

Abstract
Intermolecular B-N coordination has been recognized as a promising driving force for molecular self-organization. However, direct utilization of this intermolecular interaction as building bridge for the supramolecular self-assembly of chemical functionalities to form nano-sized architectures remains a daunting challenge. Here, we outline a multiple intermolecular B-N coordination based supramolecular system, where small boronate molecules can be brought together in solution to form nanoparticles with controllable sizes and morphologies. We not only demonstrate the intrinsic switchable fluorescence and the stimuli-responsive capabilities of the designed boronate molecule, but also show that the stabilized or surface functionalized nanoparticles are degradable in response to pH and D-glucose and able to retain the fluorescence features of the boronate molecule. Additionally, the degraded nanoparticles can repair themselves through the reformation of B-N coordination.
Introduction
The development of self-assembly depends on not only the creation of new building blocks but also the exploration of reliable driving forces. Intermolecular interactions such as hydrogen bonding,1–3 hydrophobic aggregation,4–8 π–π stacking9–12, coordination,13–16 electrostatic interaction17, 18 and host-guest recognition19–21 have been extensively employed in the self-assembly of building blocks in various scales and have been explored for a variety of applications.22–24 Among these driving forces, coordination-based interactions are interesting as they have the versatility to afford a predictable control over the directionality and strength of interactions.25 However, self-assembly driven by strong-coordination may be not preferred in the design of smart materials that should be capable of responding, adapting and evolving, because of its irreversible features. Herein, we are interested in developing a system, where small molecules are designed to self-assemble into nano-sized architectures due to a weak, but reversible intermolecular B-N coordination.
The ability of boronic acids or esters with covalently appended neighboring nitrogenous groups to form intramolecular B-N interaction (worth ~13.0 kJ/mol) has been reported.26–28 In protic solvents such as methanol, these molecules can be solvated to afford complexes in which nitrogen and boron atoms are associated through hydrogen bonds between N-H and B-OCH3.[28] This interaction is similar to the direct B-N coordination and can be denoted as methoxylated B-N bonding. Importantly, intermolecular B-N dative bonds are promising building bridges that can bring molecules together to form dimers, molecular aggregates or even submicrospheres29–32. However, self-assembly driven by intermolecular B-N coordination requires the cooperation of secondary reinforcement such as π–π stacking33, 34 and hydrogen bonding.29, 35 Also, Nishiyabu et al.32 and our previous work36 have shown that uniform particles can only be formed through a combination of condensation polymerization and B-N coordination. Thus, it is difficult to construct nano-sized assemblies purely through the intermolecular B-N coordination.
We envisioned that the formation of multiple intermolecular B-N coordination (including direct and methoxylated B-N dative bond) would reinforce the intermolecular interaction, therefore promoting the self-organization of small molecules in solutions. To test our hypothesis, we targeted a boronic molecule (BM) and a catechol molecule (CM) shown in Scheme 1. These two molecules are planar and a simple condensation reaction between them affords a planar boronate ester (BCe), which comprises three functional groups, viz. two imine moieties, a boronate ester, and two polymerizable carbon-carbon double bonds. The aim of this design is not only to facilitate the formation of multiple B-N coordination among BCe molecules, but also to endow the resultant supramolecular assemblies with responsive characteristics to pH and D-glucose. The introduction of carbon-carbon double bond into the BCe makes it possible to crosslink and functionalize the BCe assemblies, thereby overcoming the pre-mature disassembly of the BCe assemblies in the aqueous phase induced by the disassociation of B-N dative bond. We show that the degradation of BCe monomer and the polymerized nanoassemblies under the stimuli of acidic pH or D-glucose, is accompanied with a dramatic evolution of fluorescence character. Importantly, the polymerized nanoassemblies after degradation can undergo self-healing by re-bridging the B-N dative bond.
Scheme 1.
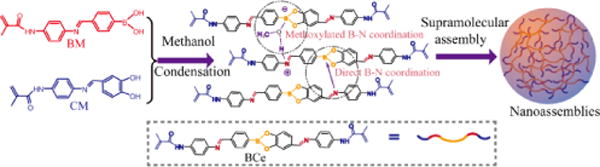
Synthesis and supramolecular assembly of BCe.
Results and Discussion
Supramolecular assembly of BCe driven by B-N coordination
Prior to the investigation of the supramolecular self-assembly of BCe, we first tested the possibility of the formation of B-N dative bond among BCe molecules. 11B NMR was used to probe the formation of B-N dative bond, as the isotropic chemical shift (iso, tested in solution) of the boron for tricoordinate species (−20~90 ppm) is usually higher than that for tetracoordinate species (−120~20 ppm) with respect to F3B·O(C2H5)2 at 0 ppm.37 As a starting material of BCe, BM shows a typical tricoordinated boron signal at δiso = 28.0 ppm in the 11B NMR spectrum with solvent methanol-D4 (Fig. S11a). The boron signal of BM in the solid state 11B NMR spectrum splits into two peaks at 33.7 and 24.3 ppm (Fig. S11b), which is consistent with the results of previous reports.37, 38 Upon formation of BCe in methanol at −10 °C, the characteristic signals of BM disappear in the solid state 11B NMR spectrum with the concurrent appearance of a sharp peak at 18.6 ppm, representing the typical tetracoordinated boron (Fig. S11b). The 11B NMR spectrum of BCe in methanol-D4 is even more interesting, a sharp peak and a broad signal appear at δiso = 18.6 and 11.1 ppm, respectively (Fig. S11a). These results indicate that BCe prefers to form direct intermolecular B-N coordination in solid state but tends to form methoxylated B-N coordination in methanol-D4. The association constant (Ka) calculated from the 11B NMR was 6.51×104 M−1. The direct intermolecular B-N coordination of BCe seems to be unstable in methanol; as evidenced by the 11B NMR spectra in methanol-D4, it evolves into methoxylated form to a large extent with the prolonging of time at room temperature (Fig. S12). This has been further verified by testing the 11B NMR spectra (solvent: methanol-D4) of BCe newly prepared at −10, 0 and 25 °C. With the increase of reaction temperature, the signal of direct B-N coordination (δiso=18.6 ppm) decays, whereas the intensity of the peak (δiso=11.1 ppm) attributed to methoxylated form increases significantly (Fig. S11c). Control experiment performed by mixing 4-formylphenyl boronate-3, 4-dihydroxybenzaldehyde ester (FHBe) with N-(4-aminophenyl)methacrylamide in methanol resulted no appearance of tetracoordinated boronate, suggesting that the B-N dative bond was indeed formed between boronate and imine moieties (Fig. S13). Differential scanning calorimetry (DSC) results indicated that the bonding energy of B-N coordination is about 12.1 kJ/mol (Fig. S11d), which is almost equal to the energy of intramolecular B-N bonding.
Based on the results above, we were interested in demonstrating whether BCe assemblies were readily formed along with the formation of B-N coordination. Indeed, when the condensation reaction between BM and CM was performed at −10 °C in methanol, the as obtained BCe was found to self-assemble into nanospheres with uniform size distribution, as characterized by the transmission electron microscopy (TEM) (Fig. 1a–c and the inset of Fig. 1c). We noted that there is a strong correlation between the particle size of the assemblies and the concentration of BM and CM. BCe assemblies prepared from 10.0, 20.0 and 40.0 mM of BM and CM show diameters from 38±4, 62±12 to 118±15 nm (statistical analysis of 100 particles), which have also been confirmed by the results of dynamic light scattering (DLS, Fig. S14). A control experiment performed with FHBe by using the same method has resulted in no assembly, probably due to the absence of intermolecular B-N coordination.
Fig. 1.

TEM images of spherical nanoparticles prepared at −10°C with (a) 10.0, (b) 20.0 and (c) 40.0 mM of BM and CM. (d), (e) And (f) represent the typical TEM images of spherical supramolecular assemblies obtained at 0°C from 10.0, 20.0 and 40.0 mM of BM and CM, respectively. (g), (h) And (i) are the TEM images of layered structures formed by cooling the BCe methanol solution with 10.0 20.0 and 40.0 mM concentrations from 25 to −10°C. (j), (k) And (l) show the morphologies of the spherical assemblies after electron beam irradiation for 30.0, 60.0 and 120 s, respectively; these nanospheres were prepared by using 20.0 mM of BM and CM at 0°C. The insets are the corresponding magnified TEM images.
The BCe assemblies obtained from the condensation reaction between BM and CM at 0 °C were found to be more transparent than those prepared at −10 °C (Fig. 1d–f and the inset of Fig. 1f). The concentration of BM and CM is also crucial in tuning the size of these assemblies, as evidenced by the successful preparation of BCe nanospheres with diameters ranging from 72±10, 122±22 to 180±28 nm (Fig. 1d–f and Fig. S14). We then tried a special method for the self-assembly of BCe, in which BCe was synthesized at room temperature before cooling its methanol solution to −10 °C. Surprisingly, such a self-assembly route leads to an unexpected layered structure (Fig. 1g–i and the inset of Fig. 1i). These layered assemblies seem to be irregular with diameters ranging from 300 to 850 nm (Fig. S14). However, all the BCe nanoassemblies mentioned above are likely unstable, as they can disassemble completely in methanol solution at room temperature after three days. The DLS results shown in Fig. S14 indicates that this disassembly event can even be achieved within 1 h at 60 °C. Interestingly, only layered structures can be formed if BCe solutions are cooled back to −10 or 0 °C again (Fig. S15). Since the content of methanol inserted intermolecular B-N coordination increases with the increasing of temperature, the direct B-N coordination probably has a higher capability to bring BCe molecules together to form well-defined nanostructures than methanol inserted B-N coordination. Presumably, the methoxylated B-N dative bond can only organize the BCe molecules into dimers or molecular aggregates but not nanostructures.
11B NMR spectra of BCe (Fig. S11) have confirmed that the increase of temperature results in a higher content of methanol inserted B-N coordination. We envisaged that plenty of methanol molecules would be restricted in the BCe assemblies prepared by the condensation reaction between BM and CM at 0 °C. Thus, we focused the electron beam onto these assemblies when doing the TEM characterization, as an irradiation of electron beam with high energy can induce a fast evaporation of volatile substance. These nanospheres exhibit an interesting morphology evolution from solid to porous upon the electron beam irradiation. A prolonging of irradiation time causes the pores to grow significantly (Fig. 1j–l and their insets). However, this electron beam irradiation induced morphology evolution has not been observed to the BCe nanospheres synthesized at −10 °C and the layered structures. This is understandable because less methanol molecules are inserted in these assemblies.
Note that a BCe molecule comprises one boronate and two imine moieties, at most three intermolecular B-N dative bonds can be attached to each BCe molecule. The formation BCe nanospheres seems to be determined by kinetic assembly, as they are formed synchronously with the condensation reaction between BM and CM, thus BCe molecule in the spherical assemblies tends to form multiple B-N dative bonds with other three molecules. In contrast, the layered BCe structures are probably determined by the thermodynamic assembly, therefore forming multiple B-N coordination between two BCe molecules and resulting in a parallel stacking of the building blocks. However, all these BCe assemblies regardless of their morphologies are amorphous as indicated by the electron diffraction patterns (Fig. S16), possibly due to the formation of three-dimensional network crosslinked by multiple B-N coordination with tetrahedral geometry (Scheme 1).
Stabilization and surface functionalization of BCe assemblies
Despite the self-assembly capability of BCe in methanol solution, the as-formed nanoassemblies are extremely unstable in aqueous solution, because of the easy dissociation of B-N dative bond. Thus, we were interested in demonstrating the possibility of stabilizing the BCe assemblies through an in-situ photo polymerization at −10 °C, as this process will likely give a more robust stimuli-sensitive nanoparticle. By covalently bringing the BCe molecules together, we hypothesized that the resultant nanoparticles would keep their morphologies in water solution. The BCe assemblies with the average diameter of 118±15 nm (Fig. 1c) were used for the photo polymerization. Indeed, the polymerized nanoparticles (noted as P(BCe) NPs) are so stable that they can preserve their morphologies in water (Fig. 2a and its inset). However, an increase in BCe concentration can easily induce inter-particle polymerization, thus resulting in the aggregation of P(BCe) NPs (Fig. S17a). The disappearance of the carbon-carbon double bond signal (1621 cm−1) in the FT-IR spectrum of P(BCe) NPs (Fig. S18) reveals the successful polymerization of BCe monomer. Concurrent emergence of the absorption bands at 1320 (B–O), 1242 (C–O) and 1022 cm−1 (B–C) provides strong evidence for the formation of boronate ester. Fig. 2b shows the typical scanning electron microscopy (SEM) image of a P(BCe) NP and its corresponding energy-dispersive X-ray spectroscopy (EDX) elemental mapping. Obvious, carbon, boron and nitrogen are homogenously distributed throughout the entire nanoparticle.
Fig. 2.
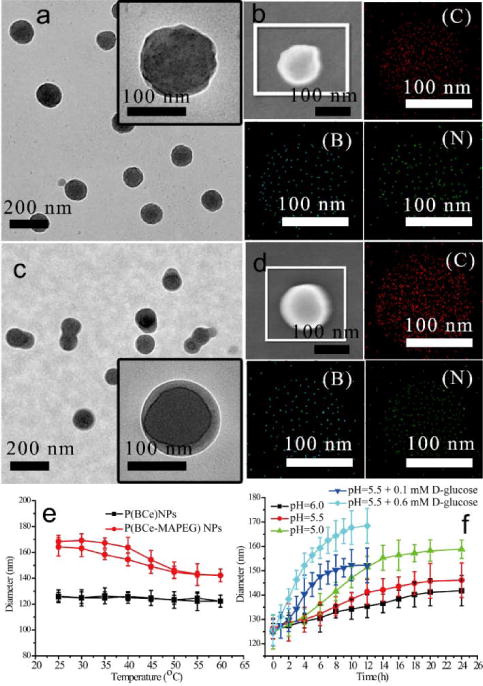
(a) TEM images of P(BCe) NPs prepared from the photo polymerization of spherical assemblies at −10°C, these assemblies were formed by using (a) 40.0 mM of BM and CM. (b) Typical SEM of a P(BCe) NP and EDX elemental mapping (C, B and N) of the nanoparticle. (C) TEM image of P(BCe-MAPEG) NPs prepared by using BCe assemblies that were obatined from (d) 40.0 mM of BM and CM. (d) Typical SEM of a P(BCe-MAPEG) N and EDX elemental mapping (C, B and N) of the nanoparticle. (e) Diameter evolution of P(BCe) NPs in water solution at neutral pH under thermal cycling. (f) Diameter evolution of P(BCe) NPs in acidic water solution or in the presence of D-glucose with pH=5.5.
The versatility of the BCe supramolecular assemblies has also been demonstrated by their facile surface functionalization. Simply adding adding oligo ethylene glycol monomethyl ether methacrylate (MAPEG) into the BCe methanol solution during photo polymerization at −10 °C affords polyethylene glycol (PEG) coated P(BCe) NPs (noted as P(BCe-MAPEG) NPs). It is well known that the polymer domains with conjugated moieties can be visually distinguished from the PEG-rich domains by the sharp color contrast in the TEM image.39, 40 If the P(BCe) NPs are truly coated by P(MAPEG), then the as prepared P(BCe-MAPEG) NPs would exhibit a clear core-shell structure. As expected, each P(BCe-MAPEG) NP comprises a darker core and a lighter shell (Fig. 2c and its inset). This core-shell structure has been further confirmed by SEM associated with EDX elemental mapping of a typical P(BCe-MAPEG) NP (Fig. 2d), as the area of carbon is evidently larger than that of boron and nitrogen. P(BCe-MAPEG) NPs prepared from a higher BCe concentration also exhibit an inter-particle crosslinked morphology (Fig. S17b), which is similar to the P(BCe) NPs. The copolymerization between BCe and PEGMA can be confirmed by the disappearance of carbon-carbon double bond signal and the concurrent emergence of the characteristic absorbance bands of both boronate ester and PEG moieties (Fig. S18).
After polymerization, P(BCe) NPs were highly stable in water solution, cycling the temperature from 25 to 60 °C resulted in no evident size evolution (Fig. 2e). P(BCe-MAPEG) NPs swelled-shrunk slightly and reversibly with the cycling of temperature (Fig. 2e), probably due to the thermal sensitivity of PEG chain. It is well known that imine bond and boronate ester are dissociable in weakly acidic solution,41, 42 and carbohydrates can specifically accelerate the degradation of the boronate ester43, 44. Therefore, we were interested in testing whether the P(BCe) NPs and P(BCe-MAPEG) NPs were degradable in response to acidic pH or D-glucose. DLS results indicated that P(BCe) NPs swelled evidently at acidic solutions (Fig. S19a). The swell speed and extent increased with the decreasing of pH value (Fig. 2f). Addition of D-glucose into the particle solution at pH=5.5 could significantly increase the swelling speed and extent (Fig. 2f). Similarly, evident swelling was observed to P(BCe-MAPEG) NPs in acidic solution or in solution with the presence of D-glucose (Fig. S19b and c). The swelling of the particles might be induced by the partial degradation of imine and boronate moieties, therefore leading to a higher hydrophilicity of the polymer chains. These results provided the first indication that the polymerized nanoassemblies were degradable.
B-N coordination induced self-healing of P(BCe) NPs
We further confirmed the degradation feature of the polymerized nanoassemblies by using TEM. Taking P(BCe) NPs as example, after degradation in water solution with pH=5.0, these nanoparticles became larger, more irregular and transparent (Fig. 3a) as compared to the undegraded particles (Fig. 2a). With the presence of D-glucose, P(BCe) NPs were even broken after degradation (Fig. 3d). However, these nanoparticles could not totally disassemble even after 3 days, possibly due to dynamic feature of both imine bond and boronate ester, as well as the crosslinked networks derived from the polymerization of carbon-carbon double bond. Consequently, it is possible to reform the B-N coordination between the residual imine bonds and boronate ester moieties under certain conditions. To test this, degraded P(BCe) NPs were collected and freeze-dried to preserve their primary morphology. Then, these nanoparticles were re-dispersed in methanol solution to promote the reformation of B-N coordination. Interestingly, after staying in methanol at 0 °C for 3.0 h, P(BCe) NPs pre-degraded in water solution at pH=5.0, became more spherical and regular (Fig. 3b). Especially, the broken P(BCe) NPs pre-degraded in water solution at pH=5.5 with 0.6 mM of D-glucose, could repair themselves to form solid nanoparticles (Fig. 3e).
Fig. 3.
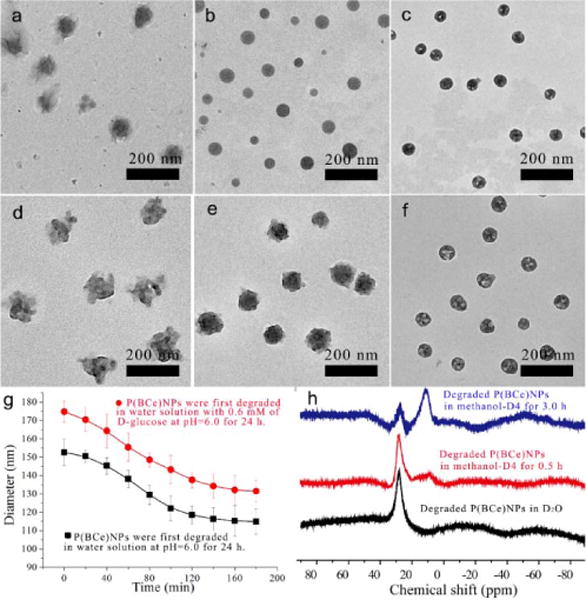
TEM image of P(BCe) NPs after degradation in water solution at pH=5.0 for 24 h (a), and their corresponding TEM images after self-healing in methanol at 0°C for 3.0 h (b) and at −10°C for 12 h (c). TEM image of P(BCe) NPs after degradation in water solution at pH=5.5 with 0.6 mM of D-glucose for 24 h (d), and their corresponding TEM images after self-healing in methanol at 0°C for 3.0 h (e) and at −10°C for 12 h (f). (g) DLS results monitoring the size evolution of degraded P(BCe) NPs in methanol at 0°C. (h) 11B NMR spectra tracking the reformation of B-N coordination in methanol at 0°C. TEM images shown in (a) and (d) were taken after freeze-drying to preserve the original morphology of the degraded nanoparticles.
If the pre-degraded P(BCe) NPs stayed in methanol solution at −10 °C for more than 12 h, they changed into hollow spherical nanoparticles (Fig. 3c and f). TEM images also showed that obvious shrinkage of the P(BCe) NPs occurred along with the morphology evolution. DLS results further confirmed that the degraded P(BCe) NPs shrink gradually in methanol solution at 0 °C with the prolong of time (Fig. 3g and Fig. S20). We then monitored the 11B NMR spectrum evolution of the degraded P(BCe) NPs in methanol solution. Obviously, the signal of tetracoordinated boron (~11.1 ppm) derived from the methoxylated B-N coordination emerged and its intensity increased with the prolonging of time (Fig. 3h). Note that the 11B NMR spectrum of degraded P(BCe) NPs in D2O displays no signal of tetracoordinated boron, it is reasonable to believe that the B-N dative bond can be reformed in methanol. Thus, the self-healing of the degraded P(BCe) NPs is probably induced by the reformation of B-N coordination. We also tested the 11B NMR spectrum of the degraded P(BCe) NPs after staying in methanol at −10 °C for 12 h (Fig. S20). A sharp peak at ~18.6 ppm appeared, revealing that direct B-N coordination was formed in the hollow nanoparticles. We consider that the direct B-N coordination can densely crosslink the nanoparticles and promote the transfer of polymer chains from interior nanoparticle to the out layer, thus resulting in the formation of the hollow structures. On the other hand, the mass loss of the degraded P(BCe) NPs induced by the cleavage of imine bond and boronate ester may also promote the particle shrinkage and the formation of hollow structure after self-healing.
Fluorescent and stimuli-responsive properties of BCe, P(BCe) and P(BCe-MAPEG) NPs
Since BM, CM and BCe are based on conjugated building blocks, we were interested in evaluating their photophysical properties as well. BM, CM and BCe are found to be unable to cause fluorescent emissions in methanol (Fig. S21), although they possess strong UV/vis absorbance (Fig. S22). We then transferred BM, CM and BCe from methanol into water with different pH values to test their fluorescent properties. While not fluorescent at higher pH, BCe shows a strong green emission with a maximum of 480 nm in water (pH≤6.5) under the excitation at wavelength of 360 nm (Fig. S23 and S24). However, neither BM nor CM exhibits fluorescent emission in aqueous solutions at different pH values (Fig. S24). The quantum yield (ϕ) of BCe in water with pH=5.5 was measured to be 0.50 by using 4′, 6-diamidino-2-phenylindole (ϕ=0.58) as standard (Fig. S25 and Table S1) and the fluorescence life time (τ) is 11.5 ns (Fig. S26). Note that the current BCe contains two different pH-sensitive moieties, viz. boronate ester and imine. The possible degradation products such as BM, CM, N-(4-aminophenyl)methacrylamide and FHBe may cause fluorescent emission in acid solution. However, this is unlikely because these compounds by themselves do not exhibit any fluorescence (Fig. S24). Additionally, the results of 1H NMR and 13C NMR spectra (Fig. S9 and S10) indicate that BCe is relatively stable in D2O (pH=5.5) with only a small amount of BCe being degraded after 4 h. Therefore, we attribute our findings to the possibility that the imine moiety is readily protonated in acid solution, thereby changing the π-conjugation of BCe, thus turning on the fluorescence in BCe.
The fluorescence of BCe in water is highly pH-dependent, reducing the pH value results in a significant increase in emission intensity (Fig. 4a and b). This emission enhancement is saturated at pH around 5.5, and thus no discernible increase of emission intensity appears with the further decrease of pH value (Fig. 4c). Notably, the fluorescence of BCe is switchable when cycling the solution pH between acidity and neutrality (Fig. 4d), further supporting a protonation-based event as the reason for the change in fluorescence. Neutralizing the BCe solution from pH=5.5 to 7.4 reduces or even quenches the fluorescence emission, whereas acidifying the neutral solution back to pH=5.5 can recover the fluorescence emission. After several cycles, the maximum emission intensity of BCe at pH=5.5 exhibits a certain decline. We attribute this to the pH induced degradation of BCe, because both boronate and the imine moieties are dynamic and degradable in acid solution. If this speculation were correct, then we would expect to observe a difference in the emission evolutions of BCe at different pH values. Indeed, when the solution pH is decreased from 6.0 to 5.0, the fluorescence attenuation of BCe becomes considerably higher (Fig. 4e–g). Also, control experiment performed by using a deceased intensity of excitation light resulted in the similar fluorescence attenuation, indicating that the fluorescence decay was not caused by photobleaching (Fig. S27). We further tested the effect of D-glucose on the emission intensity of BCe to understand the effect of boronate ester moiety on the fluorescence property. Note that the emission intensity of BCe in water solution (pH=6.0) exhibits a 16.5 % decrease within 24 h (calculated from Fig. 4e). In comparison, BCe shows a much faster decay of emission intensity upon the addition of D-glucose (Fig. 4h–j). The emission intensities of BCe solutions at pH 6.0 in the presence of 0.1, 0.2 and 0.6 mM of D-glucose show reductions of 22, 34 and 54 % (calculated from Fig. 4h–j) within 10 h, respectively.
Fig. 4.
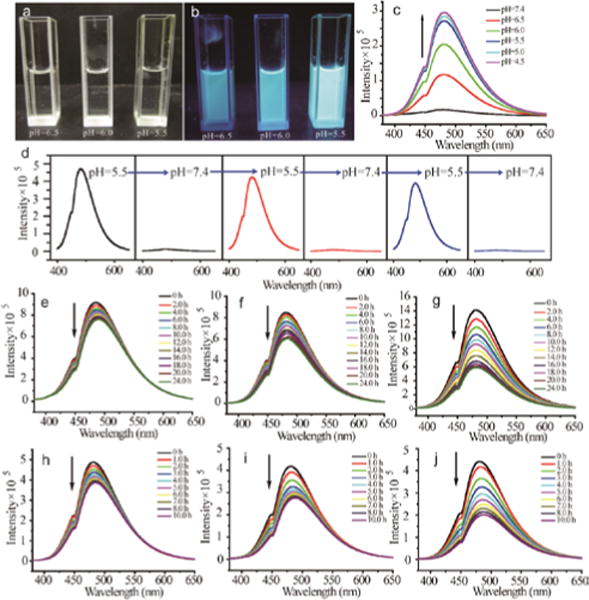
Photographs of BCe in water with different pH under (a) natural light and (b) UV irradiation (wavelength 360 nm). (c) Effect of pH on the fluorescent emission intensity of BCe in water under an excitation wavelength 360 nm. (d) Switchable fluorescent emission of BCe in water when cycling the pH between 5.5 and 7.4. (e), (f) And (g) represent the emission evolution of BCe in water with pH=6.0, 5.5 and 5.0. (h), (i) And (j) trace the emission decays of BCe in water at pH=6.0 with 0.1, 0.2 and 0.6 mM of D-glucose, respectively.
Finally, we were interested in studying whether the P(BCe) and P(BCe-PEGMA) NPs could preserve the fluorescence property of BCe. Thus, we tested the fluorescent emission of both P(BCe) and P(BCe-MAPEG) NPs in water solution with different pH values. Surprisingly, the fluorescence characteristics of these polymerized NPs are almost the same with that of the BCe in water (Fig. 5a and Fig. S28). Both P(BCe) and P(BCe-PMAPEG) NPs show no fluorescence in water at pH=7.4, whereas a strong emission peak at 480 nm is found in acidic water solution (excitation wavelength 360 nm). The emission intensity of these polymerized NPs is also highly pH dependent, a reduction of pH value results in an evident increase in emission intensity (Fig. 5b and c). These results suggest that the carbon-carbon double bond has no contribution to the fluorescence characteristic of BCe. P(BCe-MAPEG) NPs coated with biocompatible PEG segments may be promising nanocarriers for drug delivery. This potential would be largely reinforced if the degradation of P(BCe-MAPEG) NPs is accompanied with a evolution of fluorescent intensity. Indeed, the emission intensities of P(BCe-MAPEG) NPs in water solutions with pH=5.5 and 5.0 show reductions of 19 and 37 % after 24 h, respectively (Fig. 5d and e). After the addition of D-glucose, the reduction of emission intensity becomes much faster, P(BCe-MAPEG) NPs in water solutions (pH=5.5) with 0.1 and 0.6 mM of D-glucose show 26 % and 45 % decreases in emission intensity within 12 h (Fig. 5f and g). Obviously, these fluorescence emission evolutions are likely due to the cleavage of imine or boronate moieties that induces the degradation of P(BCe-MAPEG) NPs. As already confirmed by the DLS results, P(BCe-MAPEG) NPs indeed swell in acidic solution and the swelling extent can be improved over 50 % with the addition of D-glucose (Fig. S19). We consider that the switchable fluorescence and stimuli-responsive fluorescence intensity features of these nanoparticles may be attractive for biomedical imaging and sensing.45–48
Fig. 5.
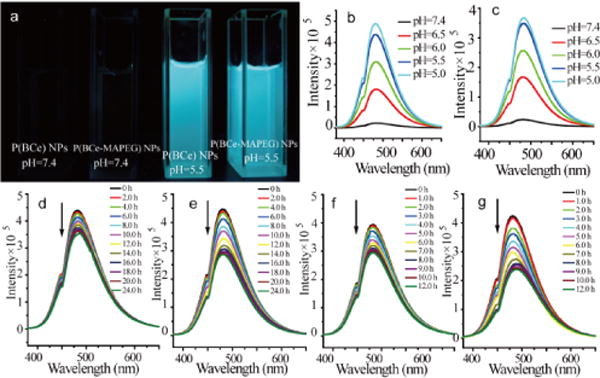
(a) Photographs of P(BCe) and P(BCe-MAPEG) NPs in water with different pH values under UV irradiation (wavelength 360 nm). (b) And (c) are the effects of pH on the emission intensity of P(BCe) and P(BCe-MAPEG) NPs in water under an excitation wavelength of 360 nm. (d) And (e) are the emission evolutions of P(BCe-MAPEG) NPs in water solution with pH=5.5 and 5.0. (f) And (g) represent the emission evolutions of P(BCe-MAPEG) NPs in water solutions (pH=5.5) with 0.1 and 0.6 mM of D-glucose.
Conclusions
In summary, we have shown that intermolecular B-N dative bonding can effectively bring small molecules together to construct supramolecular nanoassemblies. The introduction of both imine and boronate moieties into a planar BCe molecule can facilitate the formation of multiple intermolecular B-N coordination, thus providing a facile route to the supramolecular assembles with controllable morphologies. BCe assemblies can be stabilized and functionalized through a simple in-stiu photo polymerization, thereby creating water stable P(BCe) and P(BCe-MAPEG) NPs. The versatility of the polymerized assemblies have been demonstrated by not only the pH or D-glucose induced degradation, but also the capability of self-healing through the reformation of B-N dative bond. In addition, both P(BCe) and P(BCe-MAPEG) NPs can preserve the intrinsic fluorescence feature of BCe monomer by showing that (i) strong fluorescent emission can be turned on in acidic water solutions; and (ii) evident decay of fluorescent emission intensity occurs along with the particle degradation. The significance of this work is that the multiple intermolecular B-N coordination may open a new avenue to the controllable construction of nanostructures. In addition, the pH switchable fluorescent emission, stimuli-responsive property and the facile surface functionalization demonstrate the promising application potential of these supramolecular assemblies in the fields of delivery and sensing.
Experimental
Materials
Methacryloyl chloride, 3, 4-dihydroxybenzaldehyde, 4-formylphenylboronic acid, triethylamine, p-phenylenediamine, magnesium sulfate anhydrous (MgSO4), sodium hydroxide (NaOH), polyethylene glycol monomethyl ether methacrylate (MAPEG, MW ~475 Da), photo initiator benzophenone (BP) and other conventional reagents were obtained from commercial sources and were used as received.
Characterization
1H-NMR, 13C-NMR and 11B-NMR spectra in solutions were measured on a Bruker ARX 400MHz spectrometer. Normal NMR tube was used to test the 11B-NMR spectra of the molecules, as the signals of the boronates in the NMR tube do not overlap with the characteristic signals of the molecules. Since the characteristic signals of solid state 11B-NMR spectra were tested on a Bruker DSX300 spectrometer. Mass spectra were acquired from an electrospray ion trap mass spectrometer (Bruker Daltronics Esquire 3000 plus). FT-IR spectroscopy (Nicolet Avatar 360) was used to confirm the structures of the polymerized supramolecular assemblies. DLS and zeta potential measurements were performed on a Malvern Nanozetasizer. UV/vis absorption data of the samples were acquired in solutions by using UV spectrophotometry (Unico UV/vis 2802PCS). Thermograms of BCe and BM were performed on a DSC instrument (TA 2920 Modulated DSC, TA instruments). Samples were heated under 30.0 mL/min nitrogen with a heating-up speed 10 °C/min. Before testing, all the samples were vacuum dried at room temperature for at least 24 h. SEM images were taken using a Hitachi SU-70 SEM instrument. Energy-dispersive X-ray spectroscopy (EDX) analyses were taken using an EDX attachment (INCA, Oxford Instruments) on the Hitachi SU-7 SEM instrument.
The fluorescence emission spectra and the life time (τ) were measured by a FLS920 Fluorescence Lifetime and Steady State Spectrometer (stimulation slit width: 5.0 nm, emission slit width: 5.0 nm). For the measurement of the quantum yield (ϕ) of BCe in water, 4′, 6-Diamidino-2-phenylindole (DAPI, maximum excitation wavelength was 310nm, ϕ=0.58) was used as the standard. The quantum yields were calculated from the following formula (1) by using at least 3 groups of solutions with increasing concentration. Both of them were excited at wavelength 360 nm.
| (1) |
Where ϕ and ϕr are the quantum yields of BCe and DAPI; I and Ir represent the integrated fluorescence intensities of BCe and DAPI; A and Ar are the UV/vis absorbances of BCe and DAPI at 360 nm.
TEM measurements and electron diffraction experiment were performed with a JEM2100 at an acceleration voltage of 200 kV. To prepare the TEM samples, a small drop of sample solution was deposited onto a carbon-coated copper electron microscopy (EM) grid and then dried under room temperature at atmospheric pressure.
Synthesis of N-(4-aminophenyl)methacrylamide
Methacryloyl chloride (2.19 g, 21.0 mM) and p-phenylenediamine (2.16 g, 20.0 mM) were dissolved in 10 mL and 50 mL of dichloromethane (DCM), respectively. Triethylamine (2.2 g, 22.0 mM) was added to the solution of p-phenylenediamine. Then, methacryloyl chloride DCM solution was added dropwise into the p-phenylenediamine solution at 5 °C. After stirring at room temperature for 12 h, the reaction mixture was washed with NaOH solution two times and with water three times to reach a neutral pH. The oil phase containing the key product was collected and dried with efficient MgSO4. The crude product (yellow solid) was obtained after the removal of solvent by rotary evaporation. The pure product was purified by passing through a column chromatography using silica gel as stationary phase and mixture of ethyl acetate/hexane (2:1) with 2.0 vol% triethylamine as eluent. Yield: 58 %. 1H-NMR (400 MHz, DMSO-D6) δ (ppm): 9.35 (s, 1H), 7.27 (d, 2H), 6.50 (d, 2H), 5.72 (s, 1H), 5.41 (s, 1H), 4.87 (s, 2H), 1.93 (s, 3H); 13C-NMR (300 MHz, DMSO-D6) δ (ppm): 166.41, 145.48, 141.12, 128.3, 122.53, 119.42, 114.07, 19.32. ESI/MS m/z 177.5 [M+H]+ (expected m/z= 177.22), m/z 199.6 [M+23]+ (expected m/z= 199.22). Melting Point: 126 °C.
Synthesis of BM
N-(4-aminophenyl)methacrylamide (0.19 g, 1.07 mM) was dissolved in 15 mL of DCM, while 4-formylphenylboronic acid (0.15 g, 1.0 mM) was dissolved in a mixed solvent containing 0.5 mL of methanol and 5 mL of DCM. The 4-formylphenylboronic acid solution accompanied with MgSO4 (1.2 g, 10 mM) was added to the N-(4-aminophenyl)methacrylamide solution. After stirring at room temperature for 12 h, the reaction mixture was filtered to remove the solid MgSO4 and a yellow solution containing the key product was obtained. The solvent was removed by rotary evaporation, and the as obtained crude product was washed with DCM three times to get the pure yellow product. Yield: 86 %. 1H-NMR (400 MHz, DMSO-D6) δ (ppm): 9.86 (s, 1H), 8.66 (s, 1H), 8.19 (s, 2H), 7.92 (d, 2H), 7.88 (d, 2H), 7.75 (d, 2H), 7.30 (d, 2H), 5.82 (s, 1H), 5.53 (s, 1H), 1.97 (s, 3H); 13C-NMR (300 MHz, DMSO-D6) δ (ppm): 167.18, 159.82, 146.94, 140.87, 137.96, 137.92, 134.88, 127.90, 121.87, 121.23, 120.41, 114.08, 19.22. ESI/MS m/z 367.26 [M+59]− (expected m/z= 367.14).
Synthesis of CM
3, 4-Dihydroxybenzaldehyde (0.14 g, 1.0 mM) was first dissolved in mixed solvent containing 0.5 mL of methanol and 5 mL of DCM. To a solution of N-(4-aminophenyl)methacrylamide (0.19 g, 1.07 mM) in 15 mL of DCM was added the 3,4-dihydroxybenzaldehyde solution accompanied with MgSO4 (1.2 g, 10 mM). The mixture was stirred at room temperature for 12 h under an argon atmosphere and protected from light. After the removal of solid MgSO4 by filtration, the solvent was removed by using rotary evaporation. The solid crude product was further washed with DCM three times to get the pure orange product. Yield: 84 %. 1H-NMR (400 MHz, DMSO-D6) δ (ppm): 9.80 (s, 1H), 8.41 (s, 1H), 7.70 (d, 2H), 7.39 (s, 1H), 7.20 (d, 2H), 7.17 (d, 1H), 6.83 (d, 1H), 5.81 (s, 1H), 5.52 (s, 1H), 1.97 (s, 3H); 13C-NMR (300 MHz, DMSO-D6) δ (ppm): 167.09, 159.41, 149.53, 147.57, 146.09, 140.90, 137.18, 128.60, 122.81, 121.55, 121.28, 120.30, 115.93, 114.52, 19.23. ESI/MS m/z 295.7 [M-H]− (expected m/z= 295.32).
Synthesis of FHBe
4-Formylphenylboronic acid (0.15 g, 1.0 mM) and 3, 4-dihydroxybenzaldehyde (0.15 g, 1.07 mM) were dissolved in 2 and 8 mL of methanol, respectively. The 4-formylphenylboronic acid solution accompanied with MgSO4 (1.2 g, 10 mM) was added to the 3, 4-dihydroxybenzaldehyde solution. After stirring at room temperature for 24 h, solid MgSO4 was removed by filtration and solvent was removed by rotary evaporation to get crude product. The solid crude product was further washed with a mixed solvent of DCM and methanol (10:1) three times to get the pure orange product. Yield: 78 %. 1H-NMR (400 MHz, CDCl3) δ (ppm): 10.11 (s, 1H), 9.99 (s, 1H), 8.25 (d, 2H), 8.00 (d, 2H), 7.85 (s, 1H), 7.75 (d, 1H), 7.47 (d, 1H); 13C-NMR (300 MHz, CDCl3) δ (ppm): 192.23, 190.76, 153.36, 149.08, 139.18, 135.69, 133.88, 132.72, 129.23, 127.78, 113.10, 112.51. ESI/MS m/z 291.08 [M+39]+ (expected m/z= 291.03).
Preparation of BCe supramolecular assemblies
(1) Spherical supramolecular assemblies: BM and CM were firstly dissolved in methanol to form solutions with different concentrations. Then the BM solution was added dropwise into the CM solution at −10 or 0 °C under a vigorous stirring to form BCe. 10.0, 20.0 and 40.0 mM of BM and CM were used in this preparation procedure to test the influence of concentration on the particle size of the BCe supramolecular assemblies. (2) Supramolecular assemblies with layered structure: BM in methanol solution was added dropwise into the CM methanol solution at 25 °C under a vigorous stirring to form BCe. Then the BCe solution was cooled to −10 °C to promote the formation of supramolecular assemblies. 10.0, 20.0 and 40.0 mM of BM and CM were used in this preparation procedure to test the influence of concentration on the diameter of the BCe supramolecular assemblies. 1H-NMR (400 MHz, D2O/methanol-D4 (volume ratio = 9:1)) δ (ppm): 8.56 (s, 1H), 8.45 (s, 1H), 7.76 (d, 2H), 7.56 (d, 4H), 7.45 (d, 4H), 7.31 (s, 1H), 7.29 (d, 2H), 7.22 (s, 1H), 6.85 (d, 1H), 5.65 (s, 2H), 5.43 (s, 2H), 1.85 (s, 6H); 13C-NMR (300 MHz, D2O/methanol-D4 (volume ratio = 9:1)) δ (ppm): 168.62, 167.36, 162.28, 159.98, 155.64, 140.76, 138.91, 136.79, 133.80, 132.01, 128.39, 127.78, 122.69, 121.78, 121.70, 121.26, 118.49, 110.03, 17.83.
Preparation of P(BCe) NPs
Spherical supramolecular assemblies formed from the condensation reaction between 40.0 mM of BM and CM at −10 °C were chosen for the preparation of P(BCe) NPs. To a 2.0 mL of spherical supramolecular assembly methanol solution was added benzophenone (0.09 mg, 0.0005 mM). The light source was a high-pressure Hg lamp (400 W), set at a distance of 40 cm from the sample. The photopolymerization was carried out at −10 °C. The as obtained P(BCe) NPs were purified by centrifugation and redispersed in water for other characterizations.
Preparation of P(BCe-MAPEG) NPs
To a methanol solution of spherical supramolecular assemblies formed from the condensation reaction between 40.0 mM of BM and CM at −10 °C, was added benzophenone (0.09 mg, 0.0005 mM) and MAPEG (4.75 mg, 0.01 mM). A high-pressure Hg lamp (400 W) was used as light source and set at a distance of 40 cm from the sample. The photopolymerization was carried out at −10 °C. The as obtained P(BCe-MAPEG) NPs were purified by centrifugation and redispersed in water for other characterizations.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (51373142, 51403176, U1205113); the Scientific and Technical Project of Xiamen (3502Z20130033).
Footnotes
Electronic Supplementary Information (ESI) available: Synthetic and characterization details; NMR spectra, TEM images, DLS results and fluorescence properities of the resultant products. See DOI: 10.1039/b000000x/
Contributor Information
S. Thayumanavan, Email: lzdai@xmu.edu.cn.
Lizong Dai, Email: thai@chem.umass.edu.
Notes and references
- 1.Aakeröy CB, Beatty AM, Helfrich BA. Angew Chem Int Ed. 2001;40:3240–3242. doi: 10.1002/1521-3773(20010903)40:17<3240::AID-ANIE3240>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Hu C, Comotti A, Ward MD. Science. 2011;333:436–440. doi: 10.1126/science.1204369. [DOI] [PubMed] [Google Scholar]
- 3.Wang J-H, Altukhov O, Cheng C-C, Chang F-C, Kuo S-W. Soft Matter. 2013;9:5196–5206. [Google Scholar]
- 4.Zhang L, Eisenberg A. Science. 1995;268:1728–1731. doi: 10.1126/science.268.5218.1728. [DOI] [PubMed] [Google Scholar]
- 5.Sun G, Cui H, Lin LY, Lee NS, Yang C, Neumann WL, Freskos JN, Shieh JJ, Dorshow RB, Wooley KL. J Am Chem Soc. 2011;133:8534–8543. doi: 10.1021/ja200182t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters O, Ritter H. Angew Chem Int Ed. 2013;52:8961–8963. doi: 10.1002/anie.201301286. [DOI] [PubMed] [Google Scholar]
- 7.Savariar EN, Aathimanikandan SV, Thayumanavan S. J Am Chem Soc. 2006;128:16224–16230. doi: 10.1021/ja065213o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gröschel AH, Walther A, Löbling TI, Schmelz J, Hanisch A, Schmalz H, Müller AHE. J Am Chem Soc. 2012;134:13850–13860. doi: 10.1021/ja305903u. [DOI] [PubMed] [Google Scholar]
- 9.Hoeben FJM, Jonkheijm P, Meijer EW, Schenning APHJ. Chem Rev. 2005;105:1491–1546. doi: 10.1021/cr030070z. [DOI] [PubMed] [Google Scholar]
- 10.Song B, Wei H, Wang Z, Zhang X, Smet M, Dehaen W. Adv Mater. 2007;19:416–420. [Google Scholar]
- 11.Schenning APHJ, Meijer EW. Chem Commun. 2005;41:3245–3258. doi: 10.1039/b501804h. [DOI] [PubMed] [Google Scholar]
- 12.Burattini S, Greenland BW, Merino DH, Weng W, Seppala J, Colquhoun HM, Hayes W, Mackay ME, Hamley IW, Rowan SJ. J Am Chem Soc. 2010;132:12051–12058. doi: 10.1021/ja104446r. [DOI] [PubMed] [Google Scholar]
- 13.Whittell GR, Hager MD, Schubert US, Manners I. Nature Mater. 2011;10:176–188. doi: 10.1038/nmat2966. [DOI] [PubMed] [Google Scholar]
- 14.Du G, Moulin E, Jouault N, Buhler E, Giuseppone N. Angew Chem Int Ed. 2012;51:12504–12508. doi: 10.1002/anie.201206571. [DOI] [PubMed] [Google Scholar]
- 15.Yan Y, Huang J. Coord Chem Rev. 2010;254:1072–1080. [Google Scholar]
- 16.Ejima H, Richardson JJ, Liang K, Best JP, van Koeverden MP, Such GK, Cui J, Caruso F. Science. 2013;341:154–157. doi: 10.1126/science.1237265. [DOI] [PubMed] [Google Scholar]
- 17.Sun TL, Kurokawa T, Kuroda S, Ihsan AB, Akasaki T, Sato K, Haque MA, Nakajima T, Gong JP. Nature Mater. 2013;12:932–937. doi: 10.1038/nmat3713. [DOI] [PubMed] [Google Scholar]
- 18.Duarte A, Slutsky M, Hanrahan G, Mello CM, Bazan GC. Chem -Eur J. 2012;18:756–759. doi: 10.1002/chem.201103237. [DOI] [PubMed] [Google Scholar]
- 19.Wang F, Han C, He C, Zhou Q, Zhang J, Wang C, Li N, Huang F. J Am Chem Soc. 2008;130:11254–11255. doi: 10.1021/ja8035465. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Luo Y, Chen J, Dong S, Yu Y, Ma Z, Huang F. Angew Chem Int Ed. 2011;50:1397–1401. doi: 10.1002/anie.201006693. [DOI] [PubMed] [Google Scholar]
- 21.Dong S, Luo Y, Yan X, Zheng B, Ding X, Yu Y, Ma Z, Zhao Q, Huang F. Angew Chem Int Ed. 2011;50:1905–1909. doi: 10.1002/anie.201006999. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Wang C. Chem Soc Rev. 2011;40:94–101. doi: 10.1039/b919678c. [DOI] [PubMed] [Google Scholar]
- 23.Lehn JM. Science. 1993;260:1762–1763. doi: 10.1126/science.8511582. [DOI] [PubMed] [Google Scholar]
- 24.Yan X, Wang F, Zheng B, Huang F. Chem Soc Rev. 2012;41:6042–6065. doi: 10.1039/c2cs35091b. [DOI] [PubMed] [Google Scholar]
- 25.Chakrabarty R, Mukherjee PS, Stang PJ. Chem Rev. 2011;111:6810–6918. doi: 10.1021/cr200077m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosch LI, Fyles TM, James TD. Tetrahedron. 2004;60:11175–11190. [Google Scholar]
- 27.Kim KT, Cornelissen JJLM, Nolte RJM, van Hest JCM. J Am Chem Soc. 2009;131:13908–13909. doi: 10.1021/ja905652w. [DOI] [PubMed] [Google Scholar]
- 28.Zhu L, Shabbir SH, Gray M, Lynch VM, Sorey S, Anslyn EV. J Am Chem Soc. 2006;128:1222–1232. doi: 10.1021/ja055817c. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Li M, Chandrasekaran S, Gao X, Fang X, Lee H-W, Hardcastle K, Yang J, Wang B. Tetrahedron. 2007;63:3287–3292. doi: 10.1016/j.tet.2007.02.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Icli B, Sheepwash E, Riis-Johannessen T, Schenk K, Filinchuk Y, Scopellitia R, Severin K. Chem Sci. 2011;2:1719–1721. [Google Scholar]
- 31.Livant PD, Northcott DJD, Shen YP, Webb TR. J Org Chem. 2004;69:6564–6571. doi: 10.1021/jo040199o. [DOI] [PubMed] [Google Scholar]
- 32.Nishiyabu R, Teraoka S, Matsushima Y, Kubo Y. ChemPlusChem. 2012;77:201–209. [Google Scholar]
- 33.Cruz-Huerta J, Salazar-Mendoza D, Hernández-Paredes J, Ahuactzic IFH, Höpfl H. Chem Commun. 2012;48:4241–4243. doi: 10.1039/c2cc00144f. [DOI] [PubMed] [Google Scholar]
- 34.Sheepwash E, Krampl V, Scopelliti R, Sereda O, Neels A, Severin K. Angew Chem Int Ed. 2011;50:3034–3037. doi: 10.1002/anie.201007225. [DOI] [PubMed] [Google Scholar]
- 35.Adamczyk-WoŸniak A, Cyrański MK, Frączak BT, Lewandowska A, Madura ID, Sporzyński A. Tetrahedron. 2012;68:3761–3767. [Google Scholar]
- 36.Li L, Yuan C, Dai L, Thayumanavan S. Macromolecules. 2014;47:5869–5876. [Google Scholar]
- 37.Weiss JWE, Bryce DLJ. Phys Chem A. 2010;114:5119–5131. doi: 10.1021/jp101416k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh S-W, Weiss JWE, Kerneghan PA, Korobkov I, Maly KE, Bryce DL. Magn Reson Chem. 2012;50:388–401. doi: 10.1002/mrc.3815. [DOI] [PubMed] [Google Scholar]
- 39.Yuan C, Xu Y, Jiang N, Chen G, Xu B, He N, Dai L. Soft Matter. 2011;7:3366–3372. [Google Scholar]
- 40.dos Santos AM, Bris TL, Graillat C, D’Agosto F, Lansalot M. Macromolecules. 2009;42:946–956. [Google Scholar]
- 41.Gu J, Cheng WP, Liu J, Lo SY, Smith D, Qu X, Yang Z. Biomacromolecules. 2008;9:255–262. doi: 10.1021/bm701084w. [DOI] [PubMed] [Google Scholar]
- 42.Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E, Levchenko TS, Torchilin VP. Bioconjugate Chem. 2006;17:943–949. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Xiao W, Xiao K, Berti L, Luo J, Tseng HP, Fung G, Lam KS. Angew Chem Int Ed. 2012;51:2864–2869. doi: 10.1002/anie.201107144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu X, Li Z, Chen X-X, Fossey JS, James TD, Jiang Y-B. Chem Soc Rev. 2013;42:8032–8048. doi: 10.1039/c3cs60148j. [DOI] [PubMed] [Google Scholar]
- 45.Ghoroghchian PP, Frail PR, Susumu K, Blessington D, Brannan AK, Bates FS, Chance B, Hammer DA, Therien MJ. PNAS. 2005;102:2922–2927. doi: 10.1073/pnas.0409394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montalti M, Battistelli G, Cantelli A, Genovese D. Chem Commun. 2014;50:5326–5329. doi: 10.1039/c3cc48464e. [DOI] [PubMed] [Google Scholar]
- 47.Thomas SW, Joly GD, Swager TM. Chem Rev. 2007;107:1339–1386. doi: 10.1021/cr0501339. [DOI] [PubMed] [Google Scholar]
- 48.Ji X, Yao Y, Li J, Yan X, Huang F. J Am Chem Soc. 2013;135:74–77. doi: 10.1021/ja3108559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.