasmids together with the 3xIRS-luciferase or Bim-luciferase reporter gene. The neurons were subjected to luciferase assays on DIV 7. Data are mean ± SEM; n = 5. *p < 0.05.
Our previous data demonstrated that forcing a nuclear localization for Cdk5 (with an NLS) can block neuronal cell cycle reactivation but cannot stop apoptosis (Zhang et al., 2008, 2011). We find that cytoplasmic Cdk5 attenuates the neuronal death but has no effect on the cell cycle reactivation in Cdk5−/− postmitotic neurons (Fig. 8F). This cytoplasmic effect proved to be tightly correlated with FOXO1 behavior. Further, increasing Cdk5 levels in cytoplasm (Cdk5-NES) significantly decreased the FOXO1-dependent transcription of 3xIRS- and Bim reporters, whereas nuclear Cdk5 (Cdk5-NLS) was ineffective in this assay (Fig. 8G).
Discussion
Cdk5 is an atypical cyclin-dependent kinase that acts like a double-edged sword with respect to the regulation of neuronal survival. Under conditions such as oxidative stress or after challenge with an increased β-amyloid burden in AD, enhanced levels of calcium activate calpain. This drives the cleavage of p35 to p25 and reportedly hyperactivates Cdk5, resulting in abnormal phosphorylation of its substrates and eventually neuronal death (Patrick et al., 1999; Ahlijanian et al., 2000; Liu et al., 2003; Cruz and Tsai, 2004). Yet other reports have demonstrated that Cdk5 activity is indeed vital for neuronal survival. Although the two effects are not mutually exclusive, our laboratory and others have found that Cdk5-deficient neurons are more vulnerable than wild-type cells to DNA-damaging agents and to excitotoxic neuronal death (Cicero and Herrup, 2005; O'Hare et al., 2005; Turner et al., 2008; Zhang et al., 2008). Until now, the detail mechanism behind this vulnerability remained unclear. The data reported here provide fresh insights into the functions of both Cdk5 and FOXO1 in the survival of postmitotic neurons.
FOXO1 is normally maintained in the neuronal cytoplasm through its interaction with the 14-3-3β protein. This interaction requires AKT-mediated FOXO1 phosphorylation on amino acids T24, S256, and S319 (Arden, 2004). The involvement of Cdk5 in the activation of AKT has been previously reported by Li et al. (2003) and Luo et al. (2014) who demonstrated that the phosphorylation S473 on AKT (the activation site of AKT) was significantly decreased in Cdk5-deficient mouse brains. Consistent with their data, we find that suppression of Cdk5 downregulates AKT activity (Fig. 7), which allows FOXO1 to dissociate from the 14-3-3β protein, permitting FOXO1 to translocate to the nucleus. We postulate that AKT determines the localization of FOXO1when Cdk5 activity is abolished. In the absence of Cdk5, FOXO1 is dephosphorylated and freed from its cytoplasmic location. Once it translocates to the nucleus, it triggers the translation of Bim and hence drives the apoptotic process.
Nevertheless, Cdk5 can also regulate cytoplasmic localization of FOXO1 through direct phosphorylation of FOXO1 at Ser249 independent of AKT (Fig. 4; Table 1). Our data thus emphasize the complex nature of the interaction between Cdk5 and AKT in controlling FOXO1 localization and hence its activity: Cdk5 phosphorylation of S249 on FOXO1 is sufficient for its cytoplasmic retention, whereas the phosphorylation of FOXO1 by AKT is necessary for its nuclear localization.
Cyclin-dependent kinases are the catalytic subunits of a family of serine/threonine kinases that have been implicated in the control of cell-cycle progression and neuronal function (Malumbres and Barbacid, 2005). Cdk2 is reported to phosphorylate FOXO1 at Ser249 in tumor cells. This phosphorylation by Cdk2 resulted in FOXO1 cytoplasmic localization and its inhibition (Huang et al., 2006). Cdk1, another classic Cdk, has also been reported to phosphorylate FOXO1 at Ser249 when it is overexpressed in cerebellar granule cells. These conflicting results suggest caution in generalizing the effects of Cdk kinases on FOXO1 activity. In our model system, we have compared the effect of several Cdk/cyclin complexes on FOXO1. Overexpression of active Cdk1/cyclin B did increase FOXO1-dependent transcription, whereas Cdk2/cyclin A decreased FOXO1 activity as Cdk5/p35 and Cdk5/p25 did (Fig. 1). The paradoxical results between cyclin Cdk1/cyclin B and Cdk5/p25 on IRS luciferase are unlikely due to cytotoxicity because we did not observe any significant difference in cell death between cells overexpressing Cdk1/cyclin B or Cdk5/p25 (data not shown). These data emphasize that, even though all Cdk family members share the same phosphorylation motif, their choice of specific substrate is likely to be more complex. One theoretical advantage of this arrangement is that, given their potency in driving the cell cycle, Cdk1 and Cdk2 activity must be maintained at very low levels in cell cycle quiescent cells, such as postmitotic neurons. By contrast, Cdk5, which is incapable of driving cell cycle activity, can stay high and realize its full benefit in the support of neuronal maturation and survival. Indeed, as we and others have shown, its presence in the nucleus actually serves to keep neurons out of the cell cycle (Jessberger et al., 2008, 2009; Zhang et al., 2008, 2010a). From this perspective, it makes sense for Cdk5 to assume the major role as FOXO1 regulator in postmitotic neurons.
This study also contributes to our understanding of two characteristics of Cdk5−/− postmitotic neurons: spontaneous neuronal death and neuronal cell cycle reactivation. In adult neurons, cell cycle reactivation will induce neuronal death, but this cell cycle-related neuronal death is a slow progress and is distinct from classic neuronal apoptosis (Fig. 8A) (Herrup and Yang, 2007). Our new data clearly demonstrated that silencing of foxo1 by shRNA markedly diminished neuronal cell death (Fig. 8B,D). More importantly, Cdk5 deficiency-induced apoptosis was restored by forced expression of silencing-resistant FOXO1 constructs (FOXO1-WT-Res or FOXO1-S249A-Res), whereas FOXO1-S249D-Res failed to restore the neuronal apoptosis when both Cdk5 and FOXO1 were silenced. We showed that FOXO1-S249D-Res retained in the cytoplasm, which limited the activation of BIM expression in Cdk5-deficient neurons, thus attenuating neuronal death (Fig. 8C,D).
The distribution of Cdk5 in nucleus and cytoplasm correlates with the cell cycle reactivation and neuronal death, respectively. Extranuclear Cdk5 suppresses the cell cycle reactivation, but it does not attenuate the quick neuronal death (Zhang and Herrup, 2011; Zhang et al., 2010a, b). As shown in this current work, cytoplasmic Cdk5 blocks apoptosis by inactivating FOXO1 to decrease BIM levels but has no affect on the cell cycle reentry in Cdk5−/− neurons (Fig. 8G).
In conclusion, our data demonstrate that Cdk5 is a key to the cytoplasmic maintenance of FOXO1, keeping it inactive and thus protecting postmitotic cortical neurons from neuronal death. These findings offer important new insights into the roles that Cdk5 plays in the adult CNS and have implications for the etiology of neurodegenerative diseases, such as AD.
Footnotes
This work was supported by Xiamen University 985 Project, National Science Foundation of China Grant 81271421 to J. Zhang and Grants 91332114 and U1405222 to H.X., Natural Science Foundation of Fujian Province of China Grant 2013J01147 and Grant 2014J06019 to J. Zhang, Program for New Century Excellent Talents in University, Hong Kong University of Science and Technology, National Key Basic Research Program of China 2013CB530900, and Research Grants Council HKSAR HKUST12/CRF/13G and GRF660813.
The authors declare no competing financial interests.
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden KC. FoxO: linking new signaling pathways. Mol Cell. 2004;14:416–418. doi: 10.1016/S1097-2765(04)00213-8. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/S0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. Cdk5: mediator of neuronal death and survival. Neurosci Lett. 2004;361:47–51. doi: 10.1016/j.neulet.2003.12.117. [DOI] [PubMed] [Google Scholar]
- Cicero S, Herrup K. Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci. 2005;25:9658–9668. doi: 10.1523/JNEUROSCI.1773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr Opin Neurobiol. 2004;14:390–394. doi: 10.1016/j.conb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Essafi A, Fernández de Mattos S, Hassen YA, Soeiro I, Mufti GJ, Thomas NS, Medema RH, Lam EW. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene. 2005;24:2317–2329. doi: 10.1038/sj.onc.1208421. [DOI] [PubMed] [Google Scholar]
- Gan L, Zheng W, Chabot JG, Unterman TG, Quirion R. Nuclear/cytoplasmic shuttling of the transcription factor FoxO1 is regulated by neurotrophic factors. J Neurochem. 2005;93:1209–1219. doi: 10.1111/j.1471-4159.2005.03108.x. [DOI] [PubMed] [Google Scholar]
- Gilmore EC, Herrup K. Neocortical cell migration: GABAergic neurons and cells in layers I and VI move in a cyclin-dependent kinase 5-independent manner. J Neurosci. 2001;21:9690–9700. doi: 10.1523/JNEUROSCI.21-24-09690.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawasli AH, Bibb JA. Alternative roles for Cdk5 in learning and synaptic plasticity. Biotechnol J. 2007;2:941–948. doi: 10.1002/biot.200700093. [DOI] [PubMed] [Google Scholar]
- Hellmich MR, Pant HC, Wada E, Battey JF. Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci U S A. 1992;89:10867–10871. doi: 10.1073/pnas.89.22.10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci. 2007;8:368–378. doi: 10.1038/nrg2084. [DOI] [PubMed] [Google Scholar]
- Hisanaga S, Endo R. Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J Neurochem. 2010;115:1309–1321. doi: 10.1111/j.1471-4159.2010.07050.x. [DOI] [PubMed] [Google Scholar]
- Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–297. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Aigner S, Clemenson GD, Jr, Toni N, Lie DC, Karalay O, Overall R, Kempermann G, Gage FH. Cdk5 regulates accurate maturation of newborn granule cells in the adult hippocampus. PLoS Biol. 2008;6:e272. doi: 10.1371/journal.pbio.0060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Gage FH, Eisch AJ, Lagace DC. Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci. 2009;32:575–582. doi: 10.1016/j.tins.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villén J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC. Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J. 2002;21:324–333. doi: 10.1093/emboj/21.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC. Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem. 2003;278:35702–35709. doi: 10.1074/jbc.M302004200. [DOI] [PubMed] [Google Scholar]
- Liu F, Su Y, Li B, Zhou Y, Ryder J, Gonzalez-DeWhitt P, May PC, Ni B. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003;547:193–196. doi: 10.1016/S0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- Luo F, Burke K, Kantor C, Miller RH, Yang Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3beta signaling. J Neurosci. 2014;34:10415–10429. doi: 10.1523/JNEUROSCI.0710-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression: implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277:47928–47937. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- O'Hare MJ, Kushwaha N, Zhang Y, Aleyasin H, Callaghan SM, Slack RS, Albert PR, Vincent I, Park DS. Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J Neurosci. 2005;25:8954–8966. doi: 10.1523/JNEUROSCI.2899-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Gilmore EC, Longenecker G, Jacobowitz DM, Brady RO, Herrup K, Kulkarni AB. Migration defects of cdk5(−/−) neurons in the developing cerebellum is cell autonomous. J Neurosci. 1999;19:6017–6026. doi: 10.1523/JNEUROSCI.19-14-06017.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Real PJ, Benito A, Cuevas J, Berciano MT, de Juan A, Coffer P, Gomez-Roman J, Lafarga M, Lopez-Vega JM, Fernandez-Luna JL. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res. 2005;65:8151–8157. doi: 10.1158/0008-5472.CAN-05-1134. [DOI] [PubMed] [Google Scholar]
- Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Ohshima T, Hirasawa M, Pareek TK, Bugge TH, Morozov A, Fujieda K, Brady RO, Kulkarni AB. Conditional deletion of neuronal cyclin-dependent kinase 5 in developing forebrain results in microglial activation and neurodegeneration. Am J Pathol. 2010;176:320–329. doi: 10.2353/ajpath.2010.081158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Veeranna, Ohshima T, Rajan P, Amin ND, Cho A, Sreenath T, Pant HC, Brady RO, Kulkarni AB. Neuronal cyclin-dependent kinase 5 activity is critical for survival. J Neurosci. 2001;21:550–558. doi: 10.1523/JNEUROSCI.21-02-00550.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–1377. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380:297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Hao Y, Tao L, Peng S, Rao C, Chen H, You H, Dong MQ, Yuan Z. Lysine methylation of FOXO3 regulates oxidative stress-induced neuronal cell death. EMBO Rep. 2012;13:371–377. doi: 10.1038/embor.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y, Schaefer EM, Bonni A. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science. 2008;319:1665–1668. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Lehtinen MK, Merlo P, Villén J, Gygi S, Bonni A. Regulation of neuronal cell death by MST1-FOXO1 signaling. J Biol Chem. 2009;284:11285–11292. doi: 10.1074/jbc.M900461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Herrup K. Cdk5 and the non-catalytic arrest of the neuronal cell cycle. Cell Cycle. 2008;7:3487–3490. doi: 10.4161/cc.7.22.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Herrup K. Nucleocytoplasmic Cdk5 is involved in neuronal cell cycle and death in post-mitotic neurons. Cell Cycle. 2011;10:1208–1214. doi: 10.4161/cc.10.8.15328. [DOI] [PubMed] [Google Scholar]
- Zhang J, Cicero SA, Wang L, Romito-Digiacomo RR, Yang Y, Herrup K. Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc Natl Acad Sci U S A. 2008;105:8772–8777. doi: 10.1073/pnas.0711355105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li H, Herrup K. Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation. J Biol Chem. 2010a;285:14052–14061. doi: 10.1074/jbc.M109.068262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li H, Yabut O, Fitzpatrick H, D'Arcangelo G, Herrup K. Cdk5 suppresses the neuronal cell cycle by disrupting the E2F1-DP1 complex. J Neurosci. 2010b;30:5219–5228. doi: 10.1523/JNEUROSCI.5628-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li H, Zhou T, Zhou J, Herrup K. Cdk5 levels oscillate during the neuronal cell cycle: Cdh1 ubiquitination triggers proteosome-dependent degradation during S-phase. J Biol Chem. 2012;287:25985–25994. doi: 10.1074/jbc.M112.343152. [DOI] [PMC free article] [PubMed] [Google Scholar]