

Abstract
Using a variety of animal models of Alzheimer’s disease (AD), there have been a number of recent studies reporting varying degrees of success with anti-AD therapeutics. The efficacies are often discussed in terms of the modulatory effects of the compounds tested on identified or assumed targets among the known (or proposed) pathogenic and neuroprotective mechanisms, largely within the context of the dominant amyloid cascade hypothesis. However, it is clear that several of the relatively more efficacious treatments tend to be multifunctional and target multiple pathological processes associated with AD including most commonly, oxidative and metabolic stress and neuroinflammation. Increasing evidence suggests that vascular and neurodegenerative pathologies often co-exist and that neurovascular dysfunction plays a critical role in the development or progression of AD. In this review, we will discuss the significance of vasculoprotection or neurovascular unit (NVU) integrity as a common, multi-targeted mechanism underlying the reported efficacy of a majority of anti-AD therapeutics - amyloid-targeted or otherwise - while providing a strong support for future neurovascular-based treatment strategies and interventions.
Keywords: Neurovascular unit, cerebrovascular dysfunction, vascular risk factors, Type 2-diabetes, Blood brain barrier, anti-AD therapeutics
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia among the elderly accounting for two-thirds of all dementia cases, currently estimated to be over 35 million worldwide (http://www.alz.co.uk/research/world-report-2014). While the early-onset familial forms are relatively rare, the late-onset sporadic disease accounts for the majority (over 95%) of cases. Of particular concern, the incidence of this age-related neurodegenerative disease is set to increase exponentially with increased population growth and life expectancy. AD is associated with selective neuronal and synaptic loss along with characteristic pathological hallmarks including extracellular senile plaques containing beta-amyloid and intracellular neurofibrillary tangles (NFT) composed of hyperphosphorylated tau. Although there has been an impressive progress in our understanding of the genetics, neurobiology and neuropathological characteristics of the disease, clinical trials based on the dominant pathogenic mechanisms proposed i.e., ‘amyloid cascade’, have mostly failed (1). The only approved drugs available are symptomatic i.e., acetylcholine esterase inhibitors and an NMDA receptor antagonist, memantine. The lack of availability so far of any disease-modifying drugs is an indication that the precise disease mechanism for this complex condition remains unclear (2). Increasing evidence suggests that cognitive impairment in sporadic AD can occur independent of amyloid deposition, which may in fact, represent a down-stream result and not the cause of the disease (3). Although tau pathology seems to correlate better with cognitive decline suggesting the potential value of tau-targeted therapy (4), there have been no promising candidates so far.
In the wake of failed amyloid-targeted drug trials and immune therapies, recent efforts are directed towards a broad range of alternative mechanisms of AD including mitochondrial dysfunction, metabolic stress, altered insulin signaling and, related to the ‘vascular hypothesis of AD’ (5), cerebrovascular dysfunction. Accumulating evidence in fact supports the notion that cerebro- or neurovascular dysfunction may represent a primary initiator of a cascade of pathogenic events leading to neurodegeneration in AD (5–7). This mechanism takes on added significance when one considers increasing evidence linking sporadic AD with a number of vascular disorders including hypertension, hypercholesterolemia, obesity and type 2-diabetes (8–11). A pathological convergence between vascular cognitive impairment (VCI) associated with such metabolic disorders as well as stroke, and AD-type dementia recently referred to as ‘vascular contributions to cognitive impairment and dementia’ (VCID) (12, 13), likely occurs at the level of neurovascular dysfunction. Neurovascular dysfunction comprises the loss of neurovascular unit (NVU) integrity (-structural and functional) including blood brain barrier (BBB) disruption due to oxidative/metabolic stress and inflammation under a variety of brain injury and disease conditions (14). The concept of NVU originally proposed in the context of stroke has been expanded to incorporate its critical role in both health and disease and there is increasing evidence in support of its dysfunction associated with many neurodegenerative diseases, in particular AD (15, 16). With increasing emphasis on combination, drug repositioning or polypharmacology approaches to combat complex diseases, the attention has shifted towards functional units such as the NVU and other cellular networks commonly affected in neurodegenerative diseases. In a systems biology perspective, network perturbation or dysfunction can be viewed as a disease phenotype (17) and altered nodes and modules of the network represent potential multifunctional targets. In this review, we will make the case that the vascular component (-a key module) of the NVU represents a shared target for a variety of drug candidates with multitarget activity and hence vasculoprotection per se could be an effective and multi-targeted approach to treat AD. In support of this perspective, we will include a brief account of the recent literature on specific compounds that have shown efficacy in models of AD that may have in common, neurovascular dysfunction as a multifunctional target for treatment.
LOSS OF NVU INTEGRITY AND METABOLIC UNCOUPLING IN AD PATHOGENESIS
Evolving out of its original concept of a coupling between neuronal activity (energy demand) and local blood flow (energy supply), the term NVU now embodies an integrated multicellular system comprising cerebrovascular cells, glia and neurons and their milieu that tightly regulates brain homeostasis and function in health and disease (14, 18, 19). In the healthy brain, well-regulated interactions among components of the NVU i.e., vascular cells (endothelial cells, pericytes and artery/arteriole-associated smooth muscle cells), glia (astrocytes and microglia) and neurons maintain the structural and functional integrity of the unit thereby ensuring cerebrovascular autoregulation, functional hyperemia and intact BBB. Most significantly, an intact BBB ensures immune surveillance and regulated solute exchange at the barrier, energy (glucose and oxygen) supply to neurons through regulated CBF, trophic support and overall homeostatic balance (18, 20). As part of the BBB organization lining the brain capillaries is a specialized endothelium sealed by tight junctions that communicates with surrounding brain cells through a continuous basement membrane. This structure also embeds pericytes with long processes extending along the vessel wall as well as end-feet of perivascular astrocytes (14). The vascular hypothesis of AD proposes a loss of structural and functional integrity of the NVU as an early event leading to BBB deregulation, chronic cerebral hypoperfusion, hypoxia, neurovascular uncoupling, neuronal and glial hypometabolism or metabolic failure preceding neurodegeneration and cognitive impairment (7, 9) (Fig 1).
Fig 1.
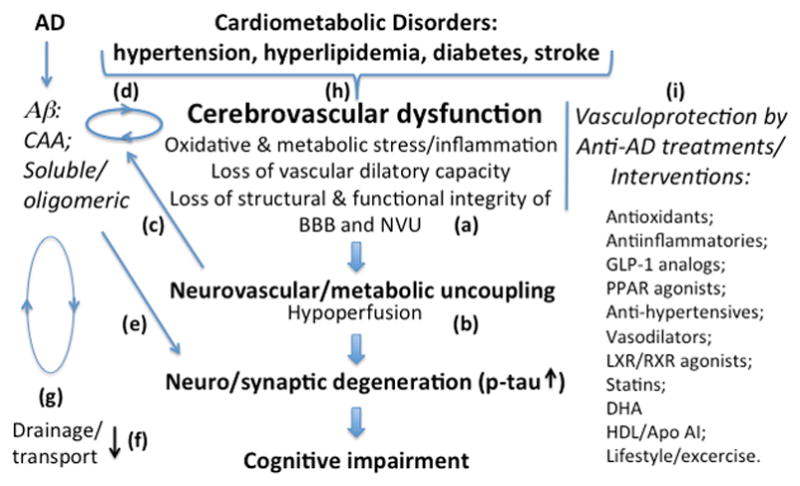
Proposed mechanisms and mediators of cerebrovascular dysfunction including neurovascular uncoupling in AD. Cerebrovascular dysfunction in AD is associated with compromised NVU integrity characterized by structural and functional abnormalities (a). Such changes include endothelial and smooth muscle cell damage, pericyte fall out, loss of astrocyte polarization, BBB breakdown, loss of vascular dilatory function and altered blood flow. While blood-borne toxins extravasating through broken BBB can be neurotoxic either directly or through inflammation (not shown), compromised function of uncoupled NVU can lead to neuronal and synaptic degeneration due to hypoperfusion and reduced energy supply (b). A state of hypoxia can result in increased amyloid processing thereby exacerbating amyloid pathology (c) and contributing to a vicious cycle at the cerebrovasculature of vascular damage and CAA formation (d). The oligomeric Aβ can elicit a direct neuro/synaptotoxicity (e). BBB dysfunction may have dual pathological consequences i.e., loss of amyloid transport out of the brain (f) and compromised transport into the CNS of nutrient and trophic factors. The loss of vascular integrity can also impede peri/paravascular drainage of excess amyloid (g). The scheme also depicts an interaction between AD and metabolic/vascular factors at the level of cerebrovasculature (h). A list of anti-AD treatments and interventions discussed in the review in the context of their vasculoprotective roles is shown on the right (i).
As an indicator of early NVU dysfunction in AD, it has been demonstrated that cerebral hypometabolism exists years before clinical symptoms of dementia and in animal models of AD, decreased cerebral glucose metabolism ensues even preceding Aβ deposition (21, 22). Since vascular insufficiency with underlyigng tissue hypoxia can accelerate amyloid production, the above findings suggest a vicious cycle involving amyloid-induced damage to already compromized NVU integrity and hence reduced CBF (23). Several studies have recorded specific pathological changes in the cellular and molecular components of the glio-vascular network that are in strong support of a vascular dysfunction hypothesis of AD (24). Structural and morphological abnormalities observed in both AD and aging including endothelial atrophy, thickened and irregular basement membranes, microvessel thinning (string vessels), their increased tortuosity and fragmentation would have adverse functional consequences of hypoperfusion and increased BBB leakage (6, 22, 24), especially when associated with cerebral amyloid angiopathy (CAA, below). Capillary rarefaction (decreased microvascular density) and degeneration are a common feature of AD brain (24, 25), most severe changes marking the zones of overt neuronal loss. Other observed cellular/structural changes of NVU in AD include pathological changes in astrocytes and loss of pericytes (26, 27) - both critical players in the maintenance of BBB integrity and function. Studies with mouse models of AD demonstrate astrocyte end-feet disruption (28, 29) that may adversely afftect vasodilation, BBB regulation and capillary blood flow. Age-dependent loss of brain pericytes in a mouse model results in BBB breakdown and reduced cerebral microcirculation preceding neurodegeneration and cognitive impairment (30) while pericyte deficiency in a transgenic AD model (APP Tg) leads to accelerated Aβ and CAA formation and the development of tau pathology along with cognitive decline (31). A recent study provides further evidence for age-dependent BBB breakdown in the human hippocampus correlated with pericyte injury that potentially contributes to observed mild cognitve impairment (32). Compromised barrier is thought to allow infiltration of blood-borne neurotoxins while perturbing the regulated entry of essential nutrients and trophic factors. Thus, recent studies have uncovered dual functions of a BBB endothelium-expressed protein i.e., major facilitator superfamily domain containing 2a (mfsd2a) in the formation and maintenance of BBB integrity (33) and the transport into the brain of docosahexaenoic acid (DHA) (34), an omega-3 fatty acid that, besides being vasculoprotective (35), has essential functions in brain growth and cognition. A loss of this protein in AD due to pericyte deficiency can result in reduced brain DHA content (36). Another recent report shows that deficiency of glucose transporter (GLUT1) in endothelium exacerbates AD-associated ‘vasculo-neuronal’ dysfunction and neurodegeneration in a mouse model (37). The transport of other trophic factors i.e., IGF-I and insulin also depends on specific transporters. These transporters are subject to down-regulation under conditions of BBB dysfunction, especially in metabolic disorders such as type 2 diabetes (38, 39) that are known to increase AD risk (below).
There is now strong evidence for increased AD risk in subjects with vascular conditions including hypertension, hypercholesterolemia and type-2 diabetes (T2DM) (8–11). In fact, the term, ‘Type 3 diabetes’ has been used to describe AD and there is evidence for altered metabolic changes indicative of ‘insulin resistant brain state’ (IRBS) in human AD and animal models (40, 41). While increased risk of dementia in T2DM can be attributed to cerebrovascular disease (CVD) (42–44) involving vascular injury, it has been suggested that AD and CVD may work synergistically to cause cognitive decline since for many patients markers of vascular injury co-exist with traditional AD hallmarks (44, 45). The frequent vascular (-arteries, arterioles and capillaries) amyloid deposition seen in AD termed cerebral amyloid angiopathy (CAA) (46–48) is associated with vascular injury (48). It is likely that the presence of CVDs such as T2DM would exacerbate the pathological changes in NVU described above via endothelial cell damage due to hyperlipidemia, hyperglycemia, oxidative stress and inflammation (7). Since pericyte-endothelial cross-talk and trophic interactions maintain each-other’s health and integrity (49), it is likely that endothelial dysfunction due to vascular factors (i.e., T2DM) could lead to increased pericyte injury/loss as well. Both cell types are highly vulnerable to oxidative stress and inflammatory mediators. With their expressed innate immune cell characteristics (50), these cells can also elicit inflammatory response thereby contributing to accelerated vascualr damage.
An intact NVU is also important for regulated amyloid clearance. It is becoming evident that ineffective clearance of brain amyloid rather than its increased production is the major cause of amyloid accumulation, especially in sporadic AD. Components of NVU are critically involved in multiple mechanisms of amyloid clearance including transport through the barrier (51, 52) and perivascular drainage (53, 54). Enzymatic/phagocytic degradation of Aβ represents the third mechanism, which engages the activity of several proteases and phagocytic glia/macrophages (55–57). Even here, it is thought that infiltrating blood-borne monocytes or perivascular macrophages are better able to engulf amyloid than resident microglia, which are incapacitated by senescene and phenotypic heterogeneity. The transcytotic delivery system has been extensively studied including the LRP-1/RAGE tandem at the BBB. Thus, while LRP-1 transports Aβ out of the CNS into the blood stream, RAGE acts in the opposite way. A specific inhibitor has been shown to block RAGE-mediated Aβ import thereby reducing amyloid pathology in a mouse model of AD (58). The transport and perivascular drainage systems are regulated by Apo E in an isoform-specific manner so that the pathogenic Apo E4 isoform may interfere with Aβ clearance across the BBB and via the drainage pathway (59). A proposed mechanism for the toxicity of Apo E4 on the cerebrovascular system involves activation of a proinflammaory cyclophyllin A-mediated pathway in pericytes leading to BBB breakdown (60). The cerebrovascular abnormalities can further interfere with the drainage system, which comprises the newly defined ‘glymphatic pathway’ –a CNS equivalent to peripheral lymphatic system. In this brain-wide paravascular pathway, subarachnoid CSF recirculates through the brain parenchyma. The CSF-interstitial fluid (ISF) exchange ensures an efficient clearance of the solutes and other waste products including beta-amyloid (61). The convective flow process is dependent on water transport via astrocytic aquaporin-4 (Aqp-4) water channels localized to their endfeet. A recent study by Kress et al demonstrates an age-dependent impairment of this pathway that correlated with the loss of Aqp-4 polarization (62). It is thought that arterial pulsatility drives perivascular CSF-ISF exchange (63) thereby suggesting the potential utility of vasoactive drugs to facilitate amyloid clearance. In fact, in a mouse model of cerebrovascular β-amyloidosis (i.e., Tg-SwDI), administration of cilostazol, an inhibitor of phosphodiesterase III with vasodilator activity was found to restore vasoreactivity and facilitate perivascular drainage of Aβ while preventing cognitive decline (64). Notably, cilostazol despite being BBB impermeable, was able to attenuate degradation of vascular walls with Aβ deposit and preserve NVU integrity.
It is important to note however, that improvement in vascular function in mouse models of AD in certain other cases has been reported to occur independently from Aβ reduction or improved memory deficits (below).
NVU AS A CONVERGENT, MULTIFUNCTIONAL TARGET FOR DISEASE MODIFICATION IN AD
From the above discussion it is clear that an intact NVU with its multifaceted roles, is critical for the maintenance of brain health and homeostasis -metabolic and ionic. Hence, the loss of its structural and functional integrity represents a key target of intervention in AD as well as in a variety of other cerebrovascular disorders and conditions. It is important to note that most, if not all of the alternative pathogenic processes (-amyloid related or otherwise) thought to contribute to AD and hence considered the targets of treatment, commonly and adversely affect the cerebrovasculature as well. These include oxidative, metabolic and endoplasmic reticulum (ER) stress, mitochondrial dysfunction, inflammation, insulin resistance etc. Hence, one can argue that the effectiveness of several of the preclinically tested anti-AD candidates may to a large extent draw from their vasculoprotective activities potentially targeting NVU-associated multiple mechanisms (specific examples to follow). Along this line, shared risk factors and mechanisms between AD and CVDs have prompted repurposing of approved drugs that may find NVU as the primary target. Drug repositioning approach usually takes advantage of the multiple beneficial properties of the drug related to common cellular processes that are either compromised or exacerbated. An alternative is to identify novel targets for the approved drugs. The current drug repositioning approach in AD mostly focuses on co-morbidities and common risk factors including diabetes, atherosclerosis and hypertension (65). Below, we will discuss with specific examples, how observed effectiveness of such re-purposed drugs and other multifunctional agents can be viewed in the context of vasculoprotection. The discussion is restricted to preclinical models only since for most candidates, clinical trials are either incomplete or have indicated questionable benefit and/or adverse side effects (longer term). Another caveat pertains to the shortcomings of the animal models per se as noted by Cavanaugh et al. (66) since a number of candidate therapeutics, despite showing great promise in such studies, have rarely translated into clinical benefits for patients.
Anti-diabetic and anti-hypertensive drugs
Some of the priority candidates that are being tested under drug repositioning strategy include anti-diabetics based on the link between diabetes and AD and the evidence for AD-associated insulin resistance, as noted above. Although the mechanistic link(s) between peripheral and brain insulin resistance in AD remains unclear, studies suggest that T2DM is associated with reduced brain insulin signaling (8, 41). Insulin and IGF-I have multiple physiological roles in the brain including trophic support, energy metabolism and synaptic activity (38, 67) thereby underscoring the importance of insulin-based therapies. In fact, promising clinical trials have been implemented involving intranasal administration of insulin in AD patients [reviewed in (68)]. A related insulin-targeted approach involves the use of insulin sensitizers or agents that stimulate insulin release, most commonly glucagon-like peptide-1 (GLP-1) analogues i.e., exendin-4 and liraglutide. In preclinical studies, different GLP-1 receptor agonists have shown neuro- and synaptic protection and in some cases, to reduce plaque burden (41, 68, 69). Multiple ‘neurocentric’ mechanisms have been proposed for their effectiveness including improved axonal transport and synaptic plasticity. However, a review of the literature suggests that GLP-1 analogues also elicit vasculoprotective effects. Thus, for example, GLP-1 enhances endothelial function in hypertension (70) and promotes endothelial barrier activity (71). GLP-1 analogs can also protect against ischemia/reperfusion injury as shown in a rat model of T2DM (72). With respect to AD, while the GLP-1 analog, liraglutide could reduce memory impairment, synaptic loss and plaque load in aged APP-PS1 Tg mice (73), a recent follow-up study indicates that the compound can restore cerebral and peripheral microvascular architecture in these mice while reducing the incidence of cerebral microaneurysms and leakage (74). The prediction is that other analogs of this class of drugs will have similar vasculoprotective effects (75).
Members of a class of nuclear receptors i.e., peroxisome proliferator-activated receptors (PPARs) that regulates glucose and lipid metabolism, represent another target of treatment in AD as well as in T2DM. In particular, PPARγ is known to play neuroprotective roles in models of neurodegeneration and agonists of PPARγ such as rosiglitazone and pioglitazone used in the treatment of T2DM, have been shown to improve memory while reducing AD-like pathology in animal models (76). The mechanisms targeted could be multifaceted including oxidative stress, mitochondrial dysfunction and inflammation. With respect to their potential vasculoprotective effects, in old AD Tg mice, a thiazolidine (TZD) agonist of PPAR-gamma i.e., pioglitazone improved cerebrovascular function without any benefit on the amyloid pathology (77). The vasculoprotective effects were attributed to the recovery of dilatory function through antioxidant and antiinflammatory mechanisms. A literature survey would reveal that similar to PPARγ, other members i.e., α and δ also have vasculoprotective roles and hence, may benefit cerebrovascular dysfunction.
Hamel et al. have also tested the effects of several other ‘repurposed’ compounds including statins and anti-hypertensive drugs in the AD Tg mice. Thus, the statin, simvastatin restored vascular reactivity, dilatory function, neurovascular coupling and memory in adult AD Tg mice, without reducing the amyloid pathology (78). The cerebrovascular benefits were associated with anti oxidant effects and activation of eNOS. Recovery of memory did not occur in aged AD Tg mice, suggesting a therapeutic window as is likely to be the case in man. Of note, statins have been extensively studied preclinically and in AD subjects but without clear clinical benefit. Nonetheless, the primary target of their pleiotropic activity in most metabolic disorders seems to involve the vascular system, activation of endothelial nitric oxide (eNOS) being a key mechanism as also seen in a model of cerebrovascualr disease i.e., the double Tg AD mice expressing active form of TGFβ (79). Midlife hypertension is another co-morbid vascular condition associated with AD development and there have been studies using approved anti-hypertensive drugs. Thus, the AT1 receptor blocker, losartan was able to reduce cerebrovascular and neuropathological changes and cognitive deficit in the APP Tg mice without an effect on soluble Aβ or plaque load (80).
Another class of anti-hypertensives with vasodilatory effects but capable of eliciting direct neuroprotection, is calcium channel blockers (CCBs). Specific CCBs have shown beneficial effects in animal models and in clinical/epidemiological studies; one of these (i.e., dihydropyridine CCB) seems to reduce or delay the development of AD [reviewed in (65)]
Other nuclear receptor agonists
Similar to PPARs discussed above, other nuclear receptors including liver x receptor (LXR) (81) and retinoic acid receptor (RXR) have been investigated as potential targets of treatment in AD. Of particular interest, Cramer et al. (82) reported that an RXR agonist, bexarotene (Bex), an approved skin cancer drug, showed remarkable effects on cognition and soluble amyloid clearance in the APP-PS1 Tg mice. However, this study was followed by a flurry of reports with disparate results some supporting and many others disputing the findings [discussed in (83)], although all of them confirmed target engagement i.e., increased expression of Apo E and ABCA1. In their report, Cramer et al suggested that the benefit observed was the result of lipidated Apo E-mediated rapid clearance of Aβ. It is possible that ABCA1-mediated lipidation of human Apo E4 isoform, a primary risk factor for sporadic AD, would have additional beneficial effects including reversal of Apo E4-mediated cerebrovascular toxicity.
The mechanism by which RXR agonists induce transcription involves the formation of heterodimers of the transcription factor with other nuclear receptors i.e., PPAR and LXR that are known to act in a coordinated manner to induce genes such as Apo E and ABCA1. In this regard, several studies have been published demonstrating the efficacy of LXR agonists as well as PPAR activators in AD models as noted above. However, much needs to be clarified with respect to the mechanisms by which nuclear receptor activation can be beneficial (or harmful i.e., Bex, a case in point) in AD. Within the cerebrovasculature, besides ABCA1, other transporters (including that for Aβ i.e., ABCB1 and ABCG2) and regulators of antioxidant defense and energy metabolism could be under the control of nuclear receptors (76). Alternatively, the nuclear receptors can also have non-genomic, acute actions. It is interesting that a recent mouse BBB transcriptome analysis has revealed that RxRα cascade is specifically enriched at the BBB (84). Hence, there is justification in considering cerebrovasculature as an alternative target of RxR (and other nuclear receptor) activation.
Modulators of LXR-ABCA1-Apo AI axis
It is now well established that a number of genes (e.g., cyp46, ABCA1) coding for proteins of cholesterol metabolism show disease-associated polymorphisms (85). Further, besides Apo E, alterations in several other mediators of cholesterol homeostasis including ABC transporters, Apo-AI as well as nuclear receptors (i.e., LXR, PPARs) that regulate their synthesis also influence cognitive function in AD models (86–89). These findings are in general agreement with the clinical observations that elevated levels of LDL cholesterol and reduced/dysfunctional HDL and Apo-AI seen in both atherosclerosis and T2DM (90–93), correlate well with AD incidence compared to asymptomatic cases (94, 95). In contrast, high HDL-C and Apo-AI correlate with better cognitive function in advanced age (96). The beneficial effect of the ‘good cholesterol’ in the form of HDL in cardiovascular diseases is well accepted and there have been intense efforts at boosting its circulating levels/functions as an atheroprotective therapeutic approach. In addition to its known role in lipid transport (RCT), HDL regulates vascular health via its antiinflammatory and anti-oxidant properties. The major constituent of HDL i.e., Apo AI determines the beneficial properties of HDL. Small Apo AI mimetic synthetic peptides have shown activity in models of atherosclerosis and T2DM via their anti-inflammatory, anti-oxidant and anti-atherogenic effects (90, 97, 98). Although not produced in the CNS, high levels of Apo AI are found in the CSF (99). Interestingly, genetic manipulation of Apo AI levels in a mouse model of AD, selectively affects vascular amyloid load and cognitive function i.e., its deletion exacerbates CAA and cognitive deficit (88) while its overexpression has the opposite effects (89). In support of the potential HDL/Apo AI-based vasculoprotective therapy, an Apo AI mimetic peptide, 4-DF administered to mice with genetic (LDL receptor-deficient) hypercholesterolemia mice reduced cerbebrovascular inflammation and improved cognitive performance (100) and, when administered with a statin, it was able to reduce amyloid burden and improve cognitive function in an AD model (101). Certainly, there is much value in the development of ABCA1 agonists and small molecule Apo AI modulators such as RVX208 (102), which may find use in AD as well as across a spectrum of cardio- and cerbrovascular diseases.
Finally, one can assign vasculoprotective function to several other compounds with known antioxidant, anti-inflammatory and ER stress reducing properties that have been and are being tested in AD models. Since the vasculoprotective actions of statins and PPAR-gamma agonists mostly derive from their ‘pleiotropic’ actions rather than anti-cholesterol and anti-diabetic effects, the list could be expanded to include additional pleiotropic compounds such as polyphenols, resveratrol (- SIRT1 activation) (103) and rapamycin. While rapamycin has multiple targets, in a recent study, chronic administration of this target-of-rapamycin (TOR) inhibitor was able to restore brain cerebrovascular integrity and function involving an activation of eNOS and improve memory in an AD model (104). It may not be out of place here to mention recent exciting findings of cerebrovascular and neurogenic rejuvenation of the aging brain by young systemic factors (105) perhaps, representing the ‘ultimate’ pleiotropic approach to treat age-related neurodegenerative and neurovascular diseases.
CONCLUDING REMARKS
AD is a complex disease often involving vascular pathologies and hence requires a multitargeted treatment strategy. The shared risk factors between AD and vascular disorders along with a multifactorial etiology of the disease point to potential utility of drug repositioning and multi-target or polypharmacology approaches. Pertinent to this, vasculoprotection represents a major target for existing drugs while promising to be a critical and active area of AD research to identify novel compounds. While adaptation to life-style modifications i.e., diet and exercise (106) with established benefits on vascular health in general is obvious both preventively and therapeutically, there is a need for developing novel strategies addressing neurovascular dysfunction and BBB integrity in AD. As discussed in this review, failed clinical trials focusing on pathological markers or indicators of AD, highlight the need for clearer understanding of the alternative disease mechanisms. Based on accumulating evidence, a key mechanism seems to involve a dysfunctional NVU. In fact, NVU represents a common and most effective target of treatment for AD as well as other cerebrovascular diseases, in particular stroke. The critical and multifaceted roles that NVU plays in maintaining the metabolic and ionic homeostasis are compromised in these conditions. As we discussed here with specific examples, many disease modifying therapeutics being tested in AD models –repurposed or novel- would have NVU dysfunction as the potential multi-targeted mechanism. This also means that future chemical scaffold-based design of synthetic multi-target directed drugs (MLTDs) should incorporate components of NVU as mechanistic targets. The outcome or target engagement measures should accordingly include neurovascular structure-function-based analyses, in addition to the measurement of classic AD markers i.e., Aβ and phospho-tau/NFT and, importantly their clearance, in relation to cognitive changes in preclinical studies. Some of the common vascular-based outcome measures/markers include: cerebral/microvascular blood flow, dilatory response, functional hyperemia/neurovascular coupling, cerebrovascular oxidative stress - the primary mechanism through which Aβ peptide impairs the function of the brain vasculature, vascular inflammatory markers, brain glucose/energy metabolism, markers/measures of glymphatic pathway of toxic solute (Aβ, tau etc) removal, astrocyte end feet and cerebrovascular basement membrane changes as well as pericyte loss/degeneration and measures of BBB leakage. Some of these analyses require advanced and live imaging techniques for functional measurements and structural analysis at the ultrastructure level. There is also the need for the development of novel and appropriate models incorporating co-morbid factors for proof-of-concept and proof-of-mechanism studies and preclinical testing. In this regard, the cerebrovascular disease model described by Papadopoulos et al (79) i.e., APP Tg mice expressing constitutively active form of TGFβ represents an innovative attempt at incorporating co-morbid factors. However, in this model, the therapeutic efficacy of some compounds did not survive as shown for simvastatin and the TZD pioglitazone (107). Obviously, there is need for developing more complex models incorporating vascular co-morbidities and metabolic disorders i.e., hypertension, microinfarcts/stroke, prediabetes, type-2 diabetes (genetic vs. non-genetic) etc.
Acknowledgments
This work was supported by grants from ADDF (#20131214), AHAF/Brightfocus (A2011081) and NIH (NS063183).
References
- 1.Lundkvist J, Halldin MM, Sandin J, Nordvall G, Forsell P, Svensson S, Jansson L, Johansson G, Winblad B, Ekstrand J. The battle of Alzheimer’s Disease - the beginning of the future Unleashing the potential of academic discoveries. Frontiers in pharmacology. 2014;5:102. doi: 10.3389/fphar.2014.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellani RJ, Perry G. The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochemical pharmacology. 2014;88:671–676. doi: 10.1016/j.bcp.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 3.Drachman DA. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2014;10:372–380. doi: 10.1016/j.jalz.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Giacobini E, Gold G. Alzheimer disease therapy--moving from amyloid-beta to tau. Nature reviews Neurology. 2013;9:677–686. doi: 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- 5.de la Torre JC. The vascular hypothesis of Alzheimer’s disease: bench to bedside and beyond. Neuro-degenerative diseases. 2010;7:116–121. doi: 10.1159/000285520. [DOI] [PubMed] [Google Scholar]
- 6.Ostergaard L, Aamand R, Gutierrez-Jimenez E, Ho YC, Blicher JU, Madsen SM, Nagenthiraja K, Dalby RB, Drasbek KR, Moller A, Braendgaard H, Mouridsen K, Jespersen SN, Jensen MS, West MJ. The capillary dysfunction hypothesis of Alzheimer’s disease. Neurobiology of aging. 2013;34:1018–1031. doi: 10.1016/j.neurobiolaging.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Kelleher RJ, Soiza RL. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? American journal of cardiovascular disease. 2013;3:197–226. [PMC free article] [PubMed] [Google Scholar]
- 8.Bhat NR. Linking cardiometabolic disorders to sporadic Alzheimer’s disease: a perspective on potential mechanisms and mediators. Journal of neurochemistry. 2010;115:551–562. doi: 10.1111/j.1471-4159.2010.06978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muresanu DF, Popa-Wagner A, Stan A, Buga AM, Popescu BO. The vascular component of Alzheimer’s disease. Current neurovascular research. 2014;11:168–176. doi: 10.2174/1567202611666140408105333. [DOI] [PubMed] [Google Scholar]
- 10.Akinyemi RO, Mukaetova-Ladinska EB, Attems J, Ihara M, Kalaria RN. Vascular risk factors and neurodegeneration in ageing related dementias: Alzheimer’s disease and vascular dementia. Current Alzheimer research. 2013;10:642–653. doi: 10.2174/15672050113109990037. [DOI] [PubMed] [Google Scholar]
- 11.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Archives of neurology. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke; a journal of cerebral circulation. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, Lamb BT, Montine TJ, Nedergaard M, Schaffer CB, Schneider JA, Wellington C, Wilcock DM, Zipfel GJ, Zlokovic B, Bain LJ, Bosetti F, Galis ZS, Koroshetz W, Carrillo MC. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2014 doi: 10.1016/j.jalz.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoknecht K, David Y, Heinemann U. The blood-brain barrier-Gatekeeper to neuronal homeostasis: Clinical implications in the setting of stroke. Seminars in cell & developmental biology. 2014 doi: 10.1016/j.semcdb.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Guo S, Lo EH. Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke; a journal of cerebral circulation. 2009;40:S4–7. doi: 10.1161/STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csermely P, Korcsmaros T, Kiss HJ, London G, Nussinov R. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacology & therapeutics. 2013;138:333–408. doi: 10.1016/j.pharmthera.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muoio V, Persson PB, Sendeski MM. The neurovascular unit - concept review. Acta Physiol (Oxf) 2014;210:790–798. doi: 10.1111/apha.12250. [DOI] [PubMed] [Google Scholar]
- 19.Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnar Z, O’Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR. Engaging neuroscience to advance translational research in brain barrier biology. Nature reviews Neuroscience. 2011;12:169–182. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daneman R, Prat A. The Blood-Brain Barrier. Cold Spring Harbor perspectives in biology. 2015;7 doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicolakakis N, Hamel E. Neurovascular function in Alzheimer’s disease patients and experimental models. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:1354–1370. doi: 10.1038/jcbfm.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomason LA, Stefanovic B, McLaurin J. Cerebrovascular contributions to Alzheimer’s disease pathophysiology and potential therapeutic interventions in mouse models. The European journal of neuroscience. 2013;37:1994–2004. doi: 10.1111/ejn.12181. [DOI] [PubMed] [Google Scholar]
- 23.de la Torre JC. Cerebral hemodynamics and vascular risk factors: setting the stage for Alzheimer’s disease. Journal of Alzheimer’s disease : JAD. 2012;32:553–567. doi: 10.3233/JAD-2012-120793. [DOI] [PubMed] [Google Scholar]
- 24.Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol and applied neurobiol. 2011;37:56–74. doi: 10.1111/j.1365-2990.2010.01139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter JM, Kwan J, Malek-Ahmadi M, Maarouf CL, Kokjohn TA, Belden C, Sabbagh MN, Beach TG, Roher AE. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer’s disease. PloS one. 2012;7:e36893. doi: 10.1371/journal.pone.0036893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013;23:303–310. doi: 10.1111/bpa.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014;24:371–386. doi: 10.1111/bpa.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM. Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta neuropathol. 2011;122:293–311. doi: 10.1007/s00401-011-0834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience. 2009;159:1055–1069. doi: 10.1016/j.neuroscience.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nature communications. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509:507–511. doi: 10.1038/nature13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, Wenk MR, Goh EL, Silver DL. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature. 2014;509:503–506. doi: 10.1038/nature13241. [DOI] [PubMed] [Google Scholar]
- 35.Siegel G, Ermilov E. Omega-3 fatty acids: benefits for cardio-cerebro-vascular diseases. Atherosclerosis. 2012;225:291–295. doi: 10.1016/j.atherosclerosis.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Z, Zlokovic BV. Blood-brain barrier: a dual life of MFSD2A? Neuron. 2014;82:728–730. doi: 10.1016/j.neuron.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P, Nelson AR, Sullivan JS, Zhao Z, Meiselman HJ, Wenby RB, Soto J, Abel ED, Makshanoff J, Zuniga E, De Vivo DC, Zlokovic BV. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nature neuroscience. 2015 doi: 10.1038/nn.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacology & therapeutics. 2012;136:82–93. doi: 10.1016/j.pharmthera.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aleman A, Torres-Aleman I. Circulating insulin-like growth factor I and cognitive function: neuromodulation throughout the lifespan. Progress in neurobiology. 2009;89:256–265. doi: 10.1016/j.pneurobio.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 40.de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochemical pharmacology. 2014;88:548–559. doi: 10.1016/j.bcp.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sebastiao I, Candeias E, Santos MS, de Oliveira CR, Moreira PI, Duarte AI. Insulin as a Bridge between Type 2 Diabetes and Alzheimer Disease - How Anti-Diabetics Could be a Solution for Dementia. Frontiers in endocrinology. 2014;5:110. doi: 10.3389/fendo.2014.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cechetto DF, Hachinski V, Whitehead SN. Vascular risk factors and Alzheimer’s disease. Expert Rev Neurother. 2008;8:743–750. doi: 10.1586/14737175.8.5.743. [DOI] [PubMed] [Google Scholar]
- 43.JSRF, Sa-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML, Moriguti JC, Roriz-Cruz M. (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta. 2009;1792:432–443. doi: 10.1016/j.bbadis.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Rocchi A, Orsucci D, Tognoni G, Ceravolo R, Siciliano G. The role of vascular factors in late-onset sporadic Alzheimer’s disease. Genetic and molecular aspects. Curr Alzheimer Res. 2009;6:224–237. doi: 10.2174/156720509788486644. [DOI] [PubMed] [Google Scholar]
- 45.de la Torre JC. Cerebrovascular and cardiovascular pathology in Alzheimer’s disease. Int Rev Neurobiol. 2009;84:35–48. doi: 10.1016/S0074-7742(09)00403-6. [DOI] [PubMed] [Google Scholar]
- 46.Kumar-Singh S. Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 2008;7(Suppl 1):67–82. doi: 10.1111/j.1601-183X.2007.00380.x. [DOI] [PubMed] [Google Scholar]
- 47.Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- 48.Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC medicine. 2014;12:206. doi: 10.1186/s12916-014-0206-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geevarghese A, I, Herman M. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Translational research : the journal of laboratory and clinical medicine. 2014;163:296–306. doi: 10.1016/j.trsl.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lampron A, Elali A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron. 2013;78:214–232. doi: 10.1016/j.neuron.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature rev Neuroscience. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sagare AP, Bell RD, Zlokovic BV. Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer’s disease. Journal of Alzheimer’s disease : JAD. 2013;33(Suppl 1):S87–100. doi: 10.3233/JAD-2012-129037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris AW, Carare RO, Schreiber S, Hawkes CA. The Cerebrovascular Basement Membrane: Role in the Clearance of beta-amyloid and Cerebral Amyloid Angiopathy. Frontiers in aging neuroscience. 2014;6:251. doi: 10.3389/fnagi.2014.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta neuropathologica. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 55.Pacheco-Quinto J, Herdt A, Eckman CB, Eckman EA. Endothelin-converting enzymes and related metalloproteases in Alzheimer’s disease. Journal of Alzheimer’s disease : JAD. 2013;33(Suppl 1):S101–110. doi: 10.3233/JAD-2012-129043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hickman SE, El Khoury J. The neuroimmune system in Alzheimer’s disease: the glass is half full. Journal of Alzheimer’s disease : JAD. 2013;33(Suppl 1):S295–302. doi: 10.3233/JAD-2012-129027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guillot-Sestier MV, Town T. Innate immunity in Alzheimer’s disease: a complex affair. CNS & neurological disord drug targets. 2013;12:593–607. doi: 10.2174/1871527311312050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M, Zarcone T, Fritz G, Friedman AE, Miller BL, Zlokovic BV. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. The Journal of clinical investigation. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, Carare RO. Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PloS one. 2012;7:e41636. doi: 10.1371/journal.pone.0041636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Science translational medicine. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M. Impairment of paravascular clearance pathways in the aging brain. Annals of neurology. 2014;76:845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maki T, Okamoto Y, Carare RO, Hase Y, Hattori Y, Hawkes CA, Saito S, Yamamoto Y, Terasaki Y, Ishibashi-Ueda H, Taguchi A, Takahashi R, Miyakawa T, Kalaria RN, Lo EH, Arai K, Ihara M. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular beta-amyloid. Annals of clinical and translational neurology. 2014;1:519–533. doi: 10.1002/acn3.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Broadstock M, Ballard C, Corbett A. Latest treatment options for Alzheimer’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Expert opinion on pharmacotherapy. 2014;15:1797–1810. doi: 10.1517/14656566.2014.936848. [DOI] [PubMed] [Google Scholar]
- 66.Cavanaugh SE, Pippin JJ, Barnard ND. Animal models of Alzheimer disease: historical pitfalls and a path forward. Altex. 2014;31:279–302. doi: 10.14573/altex.1310071. [DOI] [PubMed] [Google Scholar]
- 67.Fernandez AM, Torres-Aleman I. The many faces of insulin-like peptide signalling in the brain. Nature reviews Neuroscience. 2012;13:225–239. doi: 10.1038/nrn3209. [DOI] [PubMed] [Google Scholar]
- 68.Holscher C. First clinical data of the neuroprotective effects of nasal insulin application in patients with Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2014;10:S33–37. doi: 10.1016/j.jalz.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 69.Patrone C, Eriksson O, Lindholm D. Diabetes drugs and neurological disorders: new views and therapeutic possibilities. The lancet Diabetes & endocrinology. 2014;2:256–262. doi: 10.1016/S2213-8587(13)70125-6. [DOI] [PubMed] [Google Scholar]
- 70.Limei L, Jian L, Yu H. Protective effects of glucagon-like peptide-1 on endothelial function in hypertension. Journal of cardiovascular pharmacology. 2014 doi: 10.1097/FJC.0000000000000176. [DOI] [PubMed] [Google Scholar]
- 71.Li AQ, Zhao L, Zhou TF, Zhang MQ, Qin XM. Exendin-4 promotes endothelial barrier enhancement via PKA- and Epac1-dependent Rac1 activation. American journal of physiology Cell physiology. 2015;308:C164–175. doi: 10.1152/ajpcell.00249.2014. [DOI] [PubMed] [Google Scholar]
- 72.Zhao L, Xu J, Wang Q, Qian Z, Feng W, Yin X, Fang Y. Protective effect of rhGLP-1 (7–36) on brain ischemia/reperfusion damage in diabetic rats. Brain research. 2015 doi: 10.1016/j.brainres.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 73.McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76(Pt A):57–67. doi: 10.1016/j.neuropharm.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 74.Kelly P, McClean P, Ackermann M, Konerding MA, Holscher C, Mitchell CA. Restoration of cerebral and systemic microvascular architecture in APP/PS1 transgenic mice following treatment with Liraglutide. Microcirculation. 2014 doi: 10.1111/micc.12186. [DOI] [PubMed] [Google Scholar]
- 75.Avogaro A, Vigili de Kreutzenberg S, Fadini GP. Cardiovascular actions of GLP-1 and incretin-based pharmacotherapy. Current diabetes reports. 2014;14:483. doi: 10.1007/s11892-014-0483-3. [DOI] [PubMed] [Google Scholar]
- 76.Zolezzi JM, Bastias-Candia S, Santos MJ, Inestrosa NC. Alzheimer’s disease: relevant molecular and physiopathological events affecting amyloid-beta brain balance and the putative role of PPARs. Frontiers in aging neuroscience. 2014;6:176. doi: 10.3389/fnagi.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, Tong XK, Hamel E. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tong XK, Lecrux C, Rosa-Neto P, Hamel E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:4705–4715. doi: 10.1523/JNEUROSCI.0169-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Papadopoulos P, Rosa-Neto P, Rochford J, Hamel E. Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer’s disease with combined amyloid-beta and cerebrovascular pathology. PloS one. 2013;8:e68612. doi: 10.1371/journal.pone.0068612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ongali B, Nicolakakis N, Tong XK, Aboulkassim T, Papadopoulos P, Rosa-Neto P, Lecrux C, Imboden H, Hamel E. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiology of disease. 2014;68:126–136. doi: 10.1016/j.nbd.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 81.Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nature reviews Drug discovery. 2014;13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 82.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tesseur I, De Strooper B. When the dust settles: what did we learn from the bexarotene discussion? Alzheimer’s research & therapy. 2013;5:54. doi: 10.1186/alzrt218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS one. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wollmer MA. Cholesterol-related genes in Alzheimer’s disease. Biochim Biophys Acta. 1801:762–773. doi: 10.1016/j.bbalip.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 86.Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280:43236–43242. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- 88.Lefterov I, Fitz NF, Cronican AA, Fogg A, Lefterov P, Kodali R, Wetzel R, Koldamova R. Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem. 2010;285:36945–36957. doi: 10.1074/jbc.M110.127738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lewis TL, Cao D, Lu H, Mans RA, Su YR, Jungbauer L, Linton MF, Fazio S, LaDu MJ, Li L. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem. 2010;285:36958–36968. doi: 10.1074/jbc.M110.127829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- 91.Ng DS, Saw NM. The role of HDL and its modulators in the development of diabetes. Current opinion in lipidology. 2012;23:167–168. doi: 10.1097/MOL.0b013e3283518614. [DOI] [PubMed] [Google Scholar]
- 92.Patel DC, Albrecht C, Pavitt D, Paul V, Pourreyron C, Newman SP, Godsland IF, Valabhji J, Johnston DG. Type 2 diabetes is associated with reduced ATP-binding cassette transporter A1 gene expression, protein and function. PLoS One. 2011;6:e22142. doi: 10.1371/journal.pone.0022142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morgantini C, Natali A, Boldrini B, Imaizumi S, Navab M, Fogelman AM, Ferrannini E, Reddy ST. Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes. 2011;60:2617–2623. doi: 10.2337/db11-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Merched A, Xia Y, Visvikis S, Serot JM, Siest G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol Aging. 2000;21:27–30. doi: 10.1016/s0197-4580(99)00103-7. [DOI] [PubMed] [Google Scholar]
- 95.Saczynski JS, White L, Peila RL, Rodriguez BL, Launer LJ. The relation between apolipoprotein A-I and dementia: the Honolulu-Asia aging study. American journal of epidemiology. 2007;165:985–992. doi: 10.1093/aje/kwm027. [DOI] [PubMed] [Google Scholar]
- 96.Atzmon G, Gabriely I, Greiner W, Davidson D, Schechter C, Barzilai N. Plasma HDL levels highly correlate with cognitive function in exceptional longevity. The journals of gerontology Series A, Biological sciences and medical sciences. 2002;57:M712–715. doi: 10.1093/gerona/57.11.m712. [DOI] [PubMed] [Google Scholar]
- 97.Mendez AJ. The promise of apolipoprotein A-I mimetics. Current opinion in endocrinology, diabetes, and obesity. 2010;17:171–176. doi: 10.1097/MED.0b013e3283373cb5. [DOI] [PubMed] [Google Scholar]
- 98.Sherman CB, Peterson SJ, Frishman WH. Apolipoprotein A-I mimetic peptides: a potential new therapy for the prevention of atherosclerosis. Cardiology in review. 2010;18:141–147. doi: 10.1097/CRD.0b013e3181c4b508. [DOI] [PubMed] [Google Scholar]
- 99.Stukas S, Robert J, Wellington CL. High-density lipoproteins and cerebrovascular integrity in Alzheimer’s disease. Cell metabolism. 2014;19:574–591. doi: 10.1016/j.cmet.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 100.Buga GM, Frank JS, Mottino GA, Hendizadeh M, Hakhamian A, Tillisch JH, Reddy ST, Navab M, Anantharamaiah GM, Ignarro LJ, Fogelman AM. D-4F decreases brain arteriole inflammation and improves cognitive performance in LDL receptor-null mice on a Western diet. Journal of lipid research. 2006;47:2148–2160. doi: 10.1194/jlr.M600214-JLR200. [DOI] [PubMed] [Google Scholar]
- 101.Handattu SP, Garber DW, Monroe CE, van Groen T, Kadish I, Nayyar G, Cao D, Palgunachari MN, Li L, Anantharamaiah GM. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiology of disease. 2009;34:525–534. doi: 10.1016/j.nbd.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McNeill E. RVX-208, a stimulator of apolipoprotein AI gene expression for the treatment of cardiovascular diseases. Curr Opin Investig Drugs. 2010;11:357–364. [PubMed] [Google Scholar]
- 103.Kodali M, V, Parihar K, Hattiangady B, Mishra V, Shuai B, Shetty AK. Resveratrol prevents age-related memory and mood dysfunction with increased hippocampal neurogenesis and microvasculature, and reduced glial activation. Scientific reports. 2015;5:8075. doi: 10.1038/srep08075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin AL, Zheng W, Halloran JJ, Burbank RR, Hussong SA, Hart MJ, Javors M, Shih YY, Muir E, Solano Fonseca R, Strong R, Richardson AG, Lechleiter JD, Fox PT, Galvan V. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:1412–1421. doi: 10.1038/jcbfm.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ, Rubin LL. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344:630–634. doi: 10.1126/science.1251141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tarumi T, Zhang R. Cerebral hemodynamics of the aging brain: risk of Alzheimer disease and benefit of aerobic exercise. Frontiers in physiology. 2014;5:6. doi: 10.3389/fphys.2014.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Papadopoulos P, Tong XK, Hamel E. Selective benefits of simvastatin in bitransgenic APPSwe, Ind/TGF-beta1 mice. Neurobiology of aging. 2014;35:203–212. doi: 10.1016/j.neurobiolaging.2013.07.010. [DOI] [PubMed] [Google Scholar]