

Abstract
There are many limitations for conducting liver disease research in human beings due to the high cost and potential ethical issues. For this reason, conducting a study that is difficult to perform in humans using appropriate animal models, can be beneficial in ascertaining the pathological physiology, and in developing new treatment modalities. However, it is difficult to determine the appropriate animal model which is suitable for research purposes, since every patient has different and diverse clinical symptoms, adverse reactions, and complications due to the pathological physiology. Also, it is not easy to reproduce identically various clinical situations in animal models. Recently, the Guide for the Care and Use of Laboratory Animals has tightened up the regulations, and therefore it is advisable to select the appropriate animals and decide upon the appropriate quantities through scientific and systemic considerations before conducting animal testing. Therefore, in this review article the authors examined various white rat animal testing models and determined the appropriate usable rat model, and the pros and cons of its application in liver disease research. The authors believe that this review will be beneficial in selecting proper laboratory animals for research purposes.
Keywords: Liver disease, Animal model, Rat
Introduction
There are many limitations in conducting liver disease research directly in human beings due to the high cost, and particularly due to the potential ethical issues.1 In other words, conducting a study that is difficult to perform in humans using appropriate animal models, can be advantageous in ascertaining the pathological physiology and in developing new treatment modalities.2 Nevertheless, it is difficult to determine the appropriate animal model which is suitable for research purposes, since every patient has different and diverse clinical symptoms, adverse reactions, and complications due to the pathological physiology. Also, identical reproduction of various clinical situations in animal models may not be feasible. Recently, the Guidelines for the Care and Use of Laboratory Animals has become stringent, and therefore it is advisable to select the appropriate animals and decide upon the appropriate quantities through scientific and systemic considerations before conducting animal testing.
Therefore, in this review article we attempted to search various rat animal experimental models, and to determine the appropriate usable rat model, and investigate the pros and cons of its application in liver disease research. We believe that this review will be beneficial in selecting proper laboratory animals for research purposes.
Acquisition of the Experimental Animal Model
Many animals are utilized in studies of disease development process and biochemical studies, in humans. The majority of animal models have dissimilar explanations for the conditions or diseases that occur in humans, but they are absolutely essential for the advancement of physiomedical knowledge. The basic criteria for an ideal experimental animal model are as follows:3,4,5
1) It should accurately reproduce the disease that is being researched.
2) It should be accessible to many researchers.
3) It should be exportable to other laboratories.
4) It should be adequate enough to provide an appropriate quantity of samples.
5) It should be easily handled by researchers.
6) It should survive long enough to be usable.
7) It should adapt well to the breeding environment.
8) It should be available in multiple species.
9) It should be in a polytocous species.
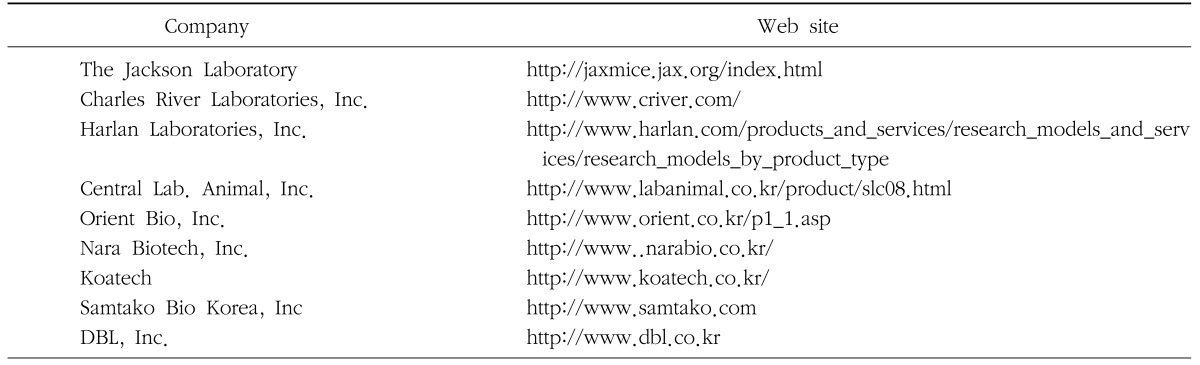
Animal disease models are divided into two types: spontaneous and experimentally induced models. Spontaneous animal models can be used as human disease models. These models resemble the human disease compared to experimentally induced models. Experimentally induced models can be created either through surgical modifications, genetic modifications, chemical modifications, and biological modifications. The classic examples of experimentally induced models are chemically induced diabetes models and neoplastic models, which have been reported to be used in many studies. Recently, the genetically engineered models have emerged as the most prevalent experimentally induced model, but their application are more common in mice than in rats. However, for surgical experimentation, rats are preferred to mice as rat is larger and therefore is more advantageous and ideal. The above mentioned ideal conditions for animal experiments are also applicable to rats, and rats which meet the animal model criteria are procured easily from supply companies (Table 1). Examples of the more common species of rats used in animal models are Wistar, Sprague-Dawley, Osborne-Mendel, Long-Evans, Holtzman, Slonaker, and Albany.
Table 1. Animal pharmacology companies and their web sites.
Rat Strains
In 1959, Billingham RHK and Silvers first defined the rat species, and more than 220 species of inbred strains and many substrains are now present.6
Inbreeding is defined as 20 or more times of breeding between offsprings or between parents and offspring, and since this is a highly genetically isogenic strain, a 'unique species' reaction may be obtained. In outbreeding, breeding is continued within a specified group which is less closely related than that in inbreeding, and was originally derived from inbreeding. Although in outbreeding, the uniformity of genetic composition is less consistent than in inbreeding animals, the reproduction rate is higher and breeding is relatively easy, and therefore outbred animals play an important function in biological examination.
Liver Disease Experimental Animal Models
1. Types of liver disease experimental animal models
The liver disease experimental animal models are divided into 2 types (Table 2).
Table 2. The classification of rat models for liver diseases.

The first category is the genetically determined disease model. Examples of genetically determined disease models are the Gunn rat in which there is deficient bilirubin metabolism (Crigler-Najjar syndrome),7 the Long-Evans Cinnamon (LEC) rat in which there is deficient copper metabolism (Wilson's disease),8 and rats with deficient albumin production; the Nagase analbuminemic rat (NAR), and the analbuminemic congenic strains of rats; ACI-alb, F344-alb and SHR-alb.9 Others examples are the spontaneously hypertensive rat (SHR), the diabetic rat (OLETF), and the obese rat (Zucker rat).
The second type of experimental animal models comprises of the drug induced or surgically induced models.
2. Metabolic liver disease animal models
Metabolic liver disease animal models were developed by genetic mutations as they share various clinical characteristics, side effects, complications in humans according to the individual pathological physiology.
1) Gunn rat (Crigler-Najjar syndrome)
Protoporphyrin moiety of heme proteins such as bilirubin and hemoglobin, is a toxic derivative. For its elimination from the serum, bilirubin conjugates with glucuronic acid, and UGT1A1 (bilirubin UDP-glucuronosyltransferase) is the sole isoform of bilirubin glucuronidating UGT1 in humans or rats.10 Genetic variations in the gene encoding UGT1A1 lead to complete or partial inactivation of the glucuronidation of bilirubin in the serum, which causes unconjugated bilirubin. This may lead to the development of hyperbilirubinemic conditions such as the Crigler-Najjar (CN) Syndrome in humans.11
The liver parenchyma and thousands of other metabolic functions are normal in the patients with this disease, but they are at risk of severe neurologic complications. Orthotopic liver transplantation is the radical treatment method,12,13 but this procedure corrects only one enzyme deficiency when all other liver functions are normal. In clinical practice, retransplantation is necessary in up to 15% of patients after liver transplantation, and progressive fibrosis of the transplanted liver over long-term is a major concern.14
Gunn rats are derived from the Wistar species and cause unstoppable elevations of serum bilirubin leading to unconjugated hyperbilirubinemia. The Gunn rats inherently lack all the glucuronidation activity catalyzed by the UGT1 isoform. Therefore, these rats are used in the animal experiment models for investigation of type I Crigler-Najjar (CN) syndrome.15,16
2) Long Evans Cinnamon (LEC) rat (Wilson's disease)
The LEC rat was discovered in 1983 at the Center for Experimental Plants and Animals, Hokkaido, Japan, and is characterized by the unique cinnamon colored fir, and is similar to the mutation form of the Long-Evans rat.17 The LEC rat is an appropriate animal model for the study of the importance of hepatitis in the development of hepatomas.18,19 The species with this mutation self expresses chronic hepatitis followed by an extremely high incidence of preneoplastic lesions and hepatomas (Fig. 1).
Fig. 1. Natural History of the LEC rat.19.

That is, the LEC rat shows a remarkable similarity to the clinical course of liver cancer development in humans. Recently, the spontaneous hepatitis shown in LEC rats is thought to be related to high copper accumulation in the liver, and this is probably due to the problems in the excretion of copper from the blood and bile by the liver. Therefore, the occurrence of liver cancer can be controlled by reducing the copper content in the feed provided to the laboratory animals, and it can theoretically be deduced whether liver cancer develops in association with hepatitis. Also, in the LEC rat model, hepatitis causes a high mortality rate and abnormal copper metabolism, which makes the animal suitable to serve as an animal model of individual human fulminant hepatitis and Wilson's disease. The spontaneous hepatitis in LEC rats is characterized by a sudden onset at 4 months after birth. Therefore, the LEC rat is the ideal model for studying the role of hepatitis in the pathogenesis of liver cancer. Excessive accumulation of copper in the liver has been confirmed to lead to hepatitis. The excretion of dietary copper into blood and bile is affected, and as a result copper accumulates in the liver cells. The treatment or control of copper in the animal feed with chelating agents, such as D-penicillamine or trientine, will accelerate hepatitis in LEC rats. In many preliminary studies, it has been shown that lowering the concentrations of copper in the animal feed adequately prevents the occurrence of hepatitis, and high concentrations of copper will lead to hepatitis induction.20 This phenomenon may explain the regulatory mechanisms of the sequential liver cancer process, by the prevention or acceleration of hepatitis.
Furthermore, the LEC rat is an appropriate animal model for Wilson's disease. It mimics the human Wilson's disease in its characteristics such as excessive copper accumulation in the liver and low levels of serum ceruloplasmin, low excretion of copper into the bile.16,21 The key defect that causes such abnormal copper metabolism has not been elucidated in Wilson's disease, and the research for the gene responsible for hepatitis in LEC rats may provide a clue to the pathogenesis of Wilson's disease. However, in LEC rats, copper deposition does not occur in the brain or cornea, and therefore the pathognomonic signs of Wilson's disease such as hepatolenticular degeneration of the brain, or Kayser-Fleischer ring of the cornea are absent in LEC rats, and hence this condition is not exactly identical with human Wilson's disease.
3) Nagase analbuminemic (NAR) rat (Analbuminemia)
Analbuminemia in humans was first described by Benhold. In 1959, and the etiology and metabolic aspects of this condition have been studied by other researchers.22 Increased levels of globulin accompanying albumin decrease have been reported in the serum of analbuminemia patients, along with high levels of cholesterol.23
In 1977, Nagase and Shimamune discovered the analbuminemic rat by mating hypercholesterolemic Sprague-Dawley rats, and named it the Nagase analbuminemia rat (NAR). NAR not only displays analbuminemia but also hyperlipidemia, which comprises of very high serum levels of cholesterol as well as triglycerides, and thus the analbuminemic rat is an appropriate model for studying hyperlipidemic conditions. Thioacetamide (TA) is usually used for induction of liver cirrhosis but sustained TA administration does not seem to elicit liver cirrhosis in the NAR.24 A the cellular level, cholangiolar proliferation occurs as a result of the mechanism of TA toxicity. In other words, it would appear that liver cirrhosis may inherently prevent hyperlipidemia and hypercholesterolemia in the NAR, which holds a potential for its clinical application in the treatment of liver cirrhosis.24
4) Other analbuminemic rats
In 1986, Nagase utilized ACl, F344, SHR (spontaneous hypertensive rat) to develop the analbuminemic syngeneic white rat. This animal was produced by repeated backcrossing, and with the exception of the albumin genes, expresses inbred fir color and biochemical genes. Compared to the rat presenting albumin, the liver and adrenals of analbuminemic syngeneic white rat show increase in weight from 30 weeks onwards. Also, the serum lipid levels appear to be higher at the same time.25
5) Eisai hyperbilirubinemic rats (Hyperbilirubinemia)
Eisai hyperbilirubinemic rat (EHBR) is a Sprague-Dawley (SD) rat mutant with conjugated hyperbilirubinemia as an autosomal recessive trait.26 EHBR manifests jaundice from birth, which is permanent except for a transient decrease in bilirubin levels at 6~8 weeks of age. EHBR is thus a useful animal model for studying the biliary excretion of bilirubin. In this animal model, there was unstable biliary excretion of organic anions such as sulfobromophthalein, indocyanine green and glutathione conjugates.26,27,28,29 In recent studies, TCA (tricarboxylic acid) cycle defect in biliary excretion has been observed in EHBR models.29,30 However, the results of this study, in which a spontaneous model was employed, may explain the altered bile secretion in EHBR models. That is, hyperbilirubinemia and high serum bile acid levels may hinder bile acid excretion in vivo.
6) Albumin-deficient and Jaundiced rat (AJR)
In 1981, Nagase and Shumiya developed the albumindeficient and jaundiced rat (AJR) strain by hybridization of albumin-deficient rats and jaundiced Gunn rats. These animals are characterized by double homozygosity and systemic jaundice and various neurological signs are observed 5~7 days after birth. Kernicterus occurs and the animal dies within 3 weeks after birth. This animal model can be used for research of human kernicterus and bilirubin metabolism.31
3. Acute liver failure animal model
Terblanche and Hickman formulated the following requisite conditions in studies with animal models of acute liver failure.5
First, the degree of acute liver failure must be reversible so that survival is likely after effective therapy. Second, identical death rates should be reproducible when inconsistent treatment is given.
Third, selective liver damage must be the cause of death. Fourth, a therapeutic window must be obtained.
Fifth, the animal must be large enough so that the treatment can be administered in humans.
Sixth, the hazard to the studying person should be minimized.
The six standards suggested above are known as the basic requisites for animal models of acute liver failure. Newsome suggested 2 additional items.4
Seventh, the animal model should have metabolic or physiological mechanisms similar to that in humans.
Eighth, animal experimental methods should adhere to appropriate ethical standards.
1) Surgical models
Surgical animal models are classified into 3 types; anhepatic model, partial hepatectomy model, and the devascularization model.
(1) Anhepatic model
In this model the liver is completely removed, and it carries the disadvantage that death of all animals ensues if transplantation is not performed. Furthermore, the duration of hepatic coma is short and is different from the pattern in human patients, and the use of this model is limited since the liver cells that are damaged or necrotized are not exposed to the vascular circulation.32
(2) Partial hepatectomy (PH) model
In the partial hepatectomy model, excellent liver regeneration and survival has been observed in 100% of models after early removal of 70% of the liver.33 Thereafter, an increase in mortality rates was demonstrated due to an increase in the resection rate which leads to increased portal circulation and endothelial cell damage, and also activation of Kupffer cells and increased secretion of cytokines that induce acute liver failure. The increased portal flow per liver tissue also increases the endotoxemic load in the gastrointestinal tract.34 In this model, late hypoglycemia occurs and the pH is maintained at 7.3±0.03. The identical death rates are reproducible. The limitation of this model is the lack of clinical patterns that manifest in humans. Late hypoglycemia develops, and therefore reproducibility is low and irreversible.
(3) Devascularization model
Mechanisms of injury due to devascularization are at present known to be very complex.35 In ischemic damage, suppression of oxidative phosphorylation leads to decreased ATP and defects in mitochondrial respiratory chain. This affects the intracellular calcium homeostasis which in turn affects the kinase activity which is the cause of injury to protein, lipid and DNA. Consequently, the reperfusion injury may cause a major injury, which produces reactive oxygen species (ROS) and hydroxyl radicals. This reaction activates the amplified cascade in the Kupffer cells, which subsequently results in lipid peroxidation, protein damage, complement activation, and finally apoptosis or necrosis.36,37,38,39
Partial devascularization can be performed by the insertion of a portacaval shunt and temporally graded clamping of the hepatic artery. Complete devascularization model of acute hepatic failure is used. But, this is an irreversible model and this is similar to the anhepatic model. This model is developed by portacaval shunting or hepatic artery ligation.
2) Bile duct ligated model
The first description of the comprehensive bile duct ligated model was in the early 1930s by Cameron and Oakley. Based on this report, comprehensive bile duct ligated model was established by Kountouras et al in 1984,40 and this model became increasingly employed in research involving other aspects of liver fibrosis. Thereafter until the early 1990s, bile duct ligation induced fibrocirrhotic disease was focused in studies on histological changes. In a recent series by Chang et al., liver cirrhosis was induced by administration of various hepatotoxins and bile duct ligation, and the comparative results were reported.41 The bile duct ligated model attempted to produce necrosis and fibrosis, and cirrhosis similar to liver cirrhosis induced by administration of many types of hepatotoxins, but it failed to produce lipid changes. Also, bile duct ligated model differs from other animal models in which liver cirrhosis was induced in all animals after 4~8 weeks. Thus, bile duct ligation is the quickest and most consistent method for inducing fibrocirrhotic disease of the liver.
3) Toxic drug model
Hepatotoxic drugs that induce hepatic failure are widely employed. Drugs that have been utilized over the past 30 years are D-galactosamine (D-Gal), acetaminophen (paracetamol/APAP), carbon tetrachloride (CCl4), and Thioacetamide (TAA). Concanavalin A (Con A) and lipopolysaccharide (LPS) are the other drugs that have emerged recently. However, toxic drug reaction differs according to different species, or even between individual models of the same species, and therefore it is difficult to attain high reproducibility rates.
(1) D-galactosamine
Keppler was the first to describe the hepatotoxic properties of D-Gal in rats in 1968.42,43 D-Gal is an amino sugar that is metabolized in the liver and causes uridine nucleotide depletion and hepatic transcriptional blockade. It cannot be said that the depressed protein synthesis by D-Gal induces hepatic failure. The cause of hepatic failure has been proposed in a study which revealed that, in hepatic transformation there are other stimuli which partially reflect the role of chemicals including uridine.44 It is thought that the administration of D-Gal induces portal endotoxemia that containes intestinal endotoxins. Also, D-Gal hepatitis in rats can be prevented by colectomy and neutralization of LPS by polymyxin B, or the induction of endotoxin tolerance. This concept continues to expand, and there is evidence that TNF-α acts as a terminal mediator.45 This is related to the significant cellular necrosis that occurs after D-Gal administration. There exists a difference in the susceptibility between different species. While the rat is sensitive to D-Gal, mice show resistance even after high-dose D-Gal administration at 1 g/kg.46
(2) Acetaminophen
In the United Kingdom, acetaminophen abuse is the most common cause of acute liver failure. In other countries such as France and the United States, the frequency of acetaminophen abuse is rapidly increasing.47 Acetaminophen metabolism occurs in the liver. Under normal conditions, acetaminophen glucuronidation and sulfation leads to biotransformation and excretion. If this mechanism becomes overloaded then acetaminophen is metabolized by the cytochrome P450 oxidative enzyme.48,49,50,51 Subsequently this results in the production of a toxic metabolite N-acetyl-p-benzoquinoneimine (NAPQI), by non-conjugation of acetaminophen with endogenous glutathione. 52,53,54 The toxic properties of NAPQI have been attributed to the formation of free radicals, reactive oxygen species, hydroxyl radicals, nitrites and nitrates, which obstruct the mitochondrial calcium channel and cell injury.55 This chain reaction is amplified by the production of cytokines and free radicals that cause apoptosis and necrosis of Kupffer cells. This is characterized particularly by the adequate presence or absence, efficiency, species diversity and senility of the cytochrome P450 oxidative enzyme.56,57,58 2E1 and 1A2 are the members of the cytochrome P450 family that are the most closely related to acetaminophen metabolism. To date many models are being researched to establish an acetaminophen model.
(3) Carbon tetrachloride (CCl4)
The hepatotoxic properties of CCl4 have been known for a long period of time.59,60 CCl4 was extensively employed in acute liver injury models in the 1970s and the early 1980s, but after it was revealed that reproducibility was not up to the desired standards, along with questions regarding species diversity,61,62 it was not used further. Recently, a model has emerged in which CCl4 is administered into the stomach and intraperitoneally to induce liver cirrhosis.63,64 The cytochrome P450 enzyme is considered to participate in the production of CCl4 metabolites.59,65,66 In 1972, it was postulated by Chopra et al that the induction process of P450 enzyme increased CCl4 toxicity, and it was further ascertained by Nayak in 1970 and 1975 who demonstrated phenobarbitone toxicity in rat fetuses, neonates, and adults by comparing P450 enzyme activity between an induction group and control group. The results were very interesting; the fetal model in the 2 groups did not demonstrate necrosis. In neonatal rats induced with P450 enzyme, the cellular characteristics observed were ballooning, centrilobular necrosis, and Councilman bodies, suggesting a small degree of injury in the control group. In contrast, injury in adult rats was extensive and was proportional to the dose of the CCl4 administered. However, this was also true for the induction group, and the production of free radicals is suggested as one of the mechanisms involved.66,67,68 Homolytic dissolution of haloalkanes occurs in the liver microsomal P450 which produces trichloromethyl that reacts with reactive oxygen species to produce active metabolites. These metabolites combine with macromolecules to increase peroxidation, and cellular injury by disruption of intracellular calcium homeostasis. In more recent studies, the mechanism of CCl4-induced cell death is explained to be the same as in necrosis, and is the consequence of apoptosis. Shi et al. explained apoptosis of cells in CCl4-only treated rats using light electron microscopy and nuclear DNA fragment patterns and immunohistochemical labeling and flow cytometry analysis. The majority of apoptotic cells are located in the centrilobular region of the liver. The kidneys are also sensitive to CCl4 and acute tubular necrosis develops after exposure to CCl4.69,70 Clinically, manifestations of acute liver failure such as early encephalopathy and late stage hepatic coma are not easily produced by CCl4 toxicity, and therefore this model is rarely used in investigating acute liver failure.
(4) Thioacetaminophen (TAA)
Thioacetamide (TAA) has also been used in the past for induction of acute hepatic failure. TAA induces TAA-S-oxide transformation by mechanisms of flavine adenine dinucleotide containing monooxygenase and causes cell necrosis by biotransformation into metabolites,71 and is thought to cause apoptosis at low concentrations. However, if the doses are increased, electrophiles and free radicals are released which contribute to lipid peroxidation and centrilobular necrosis and are then expelled.72 TAA is widely employed as an agent to induce acute liver failure in rats and mice, but it has been suggested that the use of other animals may lead to species variability. In models using rabbits there is definite variability with respect to clinical and histological change. Clinical characteristics such as encephalopathy, metabolic acidosis, high levels of transaminase, abnormal coagulopathy, and centrilobular necrosis were observed after intraperitoneal TAA administration in rats, and these manifestations reached their peak after 12~24 hours.72,73,74 Administration doses ranged from 400 mg/kg to 600 mg/kg, and intervals varied from once a day for 2 days to once a day for 3 days. Recent reports have successed to show liver cirrhosis development after chronic administration of TAA.64,75,76 The authors of this review have also experienced induction of liver cirrhosis via various routes of TAA administration.77
Conclusions
Animal model experiments have recently contributed to proof of theories and delineation of the pathophysiology. It is of utmost importance to gain accurate knowledge via various animal experiments for an appropriate selection of the ideal animal model for research. High reproducibility and simple animal models that will provide high quality research will contribute to our academic development.
Footnotes
This study was supported by a grant from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A084120).
References
- 1.Blumberg BS, Fox RC. The Daedalus effect: changes in ethical questions relating to hepatitis B virus. Ann Intern Med. 1985;102:390–394. doi: 10.7326/0003-4819-102-3-390. [DOI] [PubMed] [Google Scholar]
- 2.Mullen KD, McCullough AJ. Problems with animal models of chronic liver disease: suggestions for improvement in standardization. Hepatology. 1989;9:500–503. doi: 10.1002/hep.1840090326. [DOI] [PubMed] [Google Scholar]
- 3.Fourneau I, Pirenne J, Roskams T, Yap SH. An improved model of acute liver failure based on transient ischemia of the liver. Arch Surg. 2000;135:1183–1189. doi: 10.1001/archsurg.135.10.1183. [DOI] [PubMed] [Google Scholar]
- 4.Newsome PN, Plevris JN, Nelson LJ, Hayes PC. Animal models of fulminant hepatic failure: a critical evaluation. Liver Transpl. 2000;6:21–31. doi: 10.1002/lt.500060110. [DOI] [PubMed] [Google Scholar]
- 5.Terblanche J, Hickman R. Animal models of fulminant hepatic failure. Dig Dis Sci. 1991;36:770–774. doi: 10.1007/BF01311235. [DOI] [PubMed] [Google Scholar]
- 6.Hedrich HJ. The Laboratory Rat. 2nd ed. Amsterdam; Boston: Elsevier; 2006. Taxonomy and Stocks and strains. [Google Scholar]
- 7.Iyanagi T, Haniu M, Sogawa K, et al. Cloning and characterization of cDNA encoding 3-methylcholanthrene inducible rat mRNA for UDP-glucuronosyltransferase. J Biol Chem. 1986;261:15607–15614. [PubMed] [Google Scholar]
- 8.Terada K, Sugiyama T. The Long-Evans Cinnamon rat: an animal model for Wilson's disease. Pediatr Int. 1999;41:414–418. doi: 10.1046/j.1442-200x.1999.01089.x. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi M, Shumiya S, Maekawa A, Hayashi Y, Nagase S. High susceptibility of an analbuminemic congenic strain of rats with an F344 genetic background to induced bladder cancer and its possible mechanism. Jpn J Cancer Res. 1988;79:705–709. doi: 10.1111/j.1349-7006.1988.tb02226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cubero FJ, Arza E, Maganto P, et al. Expression of bilirubin UDP-glucuronosyltransferase (bUGT) throughout fetal development: intrasplenic transplantation into Gunn rats to correct enzymatic deficiency. Dig Dis Sci. 2001;46:2762–2767. doi: 10.1023/a:1012743916800. [DOI] [PubMed] [Google Scholar]
- 11.Iyanagi T, Emi Y, Ikushiro S. Biochemical and molecular aspects of genetic disorders of bilirubin metabolism. Biochim Biophys Acta. 1998;1407:173–184. doi: 10.1016/s0925-4439(98)00044-1. [DOI] [PubMed] [Google Scholar]
- 12.Kaufman SS, Wood RP, Shaw BW, Jr, et al. Orthotopic liver transplantation for type I Crigler-Najjar syndrome. Hepatology. 1986;6:1259–1262. doi: 10.1002/hep.1840060606. [DOI] [PubMed] [Google Scholar]
- 13.Sokal EM, Silva ES, Hermans D, et al. Orthotopic liver transplantation for Crigler-Najjar type I disease in six children. Transplantation. 1995;60:1095–1098. doi: 10.1097/00007890-199511270-00006. [DOI] [PubMed] [Google Scholar]
- 14.Evans HM, Kelly DA, McKiernan PJ, Hubscher S. Progressive histological damage in liver allografts following pediatric liver transplantation. Hepatology. 2006;43:1109–1117. doi: 10.1002/hep.21152. [DOI] [PubMed] [Google Scholar]
- 15.Chowdhury JR, Kondapalli R, Chowdhury NR. Gunn rat: a model for inherited deficiency of bilirubin glucuronidation. Adv Vet Sci Comp Med. 1993;37:149–173. [PubMed] [Google Scholar]
- 16.Yamada T, Agui T, Suzuki Y, Sato M, Matsumoto K. Inhibition of the copper incorporation into ceruloplasmin leads to the deficiency in serum ceruloplasmin activity in Long-Evans cinnamon mutant rat. J Biol Chem. 1993;268:8965–8971. [PubMed] [Google Scholar]
- 17.Yoshida MC, Masuda R, Sasaki M, et al. New mutation causing hereditary hepatitis in the laboratory rat. J Hered. 1987;78:361–365. doi: 10.1093/oxfordjournals.jhered.a110416. [DOI] [PubMed] [Google Scholar]
- 18.Enomoto K, Takahashi H, Mori M. A new rat model for the study of hepatocarcinogenesis. J Gastroenterol Hepatol. 1992;7:98–104. doi: 10.1111/j.1440-1746.1992.tb00941.x. [DOI] [PubMed] [Google Scholar]
- 19.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugawara N, Sugawara C, Katakura M, Takahashi H, Mori M. Harmful effect of administration of copper on LEC rats. Res Commun Chem Pathol Pharmacol. 1991;73:289–297. [PubMed] [Google Scholar]
- 21.Ono T, Abe S, Yoshida MC. Hereditary low level of plasma ceruloplasmin in LEC rats associated with spontaneous development of hepatitis and liver cancer. Jpn J Cancer Res. 1991;82:486–489. doi: 10.1111/j.1349-7006.1991.tb01875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennhold HH. Two cases of familial analbuminemia. Mars Med. 1959;96:1–6. [PubMed] [Google Scholar]
- 23.Cormode EJ, Lyster DM, Israels S. Analbuminemia in a neonate. J Pediatr. 1975;86:862–867. doi: 10.1016/s0022-3476(75)80215-0. [DOI] [PubMed] [Google Scholar]
- 24.David P, Alexandre E, Chenard-Neu MP, Wolf P, Jaeck D, Richert L. Failure of liver cirrhosis induction by thioacetamide in Nagase analbuminaemic rats. Lab Anim. 2002;36:158–164. doi: 10.1258/0023677021912442. [DOI] [PubMed] [Google Scholar]
- 25.Shumiya S, Nagase S. Establishment and characteristics of three analbuminemic congenic strains of rats. Jikken Dobutsu. 1986;35:409–416. doi: 10.1538/expanim1978.35.4_409. [DOI] [PubMed] [Google Scholar]
- 26.Kurisu H, Kamisaka K, Koyo T, et al. Organic anion transport study in mutant rats with autosomal recessive conjugated hyperbilirubinemia. Life Sci. 1991;49:1003–1011. doi: 10.1016/0024-3205(91)90301-q. [DOI] [PubMed] [Google Scholar]
- 27.Fernández-Checa JC, Takikawa H, Horie T, Ookhtens M, Kaplowitz N. Canalicular transport of reduced glutathione in normal and mutant Eisai hyperbilirubinemic rats. J Biol Chem. 1992;267:1667–1673. [PubMed] [Google Scholar]
- 28.Hosokawa S, Tagaya O, Mikami T, et al. A new rat mutant with chronic conjugated hyperbilirubinemia and renal glomerular lesions. Lab Anim Sci. 1992;42:27–34. [PubMed] [Google Scholar]
- 29.Takikawa H, Sano N, Narita T, et al. Biliary excretion of bile acid conjugates in a hyperbilirubinemic mutant Sprague-Dawley rat. Hepatology. 1991;14:352–360. [PubMed] [Google Scholar]
- 30.Takikawa H, Sano N, Wako Y, Yamanaka M. Effects of organic anions and bile acids on biliary lipid excretion in hyperbilirubinemic mutant Sprague-Dawley rats. J Hepatol. 1993;17:247–252. doi: 10.1016/s0168-8278(05)80046-7. [DOI] [PubMed] [Google Scholar]
- 31.Shumiya S, Nagase S. Establishment of an albumin-deficient and jaundiced strain of rats. Jikken Dobutsu. 1981;30:291–297. doi: 10.1538/expanim1978.30.3_291. [DOI] [PubMed] [Google Scholar]
- 32.Tonnesen K. Experimental liver failure. A comparison between hepatectomy and hepatic devascularization in the pig. Acta Chir Scand. 1977;143:271–277. [PubMed] [Google Scholar]
- 33.Emond J, Capron-Laudereau M, Meriggi F, Bernuau J, Reynes M, Houssin D. Extent of hepatectomy in the rat. Evaluation of basal conditions and effect of therapy. Eur Surg Res. 1989;21:251–259. doi: 10.1159/000129034. [DOI] [PubMed] [Google Scholar]
- 34.Panis Y, McMullan DM, Emond JC. Progressive necrosis after hepatectomy and the pathophysiology of liver failure after massive resection. Surgery. 1997;121:142–149. doi: 10.1016/s0039-6060(97)90283-x. [DOI] [PubMed] [Google Scholar]
- 35.Fischer M, Stotter L, Schmahl W, Gartmaier P, Erhardt W. Acute liver failure due to temporary hepatic ischemia in the pig. Acta Hepatogastroenterol (Stuttg) 1976;23:241–249. [PubMed] [Google Scholar]
- 36.Borghi-Scoazec G, Scoazec JY, Durand F, et al. Apoptosis after ischemia-reperfusion in human liver allografts. Liver Transpl Surg. 1997;3:407–415. doi: 10.1002/lt.500030408. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Valdecasas JC, Rull R, Grande L, et al. Prostacyclin, thromboxane, and oxygen free radicals and postoperative liver function in human liver transplantation. Transplantation. 1995;60:662–667. doi: 10.1097/00007890-199510150-00008. [DOI] [PubMed] [Google Scholar]
- 38.Gasbarrini A, Colantoni A, Di Campli C, et al. Intermittent anoxia reduces oxygen free radicals formation during reoxygenation in rat hepatocytes. Free Radic Biol Med. 1997;23:1067–1072. doi: 10.1016/s0891-5849(97)00141-x. [DOI] [PubMed] [Google Scholar]
- 39.Shirasugi N, Wakabayashi G, Shimazu M, et al. Up-regulation of oxygen-derived free radicals by interleukin-1 in hepatic ischemia/reperfusion injury. Transplantation. 1997;64:1398–1403. doi: 10.1097/00007890-199711270-00004. [DOI] [PubMed] [Google Scholar]
- 40.Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol. 1984;65:305–311. [PMC free article] [PubMed] [Google Scholar]
- 41.Chang ML, Yeh CT, Chang PY, Chen JC. Comparison of murine cirrhosis models induced by hepatotoxin administration and common bile duct ligation. World J Gastroenterol. 2005;11:4167–4172. doi: 10.3748/wjg.v11.i27.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keppler D, Lesch R, Reutter W, Decker K. Experimental hepatitis induced by D-galactosamine. Exp Mol Pathol. 1968;9:279–290. doi: 10.1016/0014-4800(68)90042-7. [DOI] [PubMed] [Google Scholar]
- 43.Keppler D, Decker K. Mechanism of action of D-galactosamine in the liver. Verh Dtsch Ges Inn Med. 1971;77:1182–1185. [PubMed] [Google Scholar]
- 44.Takahashi N, Ishizuya T, Mori N. In-vitro preparation of experimental models of hepatitis with D-galactosamine and their modification by liver-repairing factors. Int J Tissue React. 1990;12:263–268. [PubMed] [Google Scholar]
- 45.Gantner F, Kusters S, Wendel A, Hatzelmann A, Schudt C, Tiegs G. Protection from T cell-mediated murine liver failure by phosphodiesterase inhibitors. J Pharmacol Exp Ther. 1997;280:53–60. [PubMed] [Google Scholar]
- 46.Leist M, Gantner F, Kunstle G, et al. The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol Med. 1996;2:109–124. [PMC free article] [PubMed] [Google Scholar]
- 47.Makin AJ, Hughes RD, Williams R. Systemic and hepatic hemodynamic changes in acute liver injury. Am J Physiol. 1997;272:G617–G625. doi: 10.1152/ajpgi.1997.272.3.G617. [DOI] [PubMed] [Google Scholar]
- 48.Black M. Acetaminophen hepatotoxicity. Gastroenterology. 1980;78:382–392. [PubMed] [Google Scholar]
- 49.Boyd EM, Bereczky GM. Liver necrosis from paracetamol. Br J Pharmacol Chemother. 1966;26:606–614. doi: 10.1111/j.1476-5381.1966.tb01841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boyd EM, Hogan SE. The chronic oral toxicity of paracetamol at the range of the LD50 (100 days) in albino rats. Can J Physiol Pharmacol. 1968;46:239–245. doi: 10.1139/y68-040. [DOI] [PubMed] [Google Scholar]
- 51.Davidson DG, Eastham WN. Acute liver necrosis following overdose of paracetamol. Br Med J. 1966;2:497–499. doi: 10.1136/bmj.2.5512.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corcoran GB, Mitchell JR, Vaishnav YN, Horning EC. Evidence that acetaminophen and N-hydroxyacetaminophen form a common arylating intermediate, N-acetyl-p-benzoquinoneimine. Mol Pharmacol. 1980;18:536–542. [PubMed] [Google Scholar]
- 53.Jollow DJ, Thorgeirsson SS, Potter WZ, Hashimoto M, Mitchell JR. Acetaminophen-induced hepatic necrosis. VI. Metabolic disposition of toxic and nontoxic doses of acetaminophen. Pharmacology. 1974;12:251–271. doi: 10.1159/000136547. [DOI] [PubMed] [Google Scholar]
- 54.Mohandas J, Duggin GG, Horvath JS, Tiller DJ. Metabolic oxidation of acetaminophen (paracetamol) mediated by cytochrome P-450 mixed-function oxidase and prostaglandin endoperoxide synthetase in rabbit kidney. Toxicol Appl Pharmacol. 1981;61:252–259. doi: 10.1016/0041-008x(81)90415-4. [DOI] [PubMed] [Google Scholar]
- 55.Gardner CR, Heck DE, Yang CS, et al. Role of nitric oxide in acetaminophen-induced hepatotoxicity in the rat. Hepatology. 1998;27:748–754. doi: 10.1002/hep.510270316. [DOI] [PubMed] [Google Scholar]
- 56.Green MD, Fischer LJ. Hepatotoxicity of acetaminophen in neonatal and young rats. II. Metabolic aspects. Toxicol Appl Pharmacol. 1984;74:125–133. doi: 10.1016/0041-008x(84)90278-3. [DOI] [PubMed] [Google Scholar]
- 57.Green MD, Shires TK, Fischer LJ. Hepatotoxicity of acetaminophen in neonatal and young rats. I. Age-related changes in susceptibility. Toxicol Appl Pharmacol. 1984;74:116–124. doi: 10.1016/0041-008x(84)90277-1. [DOI] [PubMed] [Google Scholar]
- 58.Gregus Z, Madhu C, Goon D, Klaassen CD. Effect of galactosamine-induced hepatic UDP-glucuronic acid depletion on acetaminophen elimination in rats. Dispositional differences between hepatically and extrahepatically formed glucuronides of acetaminophen and other chemicals. Drug Metab Dispos. 1988;16:527–533. [PubMed] [Google Scholar]
- 59.Hübner G. Ultrastructural liver damage caused by direct action of carbon tetrachloride in vivo and in vitro. Virchows Arch Pathol Anat Physiol Klin Med. 1965;339:187–197. [PubMed] [Google Scholar]
- 60.Smith DH. Carbon tetrachloride toxicity. Br Med J. 1965;2:1434. doi: 10.1136/bmj.2.5475.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Das PK, Chopra P, Nayak NC. Hepatocellular tolerance to carbon tetrachloride induced injury in the rat: a study of its nature and possible mode of evolution. Exp Mol Pathol. 1974;21:218–236. doi: 10.1016/0014-4800(74)90091-4. [DOI] [PubMed] [Google Scholar]
- 62.Shi J, Aisaki K, Ikawa Y, Wake K. Evidence of hepatocyte apoptosis in rat liver after the administration of carbon tetrachloride. Am J Pathol. 1998;153:515–525. doi: 10.1016/S0002-9440(10)65594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dashti H, Jeppsson B, Hagerstrand I, et al. Thioacetamide- and carbon tetrachloride-induced liver cirrhosis. Eur Surg Res. 1989;21:83–91. doi: 10.1159/000129007. [DOI] [PubMed] [Google Scholar]
- 64.Nakano A, Kanda T, Abe H. Bone changes and mineral metabolism disorders in rats with experimental liver cirrhosis. J Gastroenterol Hepatol. 1996;11:1143–1154. doi: 10.1111/j.1440-1746.1996.tb01843.x. [DOI] [PubMed] [Google Scholar]
- 65.Recknagel RO, Ghoshal AK. New data on the question of lipoperoxidation in carbon tetrachloride poisoning. Exp Mol Pathol. 1966;5:108–117. doi: 10.1016/0014-4800(66)90008-6. [DOI] [PubMed] [Google Scholar]
- 66.Slater TF, Strauli UD, Sawyer BC. Changes in liver nucleotide concentrations in experimental liver injury. 1. Carbon tetrachloride poisoning. Biochem J. 1964;93:260–266. doi: 10.1042/bj0930260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benedetti A, Ferrali M, Chieli E, Comporti M. A study of the relationships between carbon tetrachloride-induced lipid peroxidation and liver damage in rats pretreated with vitamin E. Chem Biol Interact. 1974;9:117–134. doi: 10.1016/0009-2797(74)90004-0. [DOI] [PubMed] [Google Scholar]
- 68.Recknagel RO, Ghoshal AK. Lipoperoxidation of rat liver microsomal lipids induced by carbon tetrachloride. Nature. 1966;210:1162–1163. doi: 10.1038/2101162a0. [DOI] [PubMed] [Google Scholar]
- 69.Nielsen VK, Larsen J. Acute renal failure due to carbon tetrachloride poisoning. Acta Med Scand. 1965;178:363–374. doi: 10.1111/j.0954-6820.1965.tb04280.x. [DOI] [PubMed] [Google Scholar]
- 70.Sinicrope RA, Gordon JA, Little JR, Schoolwerth AC. Carbon tetrachloride nephrotoxicity: a reassessment of pathophysiology based upon the urinary diagnostic indices. Am J Kidney Dis. 1984;3:362–365. doi: 10.1016/s0272-6386(84)80084-0. [DOI] [PubMed] [Google Scholar]
- 71.Chieli E, Malvaldi G. Role of the microsomal FAD-containing monooxygenase in the liver toxicity of thioacetamide S-oxide. Toxicology. 1984;31:41–52. doi: 10.1016/0300-483x(84)90154-9. [DOI] [PubMed] [Google Scholar]
- 72.Bruck R, Oren R, Shirin H, et al. Hypothyroidism minimizes liver damage and improves survival in rats with thioacetamide induced fulminant hepatic failure. Hepatology. 1998;27:1013–1020. doi: 10.1002/hep.510270417. [DOI] [PubMed] [Google Scholar]
- 73.Peeling J, Shoemaker L, Gauthier T, Benarroch A, Sutherland GR, Minuk GY. Cerebral metabolic and histological effects of thioacetamide-induced liver failure. Am J Physiol. 1993;265:G572–G578. doi: 10.1152/ajpgi.1993.265.3.G572. [DOI] [PubMed] [Google Scholar]
- 74.Zimmermann C, Ferenci P, Pifl C, et al. Hepatic encephalopathy in thioacetamide-induced acute liver failure in rats: characterization of an improved model and study of amino acid-ergic neurotransmission. Hepatology. 1989;9:594–601. doi: 10.1002/hep.1840090414. [DOI] [PubMed] [Google Scholar]
- 75.Fontana L, Moreira E, Torres MI, et al. Serum amino acid changes in rats with thioacetamide-induced liver cirrhosis. Toxicology. 1996;106:197–206. doi: 10.1016/0300-483x(95)03177-h. [DOI] [PubMed] [Google Scholar]
- 76.Petermann H, Heymann S, Vogl S, Dargel R. Phagocytic function and metabolite production in thioacetamide-induced liver cirrhosis: a comparative study in perfused livers and cultured Kupffer cells. J Hepatol. 1996;24:468–477. doi: 10.1016/s0168-8278(96)80168-1. [DOI] [PubMed] [Google Scholar]
- 77.Cui FJ, Choi SB, Cho JA, et al. The development of an efficient rat hepatic cirrhosis model. Korean J Hepatobiliary Pancreat Surg. 2007;11:46–52. [Google Scholar]