

Key Points
Fibrin is a novel ligand for the platelet collagen receptor, GPVI, increasing platelet procoagulant activity.
Activation of GPVI by fibrin contributes to thrombus growth and stabilization.
Abstract
The glycoprotein VI (GPVI)-Fc receptor γ (FcRγ) chain is the major platelet signaling receptor for collagen. Paradoxically, in a FeCl3 injury model, occlusion, but not initiation of thrombus formation, is delayed in GPVI-deficient and GPVI-depleted mice. In this study, we demonstrate that GPVI is a receptor for fibrin and speculate that this contributes to development of an occlusive thrombus. We observed a marked increase in tyrosine phosphorylation, including the FcRγ chain and Syk, in human and mouse platelets induced by thrombin in the presence of fibrinogen and the αIIbβ3 blocker eptifibatide. This was not seen in platelets stimulated by a protease activated receptor (PAR)-4 peptide, which is unable to generate fibrin from fibrinogen. The pattern of tyrosine phosphorylation was similar to that induced by activation of GPVI. Consistent with this, thrombin did not induce tyrosine phosphorylation of Syk and the FcRγ chain in GPVI-deficient mouse platelets. Mouse platelets underwent full spreading on fibrin but not fibrinogen, which was blocked in the presence of a Src kinase inhibitor or in the absence of GPVI. Spreading on fibrin was associated with phosphatidylserine exposure (procoagulant activity), and this too was blocked in GPVI-deficient platelets. The ectodomain of GPVI was shown to bind to immobilized monomeric and polymerized fibrin. A marked increase in embolization was seen following FeCl3 injury in GPVI-deficient mice, likely contributing to the delay in occlusion in this model. These results demonstrate that GPVI is a receptor for fibrin and provide evidence that this interaction contributes to thrombus growth and stability.
Introduction
Glycoprotein VI (GPVI) is an immunoglobulin superfamily receptor expressed on the surface of platelets in association with the Fc receptor γ chain (FcRγ chain). Ligand binding of GPVI results in receptor clustering and Src kinase-dependent tyrosine phosphorylation of the FcRγ chain immunoreceptor tyrosine-based activation motif. This initiates a signaling pathway involving Src and Syk tyrosine kinases and various adapter proteins and results in activation of phospholipase-C γ2.1
GPVI is the major signaling receptor for the extracellular matrix protein, collagen,2-4 and is also activated by other endogenous ligands including laminin,5,6 adiponectin,7 and the immunoglobulin superfamily protein extracellular matrix metalloproteinase inducer (EMMPRIN; CD147/Basigin).8 GPVI is also activated by a variety of exogenous and synthetic ligands including collagen-related peptide, which consists of glycine-proline-hydroxyproline repeats9; the snake venom toxin, convulxin10,11; various artificial peptides including 4N1-1 and LSARLAF; antisense nucleotides12; and diesel exhaust particles.13
GPVI plays a critical role in hemostasis and thrombosis through integrin activation, supporting adhesion and the initial stages of platelet aggregation. Despite this, patients and mice deficient in GPVI exhibit mild or no apparent impairment in hemostasis due to compensatory pathways of initiation of platelet activation including tissue factor-driven thrombin generation14 and adenosine diphosphate release from damaged cells. GPVI also plays a critical role in the maintenance of a healthy endothelial cell layer, a process known as vascular integrity.15
The contribution of GPVI to thrombosis varies according to the experimental model. This is illustrated by its role in FeCl3 and laser injury models, which is dependent on the level of injury to the vessel wall and the degree of exposure of the subendothelial matrix.16-19 In the absence of a breach in the endothelium, thrombus formation is mediated by endothelial cell activation, leading to tissue factor exposure and thrombin generation.17,20
The focus of research on GPVI in hemostasis and thrombosis has been on its interaction with collagen. Paradoxically, however, the time to occlusion rather than initiation of thrombus formation has been shown to be prolonged in GPVI-deficient and GPVI-depleted mice following FeCl3 injury, suggesting a role for GPVI in thrombus growth,16 even though collagen is only thought to play a role at the site of lesion. In the present study, we provide an explanation for this paradox by demonstrating that GPVI is a receptor for fibrin, which is generated from fibrinogen by the action of thrombin. Thus, GPVI is a receptor for both collagen and fibrin and plays a critical role in thrombus growth and in initiating thrombus formation.
Methods
Reagents
Human fibrinogen was obtained from Enzyme Research Laboratories (Swansea, UK). The α-phospho-tyrosine (4G10) monoclonal antibody was from Millipore (Abingdon, UK). Alexa-488 fibrinogen, Alexa-488 phalloidin, and Alexa-568 phalloidin were from Molecular Probes (Life Technologies, Paisley, UK). The α-Syk antibody (SC-573) was from Insight Biotechnology (Wembley, UK). The α-CLEC-2 antibodies (AYP1 and AYP2) have been described previously.21 Fluorescein isothiocyanate (FITC) Annexin V was from BD Bioscience (Oxford, UK). FITC-conjugated α-mouse αIIbβ3, α2, CLEC-2 (C-type lectin-like receptor-2), GPIbα, GPV, GPVI, GPIX, and immunoglobulin (Ig)G antibodies were from Emfret Analytics (Würzburg, Germany). Dasatinib was from LC Laboratories (Woburn, MA). Protease activated receptor (PAR)-4 peptide (Ala-Tyr-Pro-Gly-Lys-Phe: AYPGKF) was produced by Alta Bioscience (Birmingham, UK). Horseradish peroxidase (HRP)-conjugated secondary antibodies and enhanced chemiluminescence reagent were from Amersham Biosciences (GE Healthcare, Bucks, UK). HRP-conjugated secondary antibody for enzyme-linked immunosorbent assay (ELISA) was from Dako (Ely, UK). The α-GPVI antibody (1A12) was provided by Dr Elizabeth Gardiner (Melbourne, Australia). SuperSignal ELISA Pico chemiluminescent substrate was from Pierce (Rockford, IL). All other reagents were purchased from Sigma-Aldrich (Poole, UK).
Mice
Gp6−/− mice (GPVI KO) were provided by Dr Jerry Ware.22 Itga2b−/− mice (αIIb-deficient mice) have been previously described.23 Wild-type mice (WT) were generated from breeding of heterozygotes or purchased from Harlan Laboratories (Hillcrest, UK). All procedures were undertaken with UK Home Office approval under PPL30/2721 and PPL30/8286.
Washed platelet preparation
Blood was drawn from CO2-asphyxiated mice or from consenting, healthy, drug-free volunteers into 10% acid citrate dextrose or sodium citrate, respectively. Ethical approval for the donation of blood by volunteers was granted by Birmingham University Internal Ethical Review (ERN_11-0175). Washed platelets were obtained by centrifugation using prostacyclin and resuspended in modified Tyrode’s buffer (134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 5 mM glucose, and 1 mM MgCl2; pH 7.3) as previously described.24 Platelets were used at 2 × 107/mL for static adhesion or 5 × 108/mL for other studies.
Protein phosphorylation
Washed platelets were pretreated with 9 μM eptifibatide (where stated), 10 μM indomethacin, and 2 U/mL apyrase or solvent controls. Platelets were stimulated with thrombin (1 U/mL) or PAR-4 peptide (AYPGKF; 150 μM) at 37°C with stirring (1200 rpm) in an aggregometer for 60 seconds. Where stated, stimulations were performed in the presence of fibrinogen (200 μg/mL) and GPRP (Gly-Pro-Arg-Pro; 5 μM). Activation was terminated with 2× ice-cold lysis buffer (300 mM NaCl, 20 mM Tris, 2 mM EGTA, 2 mM EDTA, and 2% IGEPAL CA-630 [NP-40 equivalent], pH 7.4, plus 2.5 mM Na3VO4, 100 μg/mL AEBSF (4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride), 5 μg/mL leupeptin, 5 μg/mL aprotinin, and 0.5 μg/mL pepstatin). Whole cell lysates (WCLs) were prepared by boiling a sample of lysate with sodium dodecyl sulfate (SDS) sample buffer. Syk was immunoprecipitated with α-Syk antibody (SC-573) and protein A-Sepharose beads for 2 hours. The beads were then washed, and proteins were eluted by boiling in SDS sample buffer. Immunoprecipitates (IPs) and WCLs were then separated by SDS-polyacrylamide gel electrophoresis, electro-transferred, and western blotted. Western blots were imaged with a Licor Odyssey-FC (Cambridge, UK) or autoradiographic film.
Platelet spreading
Coverslips were coated in the presence of 200 μg/mL fibrinogen (±5 μM GPRP) for 60 minutes, followed by washing with phosphate-buffered saline (PBS) and then blocking with 5 mg/mL heat-inactivated bovine serum albumin for 60 minutes. Fibrin-coated coverslips were generated by the addition of 1 U/mL thrombin for 15 minutes to coverslips that had been incubated with 200 μg/mL fibrinogen for 30 min. Thrombin was neutralized by addition of 5 U/mL hirudin for 30 minutes. Following coating, coverslips were washed and blocked with 5 mg/mL heat-inactivated bovine serum albumin for 60 minutes.
Platelets were spread on fibrinogen or fibrin for 45 minutes at 37°C before washing with Tyrode’s buffer followed by fixation with paraformaldehyde (3.7%). Where stated, platelets were preincubated with dasatinib (10 μM) or hirudin (5 U/mL). For actin staining, platelets were permeabilized with 0.1% Triton X-100 and stained with Alexa-488 phalloidin for 45 minutes. In some studies, Alexa-488 fibrinogen was used for fluorescent imaging to monitor conversion to fibrin, in which case platelets were counterstained with Alexa-568 phalloidin for 45 minutes. For phosphatidylserine (PS) staining, platelets were incubated with FITC-Annexin V in Tyrode’s buffer plus 2 mM CaCl2 for a further 15 minutes, followed by fixation and counterstaining for actin using Alexa-568 phalloidin for 45 minutes. Platelets were imaged on a Zeiss Axiovert 200M microscope. Platelet surface area was analyzed using ImageJ (National Institutes of Health, Bethesda, MD).
Flow cytometry
Surface glycoprotein expression was measured in WT and GPVI-deficient mouse platelets by staining whole blood with selected FITC-conjugated antibodies and analyzing with a BD Accuri C6 flow cytometer (BD Biosciences, Oxford, UK).
GPVI ELISA
Binding of GPVI to fibrin/fibrinogen was measured using an adapted version of a soluble GPVI ELISA.25 Wells of a Nunc Maxisorp micro-titer plate (Thermo Scientific, Paisley, UK) were coated with fibrin/fibrinogen using the same protocol as above. Wells were washed 6 times with 100 μL of PBS-tween (PBS-T) (0.2%), blocked with 1% (w/v) bovine serum albumin for 1 hour at room temperature, and then washed 6 times with PBS-T. GPVI ectodomain (prepared from N-ethylmaleimide [NEM]-treated plasma) was added to the wells for 1 hour at room temperature, followed by washing 6 times with PBS-T. A standard curve was generated by a serial dilution of GPVI ectodomain into GPVI-depleted plasma. Primary antibody (1A12; 1 μg/mL) was then incubated for 1 hour followed by 6 washes. Secondary α-mouse HRP-conjugated antibody (2.6 μg/mL) was then incubated for 1 hour, followed by 6 washes, followed by 100 μL SuperSignal substrate for 1 minute. Light emission was measured using a Wallac-Victor2 luminescence plate reader (PerkinElmer, Waltham, MA) for 10 s/well. Amounts of bound GPVI were calculated from the standard curve.
Intravital microscopy
Four- to 5-week-old WT or GPVI KO mice26 were anesthetized, and the mesentery was exteriorized through a midline abdominal incision. Arterioles (40-60 µm diameter) were visualized with a Zeiss Axiovert 200 inverted microscope (×10) equipped with a 100-W HBO fluorescent lamp source and a CoolSNAP-EZ camera (Visitron Systems, Puchheim, Germany). Digital images were recorded and analyzed offline using Metavue software. Injury was induced by topical application of a droplet of FeCl3 from 3-mm2 filter paper saturated with 20% FeCl3. The droplet rapidly dissipated through the vessel. Adhesion and aggregation of fluorescently labeled platelets (Dylight-488–conjugated α-GPIX Ig derivative) in arterioles were monitored for 40 minutes or until complete occlusion occurred (blood flow stopped for >1 minute).
Statistical analysis
Statistical analysis was by analysis of variance with a Bonferroni posttest, or for the ELISA, a 1-tailed Student t test. For in vivo experiments, results are shown as the mean ± standard deviation of a minimum of 6 mice. Statistical analysis was by Student t test.
Results
Fibrin stimulates tyrosine phosphorylation in human and mouse platelets through GPVI
The binding of fibrinogen to integrin αIIbβ3 induces outside-in signaling leading to activation of Src and Syk tyrosine kinases and a signaling cascade that culminates in activation of phospholipase-C γ2.27,28 In addition, αIIbβ3 is a receptor for fibrin, which binds to multiple sites in the αIIb β-propeller.29 It is not known, however, whether binding of fibrin to the integrin also induces outside-in signaling. To address this, we compared tyrosine phosphorylation in human platelets stimulated by thrombin, which stimulates fibrin formation, and by a PAR-4 peptide. The contribution of fibrin polymers to tyrosine phosphorylation in this assay can be established using GPRP, which blocks polymerization of fibrin monomers.
Thrombin stimulates tyrosine phosphorylation in human platelets under aggregating conditions, which is slightly increased in the presence of added fibrinogen (supplemental Figure 1, available on the Blood Web site). A similar increase in tyrosine phosphorylation is induced by the PAR-4 peptide. For both agonists, tyrosine phosphorylation is marginally reduced or unaltered in the presence of GPRP. These results indicate that fibrin does not play a major role in mediating tyrosine phosphorylation under aggregating conditions, which is predominantly mediated through binding of fibrinogen to integrin αIIbβ3 as previously reported.30-32
A role for fibrin in mediating tyrosine phosphorylation may, however, be masked by outside-in signaling by fibrinogen. To address this, we performed studies in the presence of eptifibatide, which blocks the binding of fibrinogen to αIIbβ3. Under these conditions, thrombin stimulates a much smaller increase in tyrosine phosphorylation in platelet lysates, which is increased significantly in the presence of fibrinogen, including a band of 72 kDa, which was identified as Syk by immunoprecipitation and western blotting (Figure 1Ai). (A decrease in Syk signal in the Syk reprobes corresponds with the increase in phosphorylated Syk. This is due to the blotting antibody not detecting phospho-Syk.) In addition, a tyrosine phosphorylated band of 14 kDa was present in Syk immunoprecipitates, which corresponds to the FcRγ chain. The marked increase in tyrosine phosphorylation, including Syk and the FcRγ chain, is blocked in the presence of GPRP, indicating that it is mediated by fibrin polymerization. In contrast, tyrosine phosphorylation induced by a PAR-4 peptide (which is unable to generate fibrin) is not altered in the presence of added fibrinogen or GPRP. A similar set of results was seen in mouse platelets stimulated by thrombin and by the PAR-4 peptide in the presence of eptifibatide (Figure 1Aii; data not shown). These results show that fibrin increases tyrosine phosphorylation in human and mouse platelets, including Syk and the FcRγ chain.
Figure 1.

Fibrin stimulates tyrosine phosphorylation in a GPVI-dependent manner. Platelets from (Ai) human, (Aii) mice, (Bi) αIIb-deficient mice, and (Bii) GPVI-deficient mice were stimulated with thrombin (1 U/mL) or PAR-4 peptide (150 μM) in the presence of eptifibatide and, where shown, fibrinogen (200 μg/mL) and GPRP (5 μM). Stimulations were stopped after (A) 1 or (B) 3 minutes with the addition of 2× lysis buffer. A sample of the WCL was removed, and the remaining lysate was used to IP Syk. WCLs and IPs were separated by SDS-polyacrylamide gel electrophoresis and western blotted for pTyr, Syk, and the FcRγ chain, which coprecipitates with Syk. The results are shown as representative of 3 experiments.
We used αIIb-deficient mouse platelets to investigate whether integrin αIIbβ3 is required for the increase tyrosine phosphorylation induced by fibrin in the presence of eptifibatide (in view of the possibility that the increase was mediated by binding of fibrin to a separate site on the integrin). As shown in Figure 1Bi, tyrosine phosphorylation, including Syk and the FcRγ chain, was again induced by thrombin in the presence of fibrinogen despite the absence of αIIb-integrin subunit, and this was blocked in the presence of GPRP. These results demonstrate that fibrin stimulates tyrosine phosphorylation of Syk and the FcRγ chain in mouse platelets independent of the major platelet integrin, αIIbβ3.
The pattern of tyrosine phosphorylation was similar to that induced by activation of GPVI, including phosphorylation of the FcRγ chain. To investigate a possible role for GPVI, we studied the effect of thrombin in mouse platelets deficient in GPVI in the presence of eptifibatide to block binding of fibrinogen to integrin αIIbβ3. Platelets from these mice have ∼50% of the WT level of the GPVI-binding partner, the FcRγ chain, whereas the level of other major platelet glycoprotein receptors was similar to that in WT platelets (supplemental Figure 2). Further, activation of platelets by thrombin is not altered in GPVI-deficient mice (data not shown), suggesting that the levels of PAR-4 are unchanged. Fibrin stimulated only a minor increase in tyrosine phosphorylation in GPVI-deficient platelets and failed to induce phosphorylation of Syk or the FcRγ chain (Figure 1Bii). The minor increase in phosphorylation in platelet lysates was not altered in the presence of GPRP (Figure 1Bii). These results demonstrate that the marked increase in tyrosine phosphorylation, including the FcRγ chain and Syk, by fibrin in platelets is mediated by GPVI.
The podoplanin receptor CLEC-2, which signals through a similar pathway to that of GPVI, was not phosphorylated (supplemental Figure 3), ruling out a role for the C-type lectin-like receptor in the response to fibrin.
Fibrin stimulates platelet spreading and procoagulant activity via GPVI
We next asked whether activation of GPVI by fibrin could induce platelet activation. To address this, we monitored spreading of mouse platelets on a fibrin surface and compared this to spreading on fibrinogen. To ensure a similar level of coating of fibrin and fibrinogen, coverslips were coated with fibrinogen prior to treatment with thrombin (hirudin was later added to block thrombin as described in the methods). We used Alexa-488 fibrinogen to visualize fibrin and fibrinogen. Fibrinogen forms a homogenous surface on the coverslip, which was converted to a heterogeneous network of fibrin following treatment with thrombin. Large areas of fibrin are easily distinguishable (supplemental Figure 4), but following longer exposures, smaller areas of fibrin can be seen. Both large and smaller coatings of fibrin are associated with platelet adhesion and spreading (Figure 2A; data not shown).
Figure 2.
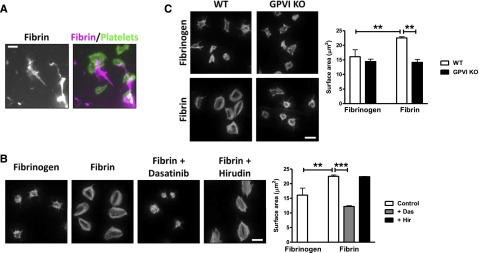
Fibrin stimulates spreading in a GPVI-dependent manner. Fibrinogen was coated onto glass coverslips and converted into fibrin by treatment with thrombin. Hirudin was used to neutralize any residual thrombin before washing and blocking. (A) Alexa-488 fibrinogen was used to visualize fibrin formation. Platelets (2 × 107/mL) were allowed to spread on coated coverslips, followed by actin staining with Alexa-568 phalloidin. Scale bar, 5 μm. (B) Platelets (2 × 107/mL) were allowed to spread on nonfluorescent fibrinogen or fibrin-coated coverslips, followed by actin staining with Alexa-488 phalloidin. Where shown, platelets were preincubated with dasatinib (10 μM) or hirudin (5 U/mL). Scale bar, 5 μm. (C) WT or GPVI KO platelets were allowed to spread on fibrinogen or fibrin-coated coverslips, followed by actin staining with Alexa-488 phalloidin. Scale bar, 5 μm. The results are shown as mean ± standard error of the mean (SEM) of 3 experiments. **P < .01; ***P < .001.
Mouse platelets form actin stress fibers and lamellipodia sheets on polymerized fibrin but not on fibrinogen, which induces formation of filopodia and partial lamellipodia (Figure 2B). Actin nodules33 and filopodia can be seen in platelets that have undergone partial spreading, which is likely to reflect an earlier stage of spreading on fibrin (data not shown). Spreading on fibrin is blocked by the Src kinase inhibitor dasatinib but unaltered in the presence of hirudin, demonstrating that it is not mediated by residual thrombin (Figure 2B). Spreading of platelets on fibrin was abolished in the absence of GPVI (Figure 2C), demonstrating that fibrin induces spreading through the collagen receptor. Fibrin also supports full spreading of human platelets (supplemental Figure 5).
Interestingly, mouse platelets underwent full spreading on fibrin in the presence of GPRP, which prevents polymerization of fibrin (Figure 3A). This demonstrates both fibrin monomers and fibrin polymers are able to induce full spreading of platelets, indicating that they share the epitope for binding to, and activation of, GPVI. The inhibitory effect of GPRP on GPVI activation in suspension studies as shown in Figure 1 is likely to be due to loss of avidity (and therefore the ability to crosslink GPVI) rather than loss of binding to fibrin. This is overcome by presentation of fibrin monomers on a 2-dimensional surface. We modified a previously described ELISA assay25 to verify that monomeric and polymerized fibrin bind to GPVI. Using this assay, we observed a similar level of binding of the ectodomain of GPVI to fibrin monomers and fibrin polymers (Figure 3B). In contrast, GPVI did not bind to immobilized fibrinogen (Figure 3B).
Figure 3.

Fibrin polymerization is not essential for platelet spreading or GPVI binding. (A) Fibrinogen was coated onto glass coverslips and converted into fibrin by treatment with thrombin. GPRP (5 μM) was used to inhibit polymerization of fibrin monomers. Hirudin was used to neutralize any residual thrombin before washing and blocking. Platelets (2 × 107/mL) were allowed to spread, followed by actin staining with Alexa-488 phalloidin. Scale bar, 5 μm. The results are representative of 3 experiments. (B) Fibrinogen was coated onto the wells of Nunc Maxisorp plates and converted into fibrin by treatment with thrombin. GPRP (5 μM) was used to inhibit polymerization of fibrin monomers. The ectodomain of GPVI was incubated, and following washing, adherent GPVI was detected with a HRP-conjugated antibody. The results are shown as mean ± SEM of 3 experiments performed in duplicate. *P < .05; **P < .01.
During the latter stages of platelet activation, phosphatidylserine (PS) is exposed on the outer leaflet of the plasma membrane and provides a negatively charged, procoagulant surface for the generation of thrombin. This process is important for thrombus formation in vivo as it generates thrombin on the platelet surface, which serves both as a positive feedback step in platelet activation and mediates conversion of fibrinogen to fibrin, thereby stabilizing the thrombus. As fibrin generation is an integral part of this positive feedback loop, and activation of GPVI is associated with PS exposure, we determined whether fibrin induces PS exposure in platelets by measurement of binding of FITC-conjugated Annexin V.34 Fibrin induced a threefold increase in number of PS-exposing platelets compared with fibrinogen, which was inhibited in GPVI-deficient platelets (Figure 4A). Thus, fibrin stimulates PS exposure through activation of GPVI.
Figure 4.

GPVI KO platelets have reduced PS exposure and reduced thrombus stability. (A) WT or GPVI-deficient platelets were allowed to spread on fibrinogen or fibrin-coated coverslips, followed by incubation with FITC-Annexin V. Actin was counterstained with Alexa-568 phalloidin to count total number of adherent platelets (data not shown). Scale bar, 20 μm. The results are shown as mean ± SEM of 3 experiments. (B) Representative fluorescent images before and after injury. Asterisk indicates vessel occlusion. Embolization rate was determined by counting embolized thrombus fragments (size > 10 µm) after an initial thrombus had formed (thrombus size ≥ 10 µm). Observation period: 40 minutes or until vessel occlusion. The results are shown as mean ± standard deviation of ≥6 mice. ***P < .001.
Time to occlusion but not initiation of thrombus formation following FeCl3 injury is diminished in GPVI-deficient platelets
The observation that GPVI is a receptor for fibrin raises the question as to whether the delayed occlusion in a FeCl3 injury model16 is due to loss of activation of GPVI by fibrin rather than by collagen, which is not present in the growing thrombus. To address this, we studied thrombus formation by intravital fluorescence microscopy following FeCl3-injured mesenteric arterioles in GPVI-deficient mice. The onset of aggregation occurred at similar times after injury in WT and GPVI-deficient mice, indicating that it was likely to be mediated by endothelial cell activation rather than by exposure to collagen (Figure 4B; supplemental Video). Complete vessel occlusion was observed within 20 minutes in WT vessels16 (data not shown). In contrast, vessel occlusion was markedly delayed or absent in GPVI-deficient mice, and constant embolization was seen (Figure 4B; supplemental Video). The rate of embolization from thrombi of >10 µm was increased by 6.6-fold in GPVI-deficient mice. Consequently, most vessels remained open or occlusion was significantly delayed. These results demonstrate a critical role for GPVI in thrombus stabilization relative to thrombus initiation in this model of FeCl3 injury.
Discussion
Until now, the focus on the role of GPVI in hemostasis and thrombosis has been on the onset of response because of expression of the ligands collagen and laminin in the subendothelial matrix. Strikingly, however, a marked delay in vessel occlusion rather than in initiation of thrombus formation is seen in mice deficient in GPVI in a FeCl3 injury model, indicating a role for GPVI in thrombus growth.16 In the present study, we identify fibrin as a novel ligand for GPVI. We speculate that loss of this interaction mediates the delay in vessel occlusion and the increase in embolization in GPVI-deficient mice following topical FeCl3 injury.
The interaction of fibrinogen with platelets is dependent on sustained activation of the integrin through the platelet adenosine diphosphate receptor P2Y1235 but becomes irreversible over time as a result of conversion to fibrin. Fibrin is formed from fibrinogen by the action of the protease thrombin. Thrombin exposes a site of interaction allowing the fibrin monomers to polymerize. These are then crosslinked by factor XIIIa, forming covalently linked polymers. Fibrin polymers stabilize the growing thrombus through crosslinking of surface receptors, including integrin αIIbβ3, and by clot retraction, which links the integrin to the platelet cytoskeleton. The interaction of fibrin and fibrinogen with αIIbβ3 is mediated through distinct epitopes. Fibrinogen binds to αIIbβ3 via the carboxy-terminal peptide sequence of the γC-peptide (GAKQAGDV). In contrast, fibrin binds to the integrin through a unique sequence in the γC-peptide, ATWKTRWYSMKK, which binds to the αIIb β-propeller.29,36 The distinct roles of these epitopes are illustrated by the differential effects of mutation of these sequences on adhesion and clot retraction.37,38 In the present study, we show that fibrin but not fibrinogen activates GPVI, thereby indicating the presence of additional functional differences between the closely related matrix proteins. Further work is required to establish the structural basis of this.
In our initial experiments, we asked whether fibrin stimulates tyrosine phosphorylation in human and mouse platelets through integrin αIIbβ3. Under aggregating conditions, fibrin polymerization is associated with a relatively minor increase in tyrosine phosphorylation, most likely because this is masked by fibrinogen-dependent αIIbβ3-outside-in signaling. Consistent with this, tyrosine phosphorylation is dramatically reduced by the αIIbβ3-blocker eptifibatide, which blocks binding of fibrinogen but not fibrin to the integrin. Tyrosine phosphorylation is markedly increased, however, in the presence of added fibrinogen and eptifibatide following stimulation by thrombin but not a PAR-4 peptide, with the FcRγ chain and Syk being identified as major phosphorylated proteins. Studies on mouse platelets deficient in the integrin subunit αIIb confirmed that phosphorylation is independent of integrin αIIbβ3.
An increase in FcRγ chain and Syk phosphorylation is indicative of GPVI activation. Consistent with this, tyrosine phosphorylation of the FcRγ chain and Syk by fibrin was abolished in GPVI-deficient platelets, which express 50% of the WT level of the FcRγ chain and normal levels of the other major platelet glycoprotein receptors. The increase in phosphorylation was blocked by GPRP, demonstrating that it is mediated by fibrin polymers and not by monomeric fibrin. However, both immobilized monomeric and polymerized fibrin induce spreading of platelets, demonstrating that polymerization is not essential for binding and activation of the glycoprotein receptor. Further, activation of GPVI by immobilized fibrin leads to PS exposure, which is also abolished in GPVI-deficient platelets. This suggests that on a monolayer, the avidity of the fibrin surface is able to support GPVI activation in the absence of crosslinking of fibrin. In solution, fibrin requires polymerization to induce clustering and activation of GPVI. Confirmation of binding of GPVI to both monomeric and polymerized fibrin was shown using a modified ELISA.
This study therefore adds to the growing number of ligands of the collagen receptor on platelets, which includes the endogenous proteins, laminin, adiponectin, and EMMPRIN, as well as a range of miscellaneous exogenous ligands including small charged peptides, sulfated sugars, antisense nucleotides, and diesel particles.12,13
This work is of significance with regard to the delay in occlusion and increased embolization that is seen following FeCl3 injury in GPVI-deficient mouse platelets, which we speculate is due to loss of platelet activation by fibrin and a corresponding reduction in aggregation and PS exposure and therefore reduced thrombin generation. Following submission of this manuscript, Mammadova-Bach et al39 demonstrated that fibrin supports thrombin generation in human platelets through activation of GPVI. We therefore propose that activation of GPVI by fibrin plays a key feedback role in promoting coagulation and thrombin formation during hemostasis and thrombosis.
These findings add to the potential significance of GPVI as an antithrombotic target40,41 by extending its role to the propagation of an occlusive thrombus. This unexpected role, coupled with its relatively minor role in hemostasis as shown by the mild bleeding seen in GPVI-deficient human and mouse platelets,22,42,43 indicates that targeting the binding of fibrin to GPVI may prevent vessel occlusion at sites of arterial thrombosis, without causing a major bleeding diathesis.
Acknowledgments
The authors thank Dr Elizabeth Gardiner for providing assistance and reagents for the ELISA, Dr Ina Hagedorn for assistance with in vivo studies, and Dr Beata Grygielska for genotyping of mice.
This work was supported by the Wellcome Trust (073107 and 088410), the British Heart Foundation (PG/07/116 and PG/05/134), Najran University (grant DLAB ROEH16283), and by an Emmy Noether grant (BE5084/3-1) from the Deutsche Forschungsgemeinschaft. S.P.W. holds a British Heart Foundation Chair (CH/03/003).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: O.M.A. and C.E.H. designed and performed research, collected data, analyzed and interpreted data, made the figures, and wrote the manuscript; S.M. and S.K.W. designed and performed research, collected data, analyzed and interpreted data, and edited the manuscript; J.F. provided a knockout mouse; M.B. designed and performed research, collected data, analyzed and interpreted data and wrote the manuscript; and S.P.W. designed research, interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Current affiliation for C.E.H. is Institute for Cardiovascular and Metabolic Research, School of Biological Sciences, University of Reading, Reading RG6 6UB, United Kingdom.
Correspondence: Steve P. Watson, Centre for Cardiovascular Sciences, Institute for Biomedical Research, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, United Kingdom; e-mail: s.p.watson@bham.ac.uk; or Craig E. Hughes, Institute for Cardiovascular and Metabolic Research, School of Biological Sciences, University of Reading, Reading RG6 6UB, United Kingdom; e-mail: c.e.hughes@bham.ac.uk.
References
- 1.Watson SP, Herbert JM, Pollitt AY. GPVI and CLEC-2 in hemostasis and vascular integrity. J Thromb Haemost. 2010;8(7):1456–1467. doi: 10.1111/j.1538-7836.2010.03875.x. [DOI] [PubMed] [Google Scholar]
- 2.Ryo R, Yoshida A, Sugano W, et al. Deficiency of P62, a putative collagen receptor, in platelets from a patient with defective collagen-induced platelet aggregation. Am J Hematol. 1992;39(1):25–31. doi: 10.1002/ajh.2830390107. [DOI] [PubMed] [Google Scholar]
- 3.Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. 1989;84(5):1440–1445. doi: 10.1172/JCI114318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arai M, Yamamoto N, Moroi M, Akamatsu N, Fukutake K, Tanoue K. Platelets with 10% of the normal amount of glycoprotein VI have an impaired response to collagen that results in a mild bleeding tendency. Br J Haematol. 1995;89(1):124–130. doi: 10.1111/j.1365-2141.1995.tb08900.x. [DOI] [PubMed] [Google Scholar]
- 5.Inoue O, Suzuki-Inoue K, McCarty OJ, et al. Laminin stimulates spreading of platelets through integrin alpha6beta1-dependent activation of GPVI. Blood. 2006;107(4):1405–1412. doi: 10.1182/blood-2005-06-2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaff M, Tang C, Maurer E, et al. Integrin α6β1 is the main receptor for vascular laminins and plays a role in platelet adhesion, activation, and arterial thrombosis. Circulation. 2013;128(5):541–552. doi: 10.1161/CIRCULATIONAHA.112.000799. [DOI] [PubMed] [Google Scholar]
- 7.Riba R, Hughes CE, Graham A, Watson SP, Naseem KM. Globular adiponectin induces platelet activation through the collagen receptor GPVI-Fc receptor gamma chain complex. J Thromb Haemost. 2008;6(6):1012–1020. doi: 10.1111/j.1538-7836.2008.02982.x. [DOI] [PubMed] [Google Scholar]
- 8.Seizer P, Borst O, Langer HF, et al. EMMPRIN (CD147) is a novel receptor for platelet GPVI and mediates platelet rolling via GPVI-EMMPRIN interaction. Thromb Haemost. 2009;101(4):682–686. doi: 10.1160/th08-06-0368. [DOI] [PubMed] [Google Scholar]
- 9.Knight CG, Morton LF, Onley DJ, et al. Collagen-platelet interaction: Gly-Pro-Hyp is uniquely specific for platelet Gp VI and mediates platelet activation by collagen. Cardiovasc Res. 1999;41(2):450–457. doi: 10.1016/s0008-6363(98)00306-x. [DOI] [PubMed] [Google Scholar]
- 10.Polgár J, Clemetson JM, Kehrel BE, et al. Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (tropical rattlesnake) venom via the p62/GPVI collagen receptor. J Biol Chem. 1997;272(21):13576–13583. doi: 10.1074/jbc.272.21.13576. [DOI] [PubMed] [Google Scholar]
- 11.Jandrot-Perrus M, Lagrue AH, Okuma M, Bon C. Adhesion and activation of human platelets induced by convulxin involve glycoprotein VI and integrin alpha2beta1. J Biol Chem. 1997;272(43):27035–27041. doi: 10.1074/jbc.272.43.27035. [DOI] [PubMed] [Google Scholar]
- 12.Flierl U, Nero TL, Lim B, et al. Phosphorothioate backbone modifications of nucleotide-based drugs are potent platelet activators. J Exp Med. 2015;212(2):129–137. doi: 10.1084/jem.20140391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alshehri OM, Montague S, Watson S, et al. Activation of Glycoprotein VI (GPVI) and C-type Lectin-like receptor-2 (CLEC-2) underlies platelet activation by diesel exhaust particles and other charged/hydrophobic ligands. Biochem J. 2015;468(3):459–473. doi: 10.1042/BJ20150192. [DOI] [PubMed] [Google Scholar]
- 14.Bynagari-Settipalli YS, Cornelissen I, Palmer D, et al. Redundancy and interaction of thrombin- and collagen-mediated platelet activation in tail bleeding and carotid thrombosis in mice. Arterioscler Thromb Vasc Biol. 2014;34(12):2563–2569. doi: 10.1161/ATVBAHA.114.304244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boulaftali Y, Hess PR, Getz TM, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123(2):908–916. doi: 10.1172/JCI65154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender M, Hagedorn I, Nieswandt B. Genetic and antibody-induced glycoprotein VI deficiency equally protects mice from mechanically and FeCl(3) -induced thrombosis. J Thromb Haemost. 2011;9(7):1423–1426. doi: 10.1111/j.1538-7836.2011.04328.x. [DOI] [PubMed] [Google Scholar]
- 17.Dubois C, Panicot-Dubois L, Merrill-Skoloff G, Furie B, Furie BC. Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo. Blood. 2006;107(10):3902–3906. doi: 10.1182/blood-2005-09-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konstantinides S, Ware J, Marchese P, Almus-Jacobs F, Loskutoff DJ, Ruggeri ZM. Distinct antithrombotic consequences of platelet glycoprotein Ibalpha and VI deficiency in a mouse model of arterial thrombosis. J Thromb Haemost. 2006;4(9):2014–2021. doi: 10.1111/j.1538-7836.2006.02086.x. [DOI] [PubMed] [Google Scholar]
- 19.Massberg S, Gawaz M, Grüner S, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197(1):41–49. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eckly A, Hechler B, Freund M, et al. Mechanisms underlying FeCl3-induced arterial thrombosis. J Thromb Haemost. 2011;9(4):779–789. doi: 10.1111/j.1538-7836.2011.04218.x. [DOI] [PubMed] [Google Scholar]
- 21.Gitz E, Pollitt AY, Gitz-Francois JJ, et al. CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood. 2014;124(14):2262–2270. doi: 10.1182/blood-2014-05-572818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato K, Kanaji T, Russell S, et al. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102(5):1701–1707. doi: 10.1182/blood-2003-03-0717. [DOI] [PubMed] [Google Scholar]
- 23.Emambokus NR, Frampton J. The glycoprotein IIb molecule is expressed on early murine hematopoietic progenitors and regulates their numbers in sites of hematopoiesis. Immunity. 2003;19(1):33–45. doi: 10.1016/s1074-7613(03)00173-0. [DOI] [PubMed] [Google Scholar]
- 24.Hughes CE, Finney BA, Koentgen F, Lowe KL, Watson SP. The N-terminal SH2 domain of Syk is required for (hem)ITAM, but not integrin, signaling in mouse platelets. Blood. 2015;125(1):144–154. doi: 10.1182/blood-2014-05-579375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Tamimi M, Mu FT, Moroi M, Gardiner EE, Berndt MC, Andrews RK. Measuring soluble platelet glycoprotein VI in human plasma by ELISA. Platelets. 2009;20(3):143–149. doi: 10.1080/09537100802710286. [DOI] [PubMed] [Google Scholar]
- 26.Bender M, May F, Lorenz V, et al. Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol. 2013;33(5):926–934. doi: 10.1161/ATVBAHA.112.300672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wonerow P, Pearce AC, Vaux DJ, Watson SP. A critical role for phospholipase Cgamma2 in alphaIIbbeta3-mediated platelet spreading. J Biol Chem. 2003;278(39):37520–37529. doi: 10.1074/jbc.M305077200. [DOI] [PubMed] [Google Scholar]
- 28.Clark EA, Shattil SJ, Ginsberg MH, Bolen J, Brugge JS. Regulation of the protein tyrosine kinase pp72syk by platelet agonists and the integrin alpha IIb beta 3. J Biol Chem. 1994;269(46):28859–28864. [PubMed] [Google Scholar]
- 29.Podolnikova NP, Yakovlev S, Yakubenko VP, Wang X, Gorkun OV, Ugarova TP. The interaction of integrin αIIbβ3 with fibrin occurs through multiple binding sites in the αIIb β-propeller domain. J Biol Chem. 2014;289(4):2371–2383. doi: 10.1074/jbc.M113.518126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost. 2005;3(8):1752–1762. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 31.Huang MM, Lipfert L, Cunningham M, Brugge JS, Ginsberg MH, Shattil SJ. Adhesive ligand binding to integrin alpha IIb beta 3 stimulates tyrosine phosphorylation of novel protein substrates before phosphorylation of pp125FAK. J Cell Biol. 1993;122(2):473–483. doi: 10.1083/jcb.122.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haimovich B, Lipfert L, Brugge JS, Shattil SJ. Tyrosine phosphorylation and cytoskeletal reorganization in platelets are triggered by interaction of integrin receptors with their immobilized ligands. J Biol Chem. 1993;268(21):15868–15877. [PubMed] [Google Scholar]
- 33.Calaminus SD, Thomas S, McCarty OJ, Machesky LM, Watson SP. Identification of a novel, actin-rich structure, the actin nodule, in the early stages of platelet spreading. J Thromb Haemost. 2008;6(11):1944–1952. doi: 10.1111/j.1538-7836.2008.03141.x. [DOI] [PubMed] [Google Scholar]
- 34.Munnix IC, Strehl A, Kuijpers MJ, et al. The glycoprotein VI-phospholipase Cgamma2 signaling pathway controls thrombus formation induced by collagen and tissue factor in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2005;25(12):2673–2678. doi: 10.1161/01.ATV.0000193568.71980.4a. [DOI] [PubMed] [Google Scholar]
- 35.Stefanini L, Paul DS, Robledo RF, et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J Clin Invest. 2015;125(4):1419–1432. doi: 10.1172/JCI77993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Podolnikova NP, Gorkun OV, Loreth RM, Yee VC, Lord ST, Ugarova TP. A cluster of basic amino acid residues in the gamma370-381 sequence of fibrinogen comprises a binding site for platelet integrin alpha(IIb)beta3 (glycoprotein IIb/IIIa). Biochemistry. 2005;44(51):16920–16930. doi: 10.1021/bi051581d. [DOI] [PubMed] [Google Scholar]
- 37.Rooney MM, Farrell DH, van Hemel BM, de Groot PG, Lord ST. The contribution of the three hypothesized integrin-binding sites in fibrinogen to platelet-mediated clot retraction. Blood. 1998;92(7):2374–2381. [PubMed] [Google Scholar]
- 38.Cohen I, Burk DL, White JG. The effect of peptides and monoclonal antibodies that bind to platelet glycoprotein IIb-IIIa complex on the development of clot tension. Blood. 1989;73(7):1880–1887. [PubMed] [Google Scholar]
- 39.Mammadova-Bach E, Ollivier V, Loyau S, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126(5):683–691. doi: 10.1182/blood-2015-02-629717. [DOI] [PubMed] [Google Scholar]
- 40.Zahid M, Mangin P, Loyau S, et al. The future of glycoprotein VI as an antithrombotic target. J Thromb Haemost. 2012;10(12):2418–2427. doi: 10.1111/jth.12009. [DOI] [PubMed] [Google Scholar]
- 41.Dütting S, Bender M, Nieswandt B. Platelet GPVI: a target for antithrombotic therapy?! Trends Pharmacol Sci. 2012;33(11):583–590. doi: 10.1016/j.tips.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Hermans C, Wittevrongel C, Thys C, Smethurst PA, Van Geet C, Freson K. A compound heterozygous mutation in glycoprotein VI in a patient with a bleeding disorder. J Thromb Haemost. 2009;7(8):1356–1363. doi: 10.1111/j.1538-7836.2009.03520.x. [DOI] [PubMed] [Google Scholar]
- 43.Matus V, Valenzuela G, Sáez CG, et al. An adenine insertion in exon 6 of human GP6 generates a truncated protein associated with a bleeding disorder in four Chilean families. J Thromb Haemost. 2013;11(9):1751–1759. doi: 10.1111/jth.12334. [DOI] [PubMed] [Google Scholar]