

Key Points
BA reduces MYC, CDK4/6, nuclear RelA, and BTK expression and is synergistically lethal with ibrutinib in MCL cells.
Cotreatment with BA and inhibitor of BCL2, CDK4/6, or histone deacetylases is synergistically lethal against ibrutinib-resistant MCL cells.
Abstract
Mantle cell lymphoma (MCL) cells exhibit increased B-cell receptor and nuclear factor (NF)-κB activities. The bromodomain and extra-terminal (BET) protein bromodomain 4 is essential for the transcriptional activity of NF-κB. Here, we demonstrate that treatment with the BET protein bromodomain antagonist (BA) JQ1 attenuates MYC and cyclin-dependent kinase (CDK)4/6, inhibits the nuclear RelA levels and the expression of NF-κB target genes, including Bruton tyrosine kinase (BTK) in MCL cells. Although lowering the levels of the antiapoptotic B-cell lymphoma (BCL)2 family proteins, BA treatment induces the proapoptotic protein BIM and exerts dose-dependent lethality against cultured and primary MCL cells. Cotreatment with BA and the BTK inhibitor ibrutinib synergistically induces apoptosis of MCL cells. Compared with each agent alone, cotreatment with BA and ibrutinib markedly improved the median survival of mice engrafted with the MCL cells. BA treatment also induced apoptosis of the in vitro isolated, ibrutinib-resistant MCL cells, which overexpress CDK6, BCL2, Bcl-xL, XIAP, and AKT, but lack ibrutinib resistance-conferring BTK mutation. Cotreatment with BA and panobinostat (pan-histone deacetylase inhibitor) or palbociclib (CDK4/6 inhibitor) or ABT-199 (BCL2 antagonist) synergistically induced apoptosis of the ibrutinib-resistant MCL cells. These findings highlight and support further in vivo evaluation of the efficacy of the BA-based combinations with these agents against MCL, including ibrutinib-resistant MCL.
Introduction
Among the genetic alterations described in mantle cell lymphoma (MCL) cells are those that involve p53, cyclin-dependent kinase (CDK)4, CDKN2A, MYC, B-cell lymphoma (BCL)2, B-cell receptor (BCR), and nuclear factor (NF)-κB signaling genes.1-3 These genetic alterations confer a cell autonomous pro-growth and pro-survival advantage on the MCL cells, which is especially dependent on NF-κB, BCL2, and MYC activities.2-4 Next generation sequencing has also disclosed new targets for therapeutic intervention in the deregulated molecular signaling through BCR, toll-like receptor, NOTCH, NF-κB, and mitogen-activated protein kinase signaling pathways in the MCL cell lines and patient-derived primary MCL.3-7 Pre-clinical and clinical studies have shown that ibrutinib, a selective, orally bioavailable, irreversible inhibitor of Bruton tyrosine kinase (BTK) in the BCR, also inhibits NF-κB activity and is active against B-cell neoplasms, including chronic lymphocytic leukemia (CLL) and MCL.6,8 Ibrutinib has demonstrated impressive clinical efficacy and is approved for the treatment of CLL and MCL.9-11 Despite its high level of clinical activity, primary or acquired clinical resistance to ibrutinib therapy is commonly observed.11-14 Similar to what has been described in CLL cells, a cysteine-to-serine (C481S) mutation in BTK at the binding site of ibrutinib, which results in a protein that is only reversibly inhibited by ibrutinib, has also been documented in MCL patients who relapsed while on ibrutinib.12-14 However, none of these ibrutinib resistance-associated mutations were detectable in the primary pre-ibrutinib treatment MCL tumor samples.15 Instead, mutations in MLL2, CREBBP, PIM1, and ERB4 were detected in the ibrutinib-refractory MCL cells.13,15 Additionally, as compared with the cell lines sensitive to ibrutinib exhibiting chronic activity of the classical NF-κB signaling pathway, ibrutinib-resistant MCL cell lines and primary MCL cells exhibited mutations in TRAF2/3 and MAP3K14 (NF-κB inducing kinase), activating the alternative NF-κB signaling, which would still show dependency on the NF-κB–activated “transcriptome” for growth and survival.7,16 The deregulated transcriptome in these cells would also be governed by the genetic alterations and epigenetic mechanisms that control the expressions of MYC, BCL2, and the G1 checkpoint proteins.3,7,16,17
Acetylation-deacetylation of the histone proteins regulates the transcriptome in transformed cells.18 The bromodomain and extra-terminal (BET) family of “reader” proteins, including bromodomain (BRD)2, BRD3, and BRD4 recognize and bind to the acetylated lysine residues on the histone proteins associated with the open, transcriptionally permissive chromatin through their amino-terminal double, tandem, 110 amino acids-long BRDs.19-21 BET proteins also contain the extra-terminal protein-interacting domain in the carboxyl (C) terminus, which assembles a complex of coregulatory proteins at the enhancers and promoters, thereby regulating gene transcription.20,21 The C-terminal positive transcription elongation factor b (pTEFb)-interacting domain of BRD4 interacts with and recruits the to the “super-enhancers” and promoters, thereby regulating the activity of RNA pol II (RNAP2) and gene expressions of important MCL-relevant oncogenes.21-24 Among these are MYC, CDK4/6, cyclin D1, and BCL-2, which control the proliferation and survival of MCL cells.22-24 pTEFb, which is a heterodimer composed of cyclin T and CDK9, phosphorylates Ser-2 on the heptad repeats of the C-terminal domain (CTD) in the stalled RNAP2 at the transcriptional start sites, enabling the pause-release of RNAP2 and inducing productive messenger RNA (mRNA) transcript elongation.24-28 Thus, by promoting the availability of active pTEFb, BRD4 couples histone acetylation to transcript elongation, especially of the MCL-relevant oncogenes c-MYC, cyclin D1, BCL-2, and CDK6.21-24 BRD4 is also essential for the transcriptional activity of NF-κB triggered by the BCR signaling.29,30 BRD4 has also been shown to bind to the acetylated RelA and mediate the transcriptional activity of NF-κB.30 Several structure/activity-based BET protein bromodomain antagonists (BAs) have been developed, including JQ1 and I-BET151, which displace the BET proteins, including BRD4 and the associated pTEFb from the acetylated chromatin.31-33 This results in the transcriptional repression of BCL-2, c-MYC, cyclin D1, and CDK6, as well as induces growth arrest and apoptosis of leukemia cells.34-36 Pertinent to this, we had previously reported that cotreatment with JQ1 and histone deacetylase inhibitor (HDI) is synergistically lethal against acute myeloid leukemia (AML) cells.36,37 In the present study, we demonstrate the molecular basis of the activity of BAs against human MCL cells. Additionally, we demonstrate that cotreatment with BA and ibrutinib exerts superior in vitro and in vivo efficacy against MCL cells. We also demonstrate that against ibrutinib-resistant MCL cells, the combination of BA with HDI, CDK4/6 inhibitor, or BCL2 antagonist exerts synergistic lethality, which has therapeutic implication for developing effective combination therapies to achieve long-term control of MCL.
Materials and methods
Reagents
(S)-JQ1 (active enantiomer, hereafter referred to as JQ1) and its inactive enantiomer (R)-JQ1 were developed as previously described.31 Panobinostat (PS) was kindly provided by Novartis Pharmaceuticals, Inc. (East Hanover, NJ). Ibrutinib, palbociclib, ABT-199, ABT737, and carfilzomib were obtained from Selleck Chemicals (Houston, TX). I-BET151, an orally available, imidazolonoquinoline-based inhibitor of the BET family of BRD-containing proteins was obtained from Xcessbio (San Diego, CA). All antibodies were obtained from commercial sources. Detailed antibody information is provided in the supplemental Methods, available on the Blood Web site.
Cell lines and cell culture
MCL cell lines MO2058, JeKo-1, Z-138, and Mino were obtained and maintained as previously described.38 HS5 cells were obtained from American Type Culture Collection (Manassas, VA). HK stromal cells were obtained and maintained as previously described.39
Assessment of cell proliferation and percentage of nonviable MCL cells
MCL cells (MO2058, Mino, JeKo-1, and Z138) were plated in 24-well plates (0.25 × 106/mL) and treated with vehicle or JQ1 for 120 hours. Total cell numbers were measured in triplicate every 24 hours with a coulter counter. Cells treated with JQ1 and/or ibrutinib were assessed for cell viability by propidium iodide (PI) staining and flow cytometry. All of these studies were performed as previously described.38,39
RNA isolation and reverse transcription-polymerase chain reaction (PCR)
RNA was extracted from the cultured MCL cells using an RNAqueous-4 PCR Kit (Applied Biosystems, Foster City, CA). Purified total RNA was quantified, reverse transcribed, and real-time quantitative PCR (qPCR) analyses were performed on the resulting complementary DNA (cDNA) utilizing TaqMan probes from Applied Biosystems (Foster City, CA) as previously described.39 Relative mRNA expression in each treatment was normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or Actin in each treatment and to the untreated control.
Assessment of apoptosis of MCL cells
To analyze synergism between JQ1 and PS or JQ1 and ibrutinib in inducing apoptosis, cells were treated at a constant ratio of the drugs for 48 hours. The percentages of apoptotic cells were determined by flow cytometry.38 The combination index (CI) for each drug combination was obtained by median dose effect of Chou and Talalay40 utilizing the CI equation (assuming mutual exclusivity) within the commercially available software CalcuSyn (Biosoft, Ferguson, MO). CI values <1.0 indicates a synergistic interaction of the two drugs in the combination.
Chromatin immunoprecipitation (ChIP) and qPCR
MO2058 cells were treated with JQ1 for 16 hours. ChIP and qPCR was performed as previously described.38
MCL xenograft
Mino cells (5 × 106 cells/mouse) were injected into the lateral tail vein of nonobese diabetic/severe combined immunodeficiency (NOD/SCID) or NOD/SCID Gamma mice that had received a preconditioning dose of radiation (2.5 gray) 24 hours prior to the injection of cells. A detailed description is provided in supplemental Methods. The survival of mice is represented by a Kaplan–Meier survival plot.
Statistical analysis
Significant differences between values obtained in a population of MCL cells treated with different experimental conditions were determined using the Student t test. For the in vivo mouse models, a two-tailed Student t test or a Mantel–Cox rank sum test was used for group comparisons. P values of < .05 were assigned significance.
Results
BA-mediated growth inhibition and lethality of MCL cells
We first determined the effects of the BA JQ1 on growth and survival of cultured and primary MCL cells. JQ1 treatment increased the percentage of G1 and inhibited the percentage of S-phase cells in the cell cycle, as well as abrogated the suspension culture growth over 120 hours of the cultured MCL cells (Figure 1A-B and supplemental Figure 1A). Treatment with JQ1, but not the inactive enantiomer R-JQ1, also dose-dependently induced apoptosis of the cultured MCL cells (Figure 1C and supplemental Figure 1B). Treatment with I-BET151 also dose-dependently induced apoptosis of the cultured MCL cells (Figure 1D). Notably, treatment with JQ1 caused loss of viability of primary MCL cells purified from the lymph nodes of patients with MCL (Figure 1E). As shown in Figure 1F, coculture with the bone marrow (BM) stromal HS5 fibroblast reduced JQ1-induced apoptosis of the cultured MCL cells. Primary MCL cells cocultured with lymph node stromal HK cells also exhibited reduced cell death following treatment with JQ1 (Figure 1G).
Figure 1.

Treatment with the BET antagonists JQ1 and I-BET151 induces cell-cycle growth arrest and lethal effects in cultured MCL cells. (A) Cell-cycle status of MO2058 (left) and Mino (right) cells following 24 hours of treatment with JQ1, as indicated. Columns, mean of 3 independent experiments; Bars, ± standard error of the mean (SEM). (B) MO2058 and Mino cells were cultured in the presence or absence of 1.0 µM of JQ1 and cell counts were measured every 24 hours for 120 hours. Lines represent the mean cell number from 3 experiments ± standard deviation (SD). (C) MO2058, JeKo-1, Mino, and Z-138 cells were treated with the indicated concentrations of JQ1 for 48 hours. The percent of Annexin V-positive apoptotic cells was determined by flow cytometry. Columns, mean of 3 independent experiments; Bars, ± SEM. (D) MO2058 and JeKo-1 cells were treated with the indicated concentrations of I-BET151 for 48 hours. Annexin V-positive apoptotic cells were determined by flow cytometry. Columns, mean of 3 independent experiments; Bars, ± SEM. IC50 values were calculated using GraphPad Prism software (version 5). (E) Primary MCL cells were treated with the indicated concentrations of JQ1 for 48 hours. The percent of nonviable cells was determined by flow cytometry. Columns, mean percent loss of viability of 6 primary MCL samples; Bars, ± SEM. (F) MO2058 (left) and Mino (right) cells were cocultured with or without HS5 stromal cells and then treated with JQ1, as indicated, for 48 hours. The percent of apoptosis of the MO2058 and Mino or HS5 cells was determined by staining with Annexin V and TO-PRO-3 iodide and flow cytometry. Columns represent the mean apoptosis of 3 independent experiments; Bars, ± SEM. (G) Primary MCL cells were cocultured with or without HK stromal cells and then treated with JQ1 for 48 hours. The percent of nonviable cells was determined by PI staining and flow cytometry. Asterisk (*) indicates loss of viability values significantly less (P < .05) in cells cocultured with HK stromal cells compared with those without coculture.
Treatment with BA attenuates pro-growth and pro-survival while increasing the levels of growth inhibitory gene expressions in MCL cells
Next, we determined that treatment with JQ1 reduced the occupancy of BRD4 on the previously reported enhancer and promoter of MYC, and on the promoter of BCL2 in MO2058 cells (Figure 2A).36,37 This was also associated with a reduced occupancy of RNAP2 on the promoters of MYC and BCL2 (Figure 2B). Based on this, we evaluated the alterations in the mRNA levels in the MCL MO2058 cells, utilizing gene expression microarray analysis (Figure 2C and supplemental Figure 2A). Following treatment of MO2058 cells with JQ1 for 8 hours, the expression levels of c-MYC, CDK4, BCL2, and CDK6 declined, whereas hexamethylene bisacetamide-inducible protein (HEXIM1) levels increased (Figure 2C). In addition, the heat map of the gene expression alterations showed that the mRNA expression of more genes was increased, as compared with the number of genes whose mRNA expression was downregulated (supplemental Figure 2A). The fold-change of the 30 most altered mRNA expressions is shown in supplemental Table 1. Data sets of genes with the altered expression profile derived from the gene expression microarray analyses were imported into the Ingenuity Pathway Analysis (IPA) Tool (Ingenuity Systems, Redwood City, CA).39 Within this gene list, IPA identified the top 5 most perturbed gene networks in the MO2058 cells following treatment with JQ1 and assigned a score for these associated network functions (supplemental Table 2). The score (eg, a score of 57) assigned by the IPA indicates the probability (1 in 1057) that the focus-genes in the data set are grouped together in a perturbed network due to random chance alone. Additionally, the most differentially expressed genes were assessed for pathway enrichment utilizing KEGG/Reactome pathway analysis. The top 20 significantly altered pathways in MO2058 cells identified following JQ1 treatment is listed in supplemental Table 3. These two distinct analyses show similar pathway perturbations due to JQ1 treatment. Next, the qPCR analysis utilizing c-MYC, CDK6, and BCL2-specific TaqMan real-time PCR probes showed that JQ1 treatment attenuated the mRNA expression of c-MYC, CDK6, and BCL2 in the MO2058 cells (Figure 2D). In contrast, JQ1 treatment induced the mRNA expression of HEXIM1 in the MCL cells (vide infra). Similar effects of JQ1 were also observed in the MCL Mino cells (data not shown). Western analysis of the protein lysates showed that treatment with JQ1 also reduced the protein expressions of c-MYC, CDK4/6, and MCL1, while simultaneously inducing the protein expression of HEXIM1, p21, p27, and BIM in MO258 cells (Figure 2E). Similar effects of JQ1 or I-BET151 on the expression of these proteins were also documented on the cultured MCL JeKo-1 and Mino cells (supplemental Figure 2B-E and data not shown).
Figure 2.

Treatment with JQ1 reduces BRD4 and Pol II occupancy on the promoters of c-MYC and BCL2, and depletes the mRNA expression of c-MYC and BCL2 in human MCL cells. (A-B) MO2058 cells were treated with the indicated concentrations of JQ1 for 16 hours. Following this, ChIP was conducted with a BRD4-specific antibody (A) or RNAP2 antibody (B). The ChIP DNA was subjected to real-time qPCR with primers against the enhancer and promoter of c-MYC and the promoter of BCL2. The fold-change was calculated using the cycle threshold (Ct) value of the ChIP DNA compared with the Ct value of the input DNA. (C) MO2058 cells were treated with 1000 nM of JQ1 for 8 hours. Total RNA was extracted and used for gene expression analyses. A heat map of TNFAIP3, MYC, CDK4, BCL2, CDK6, and HEXIM1 is shown. (D) MO2058 cells were treated with the indicated concentrations of JQ1 for 16 hours. At the end of treatment, RNA was isolated and reverse transcribed. The resulting cDNA was used for real-time qPCR analysis of c-MYC, BCL-2, and CDK6. The relative mRNA expression was normalized to GAPDH and compared with the untreated cells. (E) Representative immunoblots of MO2058 cells treated with the indicated concentrations of JQ1 for 24 hours. Immunoblot analyses were conducted for the expression levels of c-MYC, MCL1, CDK4, CDK6, HEXIM1, p21, p27, BIM, and β-actin in the cell lysates.
BA treatment inhibits NF-κB activity and reduces BTK levels in the cultured MCL cells
Next, studies utilizing confocal immunofluorescence microscopy demonstrated that JQ1 treatment markedly attenuated the nuclear levels of RelA in the Mino and MO2058 cells (Figure 3A and supplemental Figure 3A). The levels of RelA were also reduced in the purified nuclear vs cytoplasmic protein extracts of JQ1-treated vs the control Mino cells (Figure 3B). Treatment with JQ1 dose-dependently inhibited the mRNA expression of a number of the known NF-κB activated, pro-growth and pro-survival genes, including tumor necrosis factor (TNF)AIP3, cFLIP, cIAP2, XIAP, BCL-xL, BMI, interleukin (IL)-10, PRDM1, and NF-κB2 (Figure 3C and supplemental Figure 3B). In contrast, JQ1 simultaneously induced the expression of HEXIM1 in the MCL cells (Figure 3C).6,7 Treatment with JQ1 also attenuated the protein levels of the antiapoptotic XIAP and Bcl-xL proteins in the MCL cells (Figure 3D and supplemental Figure 3C).41 BTK is also known to be activated by NF-κB and shuttles between the nucleus and cytoplasm.42,43 Consistent with this, JQ1-mediated NF-κB inhibition attenuated BTK levels both in the nucleus and cytoplasm (Figure 3B), as well as reduced the p-BTK levels in the Mino cells (Figure 3B,D). Similar effects of JQ1 were observed in MO2058 cells (Figure 3D). Treatment with I-BET151 also reduced BTK and p-BTK levels in Mino cells (supplemental Figure 3D). Notably, cotreatment with the proteasome inhibitor carfilzomib did not restore JQ1-mediated attenuation of BTK and MYC levels, thereby arguing against their degradation by the proteasome (supplemental Figure 3E).
Figure 3.
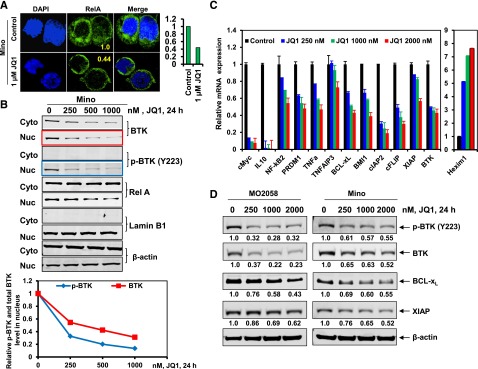
Treatment with BET antagonist reduces nuclear expression of RelA and BTK, and depletes mRNA expression of NF-kB target genes in MCL cells. (A) Confocal immunofluorescence analysis of RelA expression and cellular localization in Mino cells following treatment with JQ1 for 24 hours. Original magnification, ×63. The bar graph (right) shows quantification of the fluorescein isothiocyanate signal intensity of the JQ1-treated Mino cells relative to the untreated cells. (B) Mino cells were treated with the indicated concentrations of JQ1 for 24 hours (top). Following this, nuclear and cytoplasmic fractions were prepared and immunoblot analyses were conducted for BTK and RelA. The localization of Lamin B served as a fraction and loading control. The graph (bottom) shows the relative expression of p-BTK and BTK in the nucleus, determined by densitometry and normalized against the expression of Lamin B. Representative immunoblots are shown. (C) qPCR performed on cDNA from Mino cells treated with the indicated concentrations of JQ1 for 8 hours. Relative expression of each target was normalized against GAPDH. (D) Immunoblot analyses were conducted on the lysates of MO2058 (left) and Mino (right) cells treated with JQ1 for 24 hours, as indicated. The numbers beneath the bands represent densitometry analysis performed on the blots and normalized to the β-actin loading control. Cyto, cytoplasmic; DAPI, 4′,6 diamidino-2-phenylindole; Nuc, nuclear.
Cotreatment with BA and ibrutinib is synergistically lethal with concomitant depletion of BTK, nuclear RelA, and NF-κB activity in MCL cells
First, we confirmed that exposure to ibrutinib inhibited p-BTK levels and the downstream signaling, as well as attenuated the nuclear levels of RelA in MCL cells (supplemental Figure 4A-B).8,9 Ibrutinib (10 µM for 8 hours) inhibited the mRNA levels of the NF-κB–activated genes IL-10, TNF-α, TNFAIP3, IkBα, cIAP2, c-FLIP, PRDM1, Bcl-xL, and BMI1, and induced apoptosis of MCL cells (supplemental Figure 4C-D). Figure 4A demonstrates that, more so than treatment with each agent alone, cotreatment with JQ1 and ibrutinib markedly inhibited p-BTK, p-PLCγ2, and p-AKT levels in the MCL cells. Further, as compared with treatment with either agent alone, cotreatment with JQ1 and ibrutinib markedly reduced the nuclear levels of RelA in Mino cells (Figure 4B-C). A similar effect was also observed in MO2058 cells, along with a marked reduction in the mRNA levels of the NF-κB–activated genes (supplemental Figure 5A-C). Notably, combined treatment with JQ1 and ibrutinib synergistically induced apoptosis of the cultured MCL cells (with CI <1.0) (Figure 4D and supplemental Figure 6A). JQ1 and ibrutinib was synergistically lethal against primary MCL cells obtained from patients untreated with ibrutinib, where the CI values were lower than those observed in the cultured MCL cells (Figure 4E). This was not observed in CD19+ normal B cells (supplemental Figure 5D). In the primary MCL cells, cotreatment with JQ1 and ibrutinib also markedly reduced p-BTK and BTK levels (Figure 4F).
Figure 4.
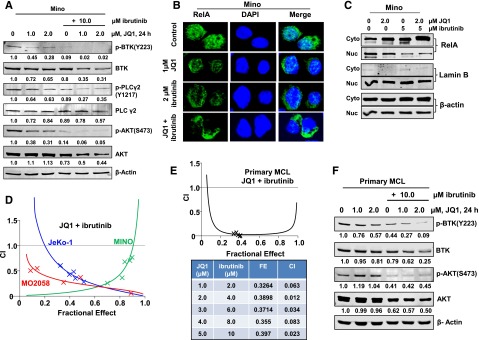
Compared with treatment with either agent alone, combined treatment with JQ1 and ibrutinib exerts synergistic lethal activity against cultured and primary MCL cells. (A) Representative immunoblots from Mino cells treated with JQ1 and/or ibrutinib, as indicated, for 24 hours. The numbers beneath the bands represent densitometry analysis performed on the blots and normalized to the β-actin loading control. (B) Mino cells were treated with JQ1 and/or ibrutinib for 24 hours. Confocal immunofluorescence microscopy was performed for RelA subcellular localization. Nuclei were stained with DAPI. Original magnification, ×63. (C) Nuclear and cytosolic fractions were prepared from Mino cells treated as indicated for 24 hours and the expression levels of RelA in each fraction were determined by immunoblot analyses. The localization of Lamin B served as a fraction and loading control. (D) MO2058, JeKo-1, and Mino cells were treated with JQ1 and ibrutinib at a constant ratio for 48 hours. The percent of apoptotic cells was determined by flow cytometry. Median dose effect and isobologram analyses were performed utilizing CalcuSyn. CI values <1.0 indicates a synergistic interaction of the two agents in the combination. Doses of drugs, fractional effect, and CI values are provided in supplemental Figure 6A. (E) Primary MCL cells were treated with JQ1 and ibrutinib at a constant ratio for 48 hours. The percent of nonviable cells was determined by flow cytometry. Median dose effect and isobologram analyses were performed utilizing CalcuSyn. CI values <1.0 indicates a synergistic interaction of the two agents in the combination. (F) Primary MCL cells were treated with the indicated concentrations of JQ1 and/or ibrutinib for 24 hours. Then, total cell lysates were prepared and immunoblot analyses were conducted as indicated. The numbers beneath the bands represent densitometry analysis performed on the blots and normalized to the β-actin loading control. Cyto, cytoplasmic; DAPI, 4′,6 diamidino-2-phenylindole; FE, fractional effect; Nuc, nuclear.
Superior in vivo activity of cotreatment with BA and ibrutinib against human MCL cells
We next determined the in vivo anti-MCL activity of JQ1 and/or ibrutinib against Mino cell xenografts in the NOD/SCID mice. Seven days after tail vein infusion and engraftment of Mino cells, treatment with vehicle alone or JQ1 and/or ibrutinib was started. In Figure 5A, the Kaplan–Meier plot depicting the survival of the mice demonstrates that, as compared with the treatment with vehicle alone, treatment with JQ1 or ibrutinib significantly improved the median survival of the mice (vehicle vs JQ1, P = .003, Student t test; vehicle vs ibrutinib, P = .0204, Student t test). However, cotreatment with JQ1 and ibrutinib significantly improved the median survival over treatment with each agent or the vehicle control alone (P = .0013, Mantel–Cox rank sum), with ∼40% of the mice treated with the combination surviving more than 50 days after the MCL cell engraftment (Figure 5A). In cohorts of 3 mice treated with the vehicle control vs treatment with JQ1 and/or ibrutinib for 5 days, the cell lysates of the BM and spleen MCL cells demonstrated that, as compared with treatment with each agent alone, cotreatment with JQ1 and ibrutinib markedly reduced the p-BTK, BTK, and p-PLCγ2 protein levels, while simultaneously increasing the levels of BIM (Figure 5B).
Figure 5.

Compared with either single agent alone, cotreatment with JQ1 and ibrutinib exerts superior in vivo anti-MCL activity against Mino xenografts. (A) NOD/SCID mice (n = 8 per cohort) were injected with Mino cells and monitored for 7 days. Following engraftment, mice were treated with JQ1 and/or ibrutinib for 3 weeks as described in supplemental Methods. Survival of the mice is represented by a Kaplan–Meier plot. Vehicle vs JQ1, P = .003, Student t test; vehicle vs ibrutinib, P = .0204, Student t test. Mantel–Cox rank sum of all groups, P .0013. (B) Immunoblot analyses conducted on the spleen and BM from NOD/SCID mice injected with Mino cells as above, and treated with JQ1 and/or ibrutinib for 1 week. Vertical line(s) have been inserted to indicate a repositioned gel lane. Ibr, ibrutinib; Veh, vehicle.
BA treatment is active and exerts synergistic lethality with HDIs, CDK4/6, and BCL2 against ibrutinib-resistant MCL cells
Next, we generated Mino/IR cells resistant to ibrutinib by culturing Mino cells in the continuous presence of escalating doses of ibrutinib. Mino/IR cells grow in the continuous presence of 30 µM of ibrutinib. Mino/IR and Mino cells displayed a similar growth rate, but Mino/IR cells exhibited significantly higher IC50 values for ibrutinib (Mino/IR: 160.6 µM vs Mino: 13.6 µM) (supplemental Figure 7A-B). Sequencing of the region around nucleotide 1443 (amino acid 481) of the BTK gene in the Mino/IR cells did not reveal any mutation in the BTK gene that had been previously discovered and reported (eg, C481S) following in vitro or in vivo exposure to ibrutinib.2,4,6,14 Notably, compared with Mino, Mino/IR cells expressed higher protein levels of BTK, TEC, AKT, CDK6, XIAP, Bcl-xL, and BCL2 levels, while simultaneously displaying lower NOXA, PUMA, and p21 levels (Figure 6A,C). In Mino/IR vs Mino cells, SOX11 expression was increased, whereas PAX5, IRF4, and BLIMP1 (PRDM1) expression levels were markedly reduced (Figure 6A).44 Compared with the expression observed in Mino cells, the Mino/IR cells lacked the expression of the plasmacytic markers CD38 or CD138 (supplemental Figure 7C). Collectively, these findings suggest that Mino/IR cells are relatively de-differentiated transformed B cells. Importantly, treatment with JQ1 inhibited the suspension culture growth of both Mino/IR and Mino cells (supplemental Figure 7D). JQ1 treatment also induced a similar level of apoptosis in Mino/IR, as compared with Mino cells (Figure 6B). JQ1-induced apoptosis of Mino/IR cells was associated with a reduction in the nuclear levels of RelA, as well as inhibition of the mRNA levels of BTK, CDK6, and BCL2. In contrast, JQ1 treatment induced the mRNA expression of BCL2L11 and HEXIM1 (supplemental Figure 7E-F). JQ1 treatment also markedly attenuated the mRNA levels of BTK and CDK6 (supplemental Figure 7F); however, a more modest inhibition of Bcl-xL, BCL2, and XIAP protein levels, and a lack of any effect on the protein levels of TEC, AKT, SOX11, or PAX5 was observed (Figure 6C). Furthermore, pretreatment with JQ1 significantly enhanced the sensitivity of Mino/IR cells to those concentrations of ibrutinib in which these cells are able to grow in suspension culture (P < .01) (supplemental Figure 7G). Consistent with our previous observation in AML cells,36,37 combined treatment with JQ1 and PS synergistically induced apoptosis of the cultured as well as of primary MCL cells, with CI <1.0 by the median dose-effect isobologram analyses (supplemental Figure 7H-I). Notably, as shown in Figure 6D, cotreatment with PS and JQ1 also synergistically induced apoptosis of Mino/IR cells. Next, we determined the effect of cotreatment of JQ1 with palbociclib (a CDK4 and CDK6 inhibitor) or with the BCL2 antagonist ABT-199.45,46 Cotreatment with JQ1 and palbociclib or ABT737 or ABT-199 was synergistically lethal against not only Mino but also Mino/IR cells (Figure 6E-G and supplemental Figure 6B and 7J). However, co-treatment with JQ1 and ibrutinib lacked synergy against Mino/IR cells (data not shown). We next determined whether pretreatment with PS, palbociclib, and ABT-199 lowers the threshold for ibrutinib-induced apoptosis of Mino/IR cells. We did not observe any significant effect of the pretreatment with these agents on ibrutinib-induced apoptosis of Mino/IR cells (data not shown). Notably, cotreatment with JQ1 and ABT-199 or palbociclib was also synergistically lethal against primary MCL cells (Figure 7A-B). Collectively, these findings demonstrate that treatment with the BA JQ1 sensitizes MCL cells to not only the histone deacetylase inhibitor but also to CDK4/6 and BCL2 antagonists.
Figure 6.
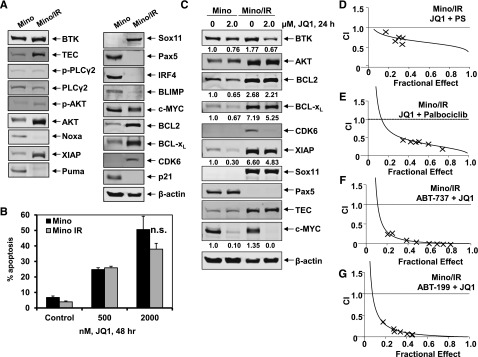
Cotreatment with JQ1 and PS or palbociclib is synergistically active in ibrutinib-resistant Mino/IR cells. (A) Representative immunoblots of basal protein expression in Mino vs Mino/IR cells. (B) Mino and Mino cells with acquired resistance to ibrutinib (Mino/IR) were treated with the indicated concentrations of JQ1 for 48 hours. The percent of Annexin V-positive apoptotic cells was determined by flow cytometry. (C) Immunoblot analyses of Mino and Mino/IR cells treated for 24 hours with JQ1, as indicated. The numbers beneath the bands represent densitometry analysis. (D-G) Mino/IR cells were treated with a fixed ratio of JQ1 and PS (D), or palbociclib (E), or ABT-737 (F), or ABT-199 (G) for 48 hours. The percent of apoptotic cells was determined by flow cytometry. Median dose effect and isobologram analyses were performed. CI values <1.0 indicates a synergistic interaction of the two agents in the combination. Doses of drugs, fractional effect, and CI values are provided in supplemental Figure 6B. n.s., not significant.
Figure 7.

Cotreatment with JQ1 and ABT-199 or palbociclib is synergistically active against primary MCL cells. (A-B) Primary MCL cells were treated with the indicated concentrations of JQ1 and the BCL2-specific inhibitor, ABT-199 or the CDK4/6 inhibitor, palbociclib at a fixed ratio for 48 hours. At the end of treatment, cells were washed with 1× phosphate-buffered saline and stained with PI. The percent of nonviable cells was determined by flow cytometry. Median dose effect and isobologram analyses were performed. CI values <1.0 indicates a synergistic interaction of the two agents in the combination. FE, fractional effect.
Discussion
Here, we demonstrate that, by inhibiting the activity of the BET protein BRD4 and the resulting inhibition of NF-κB, treatment with a BA attenuates the expression of several NF-κB–activated pro-growth and pro-survival genes, including BTK, in MCL cells. This results in the BA-mediated growth inhibition and apoptosis of MCL cells. Our findings also show that coculture with lymph node or BM stromal cells reduced BA-induced lethality of MCL cells. Whereas treatment with ibrutinib is highly effective in inducing remissions in a majority of patients, primary refractoriness or relapse with resistant disease is a common outcome in patients with MCL. We also demonstrate here that cotreatment of MCL with the BA JQ1 and ibrutinib exerts synergistic in vitro lethality and in vivo activity against MCL cells. Additionally, our findings also highlight for the first time that, although treatment with JQ1 alone is also active, cotreatment with JQ1 and PS, palbociclib, or ABT-199 is synergistically lethal against cultured MCL cells that display in vitro resistance to ibrutinib.
As reported for AML and diffuse large B-cell lymphoma (DLBCL) cells, inhibition of BRD4 by JQ1 also attenuates the levels of c-MYC, CDK4/6, and to a lesser extent BCL2 in MCL cells, which inhibits the growth and survival of MCL cells. Acetylation of the NF-κB subunit RelA at lysine 310 regulates the transcriptional activity of NF-κB. BRD4 has been shown to directly bind to acetylated RelA and maintain the constitutive activity of NF-κB.30 Additionally, DNA binding by RelA leads to increased acetylation of histone H4K5/8 to which BRD4 binds. BRD4, in turn recruits pTEFb, which phosphorylates the CTD of RNAP2 and stimulates the mRNA transcript elongation of NF-κB–target genes.21 As an acetyl-lysine mimetic antagonist of BRD4, JQ1 treatment disrupts the binding of BRD4 to the chromatin as well as to RelA, undermining the ability of BRD4 to sustain the transcriptional activity of NF-κB.21,30,47 Consistent with this, our findings demonstrate that JQ1 depletes the constitutively active nuclear RelA, and represses the NF-κB–activated genes and induces apoptosis in MCL cells. Utilizing an inducible and reversible transgenic RNA interference mouse model, inhibition of BRD4 was shown recently to cause reversible epidermal hyperplasia, alopecia, decreased cellular diversity, and stem cell depletion in the small intestine.48 However, this has not been observed following in vivo treatment of immune-depleted mice with BA.31,35,36 Among NF-κB–activated genes inhibited by JQ1 are the pro-growth and pro-survival genes, including TNFAIP3, cFLIP, cIAP2, XIAP, BCL-xL, BMI1, IL-10, PRDM1, NF-κB2, as well as BTK. Importantly, simultaneous lowering of BTK levels by JQ1 and abrogation of the activity of BTK by ibrutinib results in synergistic lethality due to co-treatment with JQ1 and ibrutinib against MCL cells. This has also been shown against the activated B-cell DLBCL, but not against the germinal center B-cell DLBCL or multiple myeloma cell lines.49 Notably, compared with each agent alone, cotreatment with JQ1 and ibrutinib also significantly improved the survival and induced a plateau in the survival curve of the immune-depleted mice engrafted with human MCL. The efficacy of cotreatment with JQ1 and ibrutinib was also associated with the in vivo attenuation of BTK, p-BTK, and p-PLCγ2 levels while inducing BIM levels in the MCL cells. These findings underscore the potential for promising in vivo efficacy of the combination of BA and ibrutinib against MCL. Because it also depletes BTK expression, cotreatment with BA and ibrutinib may prevent the emergence of the mutant versions of BTK that confer resistance against ibrutinib administered alone in patients with MCL.
JQ1 treatment attenuates the levels of the antiapoptotic proteins Bcl-xL, XIAP, MCL-1, BCL2, and p-AKT, as well as upregulates the proapoptotic proteins p27 and BIM. MYC is known to repress BIM via microRNA 17-92, which is reversed following JQ1-mediated inhibition of MYC.50 Collectively, these effects of JQ1 lower the threshold for apoptosis and sensitize MCL cells to the apoptosis induced by a variety of agents that are also individually active as single agents against MCL cells. These agents include the pan-histone deacetylase inhibitor PS, Bcl-xL and BCL2 antagonist ABT-737, BCL2-specific antagonist ABT-199, as well as the CDK4/6 antagonist palbociclib.45,46 By attenuating the levels of CDK4/6, cotreatment with JQ1 also sensitizes MCL cells to apoptosis induced by the CDK4/6 inhibitor palbociclib.46 Treatment with BA also induces HEXIM1, which binds to and sequesters pTEFb in an inhibitory complex, thereby inhibiting the phosphorylation of serine 2 on the CTD of RNAP2 and abrogating the mRNA transcript elongation by RNAP2.25-27 Consistent with this, HEXIM1 induction due to BA treatment is likely to contribute to the growth-inhibitory and apoptotic effects of BA-based combinations against MCL cells.
Similar to the ibrutinib-resistant Mino/IR cells described here, BTK or PLCγ2 mutations were also not discovered in the primary, ibrutinib-refractory MCL.7,14,15 Instead, mutations in TRAF2/3, BIRC3, and MAP3K14 (NF-κB inducing kinase), which activate the alternative (noncanonical) NF-κB activation pathway have been documented, resulting in the dependency of the ibrutinib-resistant MCL cells on the NF-κB–activated pro-growth and pro-survival genes, or on the PI3K/AKT survival signaling.7,14,16 Compared with the parental Mino cells, ibrutinib-resistant Mino/IR cells exhibit increased expression of BTK and TEC, as well as display elevated levels of the antiapoptotic proteins BCL2, Bcl-XL, XIAP, and AKT. Consistent with this, cotreatment with JQ1 and ABT-199 or ABT-737 was synergistically lethal against Mino/IR cells. Recently, the C481S missense mutation at the ibrutinib binding site of BTK was also discovered as a secondary resistance mechanism in 2 patients with MCL who progressed on ibrutinib after a durable response.14 This mutation was shown to be associated with increased BTK and AKT activation, which resulted in the proliferation of the resistant MCL cells driven by increased CDK4 activity. These cells were highly sensitive to palbociclib.14 Similar to this, increased expression and dependency of Mino/IR cells on CDK6 may make them sensitive to palbociclib and also susceptible to the synergistic lethality of the cotreatment with JQ1 and palbociclib. Collectively, these preclinical findings show promising activity of BA-based combinations, especially with ibrutinib, against MCL cells. These findings also support further in vivo testing of the combined therapy with BA and BCL2 antagonist, HDI, or CDK4/6 inhibitor against ibrutinib-resistant MCL.
Acknowledgments
The authors thank Helen Heslop at the Baylor College of Medicine for assistance in critically reviewing the manuscript.
This research was supported in part by the National Institutes of Health, National Cancer Institute, through the MD Anderson Cancer Center Support grant (P30 CA016672), and through the MD Anderson Cancer Center Leukemia Specialized Program of Research Excellence grant (CA 100632).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: B. Sun, B. Shah, W.F., S.G.T.D., D.T.S., S.K., and S.S. performed in vitro experiments with cultured and primary MCL cells, and analyzed the data; B. Shah performed the in vivo studies in the NOD/SCID mice; J.Q. and J.E.B. provided a critical new reagent for the studies; L.L. provided the HK stromal cells and critically reviewed the manuscript; K.R. and C.C. processed and normalized the microarray data, provided bio-informatics support, and IPA of the gene expression data; J.E.B. provided intellectual input for the in vitro and in vivo studies; L.Z. provided primary MCL samples for the studies; M.L.W. planned experiments and critically reviewed the manuscript; and K.N.B. conceptualized, planned the experiments, supervised the studies, analyzed the data, and prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kapil N. Bhalla, Leukemia Department, MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 428, Houston, TX 77030-4009; e-mail: kbhalla@mdanderson.org.
References
- 1.Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26–38. doi: 10.1182/blood-2010-04-189977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122(10):3416–3423. doi: 10.1172/JCI61272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beà S, Valdés-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi D, Ciardullo C, Gaidano G. Genetic aberrations of signaling pathways in lymphomagenesis: revelations from next generation sequencing studies. Semin Cancer Biol. 2013;23(6):422–430. doi: 10.1016/j.semcancer.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Kridel R, Meissner B, Rogic S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119(9):1963–1971. doi: 10.1182/blood-2011-11-391474. [DOI] [PubMed] [Google Scholar]
- 6.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92. doi: 10.1038/nm.3435. [DOI] [PubMed] [Google Scholar]
- 8.Herman SE, Mustafa RZ, Gyamfi JA, et al. Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123(21):3286–3295. doi: 10.1182/blood-2014-02-548610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maddocks K, Blum KA. Ibrutinib in B-cell Lymphomas. Curr Treat Options Oncol. 2014;15(2):226–237. doi: 10.1007/s11864-014-0274-8. [DOI] [PubMed] [Google Scholar]
- 11.Herrera AF, Jacobsen ED. Ibrutinib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2014;20(21):5365–5371. doi: 10.1158/1078-0432.CCR-14-0010. [DOI] [PubMed] [Google Scholar]
- 12.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma J, Lu P, Guo A, et al. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol. 2014;166(6):849–861. doi: 10.1111/bjh.12974. [DOI] [PubMed] [Google Scholar]
- 14.Chiron D, Di Liberto M, Martin P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–1035. doi: 10.1158/2159-8290.CD-14-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balasubramanian S, Schaffer M, Deraedt W, et al. Mutational analysis of patients with primary resistance to single-agent ibrutinib in relapsed or refractory Mantle cell lymphoma (MCL) [abstract]. Blood. 2014;124(21) Abstract 78. [Google Scholar]
- 16.Colomer D, Campo E. Unlocking new therapeutic targets and resistance mechanisms in mantle cell lymphoma. Cancer Cell. 2014;25(1):7–9. doi: 10.1016/j.ccr.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 19.Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol. 2013;14(4):211–224. doi: 10.1038/nrm3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12(7):465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chapuy B, McKeown MR, Lin CY, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma [published correction appears in Cancer Cell. 2014;25(4):545-546]. Cancer Cell. 2013;24(6):777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itzen F, Greifenberg AK, Bösken CA, Geyer M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014;42(12):7577–7590. doi: 10.1093/nar/gku449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nechaev S, Adelman K. Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochim Biophys Acta. 2011;1809(1):34–45. doi: 10.1016/j.bbagrm.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13(10):720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel MC, Debrosse M, Smith M, et al. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol Cell Biol. 2013;33(12):2497–2507. doi: 10.1128/MCB.01180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang G, Liu R, Zhong Y, et al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem. 2012;287(34):28840–28851. doi: 10.1074/jbc.M112.359505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou Z, Huang B, Wu X, et al. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene. 2014;33(18):2395–2404. doi: 10.1038/onc.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13(5):337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 33.Seal J, Lamotte Y, Donche F, et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A). Bioorg Med Chem Lett. 2012;22(8):2968–2972. doi: 10.1016/j.bmcl.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 34.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiskus W, Sharma S, Qi J, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther. 2014;13(5):1142–1154. doi: 10.1158/1535-7163.MCT-13-0770. [DOI] [PubMed] [Google Scholar]
- 37.Fiskus W, Sharma S, Qi J, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. 2014;13(10):2315–2327. doi: 10.1158/1535-7163.MCT-14-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiskus W, Rao R, Balusu R, et al. Superior efficacy of a combined epigenetic therapy against human mantle cell lymphoma cells. Clin Cancer Res. 2012;18(22):6227–6238. doi: 10.1158/1078-0432.CCR-12-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fiskus W, Saba N, Shen M, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014;74(9):2520–2532. doi: 10.1158/0008-5472.CAN-13-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 41.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 42.Yu L, Mohamed AJ, Simonson OE, et al. Proteasome-dependent autoregulation of Bruton tyrosine kinase (Btk) promoter via NF-kappaB. Blood. 2008;111(9):4617–4626. doi: 10.1182/blood-2007-10-121137. [DOI] [PubMed] [Google Scholar]
- 43.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–232. doi: 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- 44.Vegliante MC, Palomero J, Pérez-Galán P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121(12):2175–2185. doi: 10.1182/blood-2012-06-438937. [DOI] [PubMed] [Google Scholar]
- 45.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 46.Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res. 2014;20(13):3379–3383. doi: 10.1158/1078-0432.CCR-13-1551. [DOI] [PubMed] [Google Scholar]
- 47.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138(1):129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ceribelli M, Kelly PN, Shaffer AL, et al. Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci USA. 2014;111(31):11365–11370. doi: 10.1073/pnas.1411701111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolden JE, Tasdemir N, Dow LE, et al. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Reports. 2014;8(6):1919–1929. doi: 10.1016/j.celrep.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014;26(2):262–272. doi: 10.1016/j.ccr.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]