

SUMMARY
Endoplasmic reticulum (ER) stress is observed in many human diseases, often associated with inflammation. ER stress can trigger inflammation through nucleotide-binding domain and leucine-rich repeat containing (NLRP3) inflammsome, which may stimulate inflammasome formation by association with damaged mitochondria. How ER stress triggers mitochondrial dysfunction and inflammasome activation is ill defined. Here we have used an infection model to show that the IRE1α ER stress sensor regulates regulated mitochondrial dysfunction through an NLRP3-mediated feed-forward loop, independently of ASC. IRE1α activation increased mitochondrial reactive oxygen species, promoting NLRP3 association with mitochondria. NLRP3 was required for ER stress-induced cleavage of caspase-2 and the pro-apoptotic factor, Bid, leading to subsequent release of mitochondrial contents. Caspase-2 and Bid were necessary for activation of the canonical inflammasome by infection-associated or general ER stress. These data identify an NLRP3-caspase-2 dependent mechanism that relays ER stress to the mitochondria to promote inflammation, integrating cellular stress and innate immunity.
Graphical abstract
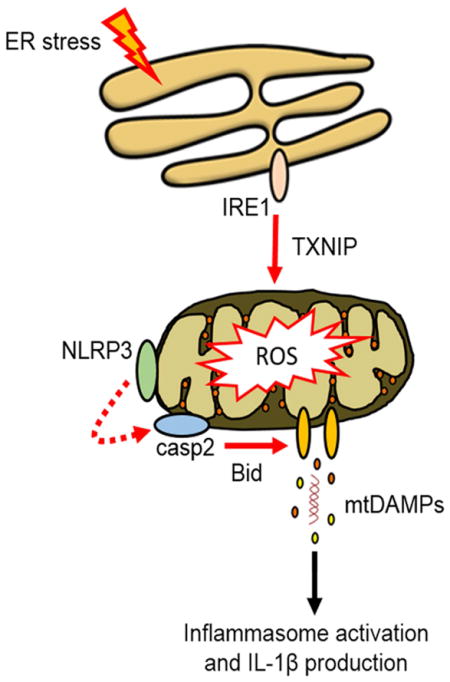
INTRODUCTION
Cellular stress provokes release of molecular danger signals that stimulate inflammatory signaling (Kono and Rock, 2008; Zhang et al., 2013), but the mechanisms linking stress with release of danger-associated molecular patterns (DAMPs) are not fully defined. Such mechanisms are relevant to human health as molecular stress and inflammation increase with age, and are associated with many acute and chronic diseases (Brown and Naidoo, 2012; Davis et al., 2011; Hao et al., 2013; Tan et al., 2013). Mitochondria can act as platforms to nucleate signaling by large molecular complexes, like the nucleotide-binding domain and leucine-rich repeat containing (NLRP3) inflammasome, and drive inflammation through release of mitochondrial DAMPs in response to diverse stressors (Subramanian et al., 2013). How such distinct stressors as infection, glucose deprivation, oxidative stress, or disruption of calcium homeostasis trigger these inflammatory events is unclear. The endoplasmic reticulum (ER) is a large endomembrane compartment that is highly sensitive to perturbation and is central to the function of many organelle networks, suggesting that ER may act as a relay station between stressors and mitochondria, linking stress and inflammatory signaling.
Three ER-resident unfolded protein sensors, IRE1α (IRE1), ATF6 and PERK control the ER stress response, an adaptive program that defines the fate of the stressed cell (Hetz, 2012). Toll-like receptors (TLR), which primarily recognize microbial ligands like lipopolysaccharide (LPS), selectively stimulate IRE1 (Martinon et al., 2010), and LPS-primed macrophages react to ER stressors by activating the NLRP3 inflammasome (Menu et al., 2012). Microbial infection is a useful model for investigating ER stress and inflammation because infection is often associated with ER stress, and animals deficient in components of the IRE1 signaling pathway are more susceptible to bacterial infection than controls (Bischof et al., 2008; Martinon et al., 2010; Pillich et al., 2012; Seimon et al., 2010). NLRP3-deficient animals exhibit increased susceptibility and decreased survival during infection by some microbial pathogens (von Moltke et al., 2013). Brucella abortus strain RB51 is an attenuated bacterial vaccine strain that infects macrophages, causes ER stress, and provokes a robust immune response without the complex effects of intracellular replication (Li and He, 2012). We therefore used RB51 as a probe to elucidate ER stress-dependent immune signaling. Virulent B. abortus mediates inflammasome activation through NLRP3 (Gomes et al., 2013), suggesting that B. abortus strains could be appropriate for studying the interplay between ER stress and NLRP3-dependent immune signaling (Davis et al., 2011). Notably, NLRP3 in resting cells is associated with ER, but upon stimulation moves to ER-mitochondrial junctions (Zhou et al., 2011). These data led us to hypothesize that upon infection, ER stress sensors could modulate NLRP3-dependent crosstalk between ER and mitochondria by an as yet unidentified mechanism, leading to inflammasome activation.
RESULTS
RB51-induced inflammasome activation requires IRE1 and TXNIP
TLR ligands (e.g., LPS) activate IRE1, but not ATF6 or PERK, activating transcription of proinflammatory cytokines (Martinon et al., 2010). To determine if IRE1 was stimulated during RB51 infection, we investigated splicing of Xbp1 transcript, a direct target of the endonuclease domain of activated IRE1 (Hetz, 2012; Sidrauski and Walter, 1997). Tunicamycin (TM), an inhibitor of protein glycosylation (Hetz, 2012; Olden et al., 1978; Zeng and Elbein, 1995), served as a positive control for ER stress assays. Robust splicing of Xbp1 was seen in TM-treated bone marrow derived macrophages (BMDM) and RB51-infected BMDM at 8h post-infection (pi) (Fig. 1A). ER stress signaling can lead to cell death in some contexts (Lerner et al., 2012; Oslowski et al., 2012), so we measured cell death by release of lactate dehydrogenase, and found that the majority of infected cells were viable by 8h pi (Fig. S1A). Thus, infection by RB51 stimulated IRE1 activation, as previously observed for other microbial ligands (Martinon et al., 2010).
Figure 1. IRE1 modulates infection-induced inflammasome activation via TXNIP.

(A) Xbp1 splicing, a marker of IRE1 activation, in RB51-infected BMDM. qRT-PCR samples were treated with PstI to distinguish between spliced (184 bp) and unspliced variants (119 bp following PstI digestion) (B) Caspase-1 cleavage in RB51-infected BMDM in absence or presence of 4μ8c. Immunoblots are representative of n≥3 independent experiments performed and imaged in parallel with identical parameters using a LiCor Odyssey imaging system. (C) IL-1β ELISA analysis of supernatants from RB51-infected BMDM treated with or without TUDCA (chemical chaperone, 300 μM) and 4μ8c (IRE1 inhibitor, 50 μM). Error bars represent mean ± SD of n≥3 independent experiments. *** represent p-value <0.0001, n.s. = not significant. (D) qRT-PCR analysis of IL-1β transcript in RB51-infected BMDM in the absence or presence of 4μ8c. (E) Serum IL-1β in mice treated with 5% DMSO (control, n = 15) or 4μ8c (n = 15) and infected with RB51 (i.p., CFU 1 × 108). Data in (E) were pooled from 2 independent experiments. (F) IL-1β ELISA analysis of supernatants from RB51-infected BMDM transfected with non-targeted and Xbp1 siRNA. (G) IL-1β ELISA analysis of supernatants from RB51-infected BMDM transfected with non-targeted and Txnip siRNA. (H) CM-H2DCFDA was used to measure ROS during RB51 infection in presence of 4μ8c or Txnip siRNA. Rotenone serves as a positive control for ROS induction. Inset in (F,G,H) demonstrates silencing efficiency by immunoblot. UNT, TM, and L+A represent untreated, tunicamycin (10 μg/mL, positive control for ER stress activation) and LPS+ATP (200 ng/mL and 1mM respectively; positive control for inflammasome activation). See also Supplemental Figure S1.
Recent studies revealed that ER stress can drive inflammasome activation (Lerner et al., 2012). RB51 stimulated robust IL-1β production at both low and high multiplicity of infection (MOI), and at high MOI was comparable to the positive control LPS+ATP treatment (Fig. S1B). We therefore tested whether ER stress was required for RB51-induced interleukin-1β (IL-1β) production. Inhibition of IRE1 with 4μ8C (Cross et al., 2012) in RB51-infected BMDM led to decreased Xbp1 splicing and caspase-1 cleavage (Fig. S1C and Fig. 1B). Treatment with tauroursodeoxycholic acid (TUDCA), a molecular chaperone that alleviates ER stress, or 4μ8C, resulted in less IL-1β production in RB51-infected BMDM, but not BMDM treated with LPS+ATP, our positive control for inflammasome activation (Fig. 1C). Neither TUDCA nor 4μ8C decreased bacterial uptake (Fig. S1D). IRE1 inhibition did not affect Il1b transcription, suggesting that IRE1 signaling was not required for priming (Fig. 1D). IRE1 inhibition also had no effect on LPS+ATP-induced caspase-1 cleavage and IL-1β production. These data suggest that unlike RB51, LPS+ATP does not rely on ER stress to induce IL-1β production. Like IRE1 inhibition, silencing of Ern1, the gene that encodes IRE1 (Fig. S1E and F), decreased Xbp1 splicing, caspase-1 cleavage and IL-1β production in RB51-infected BMDM. To determine whether IRE1 modulates inflammation in vivo, we treated C57BL/6 mice with either vehicle control or 4μ8C, and infected animals intraperitoneally with 108 CFU of RB51. Consistent with our in vitro findings, 4μ8C-treated mice showed significantly less serum IL-1β and increased bacterial burden (Fig. 1E and Fig. S1G). These data point to IRE1 as an important regulator of ER stress-induced inflammasome activation during infection.
Previous studies identified multiple targets for the IRE1 endonuclease, including Xbp1 and miR-17, a negative regulator of thioredoxin-interacting protein (TXNIP) translation (Lerner et al., 2012; Yoshida et al., 2001). The XBP1 transcription factor selectively enhances transcription of pro-inflammatory cytokine genes (Martinon et al., 2010). IRE1-dependent miR-17 degradation increases TXNIP protein, which shuttles to mitochondria and binds thioredoxin-2, raising concentrations of mitochondrial reactive oxygen species (ROS) (Lerner et al., 2012). To assess if XBP1 or TXNIP controls IRE1-driven IL-1β production, we transfected BMDM with Xbp1 or Txnip specific siRNA, infected with RB51 and measured supernatant IL-1β. Transient silencing of Xbp1 had no effect on RB51-induced IL-1β production (Fig. 1F), while silencing of Txnip led to reductions in IL-1β and ROS in infected BMDM (Fig. 1G and H). These data indicate that during infection-induced ER stress, IRE1 acts through TXNIP to induce IL-1β production.
ER stress-induced mitochondrial dysfunction aids in inflammasome activation
ER stress drives mitochondria to release mitochondrial-derived damage associated molecular patterns (mtDAMPs), which can activate the inflammasome (Iyer et al., 2013; Nakahira et al., 2011; Zhou et al., 2011). Previous studies have shown that TXNIP shuttling to mitochondria increases mtROS (Saxena et al., 2010). We observed increased ROS over time in RB51-infected BMDM, a phenotype dependent upon both IRE1 and TXNIP (Fig. 1H). To determine if mtROS was required for IRE1-induced IL-1β production, we infected BMDM derived from transgenic mice that express a mitochondrial-targeted catalase (mCAT) (Lee et al., 2010). Both ROS and IL-1β decreased in infected mCAT BMDM compared to infected WT BMDM (Fig. 2A and B). The decreased ROS in mCAT BMDM had no effect on LPS+ATP-induced IL-1β production, consistent with previous work showing that mitochondrial function is not essential for activation of the NLRP3 inflammasome (Munoz-Planillo et al., 2013). Since we observed increased ROS upon RB51 infection, possibly indicating mitochondrial dysfunction, we infected BMDM with RB51 and measured cytochrome c and mtDNA release. TM-treated and RB51-infected macrophages released mtDNA and cytochrome c into the cytosol, a process blocked by IRE1 inhibition or silencing (Fig. 2C and Fig. S2A and B). Since the presence of transfected mtDNA in the cytosol can stimulate IL-1β production (Nakahira et al., 2011; Shimada et al., 2012), we investigated if release of mitochondrial components during RB51 infection contributed to IL-1β production by treating BMDM with cyclosporin A (CsA), which prevents opening of the mitochondrial permeability transition pore (Handschumacher et al., 1984). We infected BMDM with RB51 with or without CsA, and measured IL-1β production by ELISA. CsA treatment significantly decreased IL-1β in RB51-infected macrophages, even though bacterial uptake was unaffected (Fig. 2D and S2C). In contrast, CsA treatment did not affect IL-1β production in LPS+ATP treated BMDM, indicating that our specific LPS+ATP treatment protocol induces IL-1β production independently of mitochondrial damage (Fig. 2D). However, a different LPS+ATP treatment protocol led to markedly higher amounts of mtDNA released into the cytosol (Fig. S2D), suggesting that involvement of the mitochondria in inflammasome activation by the classical activator, LPS+ATP, may depend on the extent or duration of cellular stress. Taken together, these data show that RB51-induced ER stress damages mitochondria, which leads to activation of the inflammasome through an IRE1-dependent mechanism.
Figure 2. ER stress-induced mitochondrial dysfunction drives IL-1β production.

(A) The ROS indicator dye, CM-H2DCFDA, was used to measure ROS in WT and mCAT BMDM infected with RB51. Rotenone serves as a positive control for ROS induction. (B) IL-1β concentrations in supernatants from RB51-infected WT and mCAT BMDM. (C) qPCR analysis of mtDNA release into cytosol during RB51 BMDM infection. (D) IL-1β concentrations in RB51-infected BMDM treated with or without cyclosporin A (CsA, 10 μM). Error bars in (A – D) represent mean ±SD of n≥3 independent experiments. *** represent p-value <0.0001, n.s. = not significant. UNT, TM, and L+A represent untreated and tunicamycin (10 μg/mL, positive control for ER stress activation), and 200 ng/mL LPS + 1mM ATP (positive control for inflammasome activation). See also Supplemental Figure S2.
NLRP3 mediates IRE1-induced mitochondrial stress independently of the ASC inflammasome adaptor
In macrophages infected with virulent B. abortus, both NLRP3 and the cytosolic DNA sensor, AIM2, were required for IL-1β production (Gomes et al., 2013). Moreover, the AIM2 inflammasome can regulate IL-1β production when stimulated by transfected mtDNA (Nakahira et al., 2011). We first assessed if AIM2 was required for RB51-induced IL-1β production by measuring IL-1β from RB51-infected WT and Aim2−/− BMDM (Fig. S3A). AIM2 did not contribute to IL-1β production induced by RB51-mediated ER stress. We then reasoned that NLRP3 would be a likely sensor to respond to the infection-induced ER stress signal. We first tested whether IRE1 signaling was required for Nlrp3 transcription, and found that 4μ8C treatment did not alter infection-induced priming of Nlrp3 transcription (Fig. S3B). Upon activation, NLRP3 can translocate from the ER to the mitochondria (Zhou et al., 2011), and is reported to trigger mitochondrial dysfunction and IL-1β production in the presence of oxidized mtDNA (Nakahira et al., 2011; Shimada et al., 2012). To determine if NLRP3 was involved in controlling the release of mitochondrial contents during RB51 infection, we determined if NLRP3 was recruited to mitochondria. In RB51-infected WT BMDM, NLRP3 was recruited to the mitochondrial fraction at 4h pi, which corresponds with the timing of cytochrome c and mtDNA release (Fig. 3A–B and Fig. S3C). NLRP3 recruitment to the mitochondria was abolished in mCAT transgenic and Txnip-silenced BMDM. Nlrp3−/− macrophages did not release mtDNA nor cytochrome c into the cytosol upon RB51 infection (Fig. 3C and Fig. S3D). In addition, NLRP3 deficiency abolished RB51-induced IL-1β production (Fig. 3D) and caspase-1 cleavage (Fig. S3E), but did not affect overall caspase-1 protein amounts. Since NLRP3 appeared to be a key component of RB51-induced inflammasome activation, we investigated whether ASC or caspase-1, components of the canonical inflammasome, were also crucial to inducing mitochondrial damage. Asc−/− and YVAD-CHO (caspase-1 inhibitor)-treated BMDM were not required for mtDNA and cytochrome c release into the cytosol upon infection (Fig. 3C and Fig. S3F). However, ASC and caspase-1 were still necessary for IL-1β production during RB51 infection (Fig. 3D). The inability to produce IL-1β in YVAD-CHO-treated macrophages was not due to decreased bacterial uptake (Fig. S3G). Together, these results show that during RB51-induced inflammasome activation, NLRP3 plays a critical role in mediating release of mitochondrial contents, independently of ASC and caspase-1.
Figure 3. NLRP3 is required for an ER stress-induced feed-forward loop of mitochondria damage.
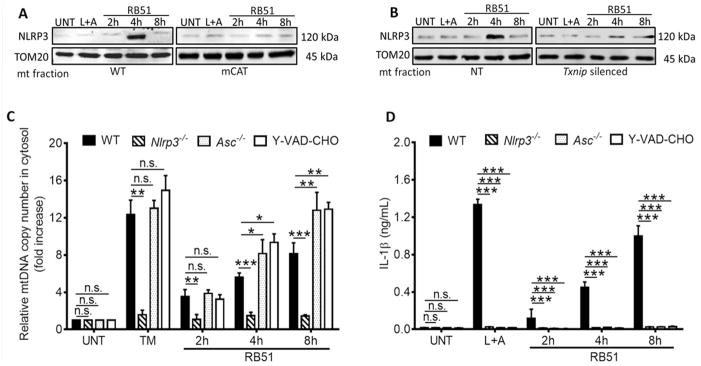
≥ Immunoblot of NLRP3 at the mitochondrial fraction of (A) WT and mCAT BMDM and (B) non-targeted (NT) and Txnip-silenced BMDM. (C) Quantitative PCR of mtDNA in cytosolic extracts from RB51-infected WT and Nlrp3−/−, Asc−/−, and z-YVAD-CHO (caspase-1 inhibitor, 2 μM) treated BMDM. (D) IL-1β concentrations in RB51-infected WT and Nlrp3−/−, Asc−/−, and z-YVAD-CHO (caspase-1 inhibitor, 2 μM) inhibited BMDM. Error bars represent mean ± SD of n≥3 independent experiments. *, **, and *** represent p-values of <0.05, <0.001, and <0.0001 respectively. n.s. = not significant. UNT, TM, and LPS+ATP represent untreated, tunicamycin (positive control for ER stress induction, 10 μg/mL), and LPS+ATP (positive control for inflammasome activation, 200 ng/mL and 1mM) respectively. Immunoblots in (A) and (B) are representative of n≥3 independent experiments that were performed and imaged in parallel with identical parameters using a LiCor Odyssey imaging system. See also Supplemental Figure S3.
NLRP3 drives mitochondrial dysfunction through caspase-2
Under ER stress conditions, NLRP3 might facilitate release of mitochondrial contents through the cysteine protease, caspase-2. During genotoxic stress, caspase-2 can cause mitochondrial dysfunction leading to cytochrome c release (Robertson et al., 2002). Moreover, caspase-2 is activated by ER stress or RB51 infection (Chen and He, 2009; Upton et al., 2008; Upton et al., 2012), and regulates caspase-1 activation (Bronner et al., 2013; Jesenberger et al., 2000). We reasoned that under conditions of ER stress, NLRP3 might be inducing activation of caspase-2 leading to release of mtDNA and cytochrome c into the cytosol. We probed lysates from control, 4μ8C-treated, and Ern1-silenced (IRE1 depleted) infected macrophages, for full-length or cleaved active caspase-2 (Fig. 4A and Fig. S4A). IRE1 was required for caspase-2 cleavage during RB51 infection, but not for caspase-2 cleavage triggered by the genotoxic agent, etoposide (ET). To determine if NLRP3 acted upstream or downstream of caspase-2, C57BL/6 and Nlrp3−/− BMDM were infected with RB51, and lysates probed for cleaved caspase-2 (Fig. 4B). Caspase-2 cleavage was nearly absent in infected Nlrp3−/− macrophages, whereas caspase-2 cleavage in Asc−/− or caspase-1 inhibitor-treated BMDM was comparable to WT (Fig. S4B). We next assessed caspase-2 mitochondrial recruitment and its role in mitochondrial damage. During RB51 infection, caspase-2 was recruited to mitochondria, in an IRE1- and NLRP3-dependent matter (Fig. 4C and D). Infected Casp2−/− BMDM released less mtDNA and cytochrome c into the cytosol (Fig. 4E and S4C). Moreover, caspase-2 deficiency abolished IL-1β production and caspase-1 activation without affecting bacterial uptake (Fig. 4F, Fig. S4D and E). Although Casp2−/− deficient BMDM were comparably infected to WT, it is possible that they were deficient in transcription of nlrp3 or Il1b. We performed quantitative RT-PCR analysis to measure nlrp3 and Il1b transcript in WT and Casp2−/− BMDM. Infected Casp2−/− BMDM produced similar amounts of nlrp3 and Il1b transcript as WT BMDM (Fig. 4G and Fig. S4F), suggesting that the defect in IL-1β production is not in priming, but in activating the inflammasome. Similar to our in vitro data, Casp2−/− mice infected with RB51 exhibited low concentrations of serum IL-1β and higher bacterial burden (Fig. 4H). These data suggest that during infection, NLRP3 can mediate ER stress-induced mitochondrial damage and inflammasome activation by a caspase-2-dependent mechanism.
Figure 4. NLRP3 and caspase-2 are required for ER stress-induced inflammasome activation.

Immunoblot analysis of caspase-2 in (A) with or without 4μ8c (IRE1 inhibitor, 50 μM) and (B) RB51-infected WT and Nlrp3−/− BMDM. Immunoblot analysis of caspase-2 in the mitochondrial fraction (C) with or without 4μ8c (IRE1 inhibitor, 50 μM) and (D) RB51-infected WT and Nlrp3−/− BMDM – ET╪ identifies duplicate lanes of the same sample. (E) qRT-PCR analysis of mtDNA in cytosolic fractions of infected WT and Casp2−/− BMDM. IL-1β concentrations in (F) WT and Casp2−/− RB51-infected (i.p., CFU 1 × 108) BMDM. (G) qRT-PCR analysis of Il1b transcript in WT and Casp2−/− RB51-infected BMDM. (H) Serum IL-1β serum by ELISA in WT (n = 15) and Casp2−/− mice (n = 15). Data in (H) were pooled from 2 independent experiments. Error bars represent mean ± SD of n≥3 independent experiments. *, **, and *** represent p-values of <0.05, <0.001, and <0.0001 respectively. n.s. = not significant. UNT, ET, TM, and L+A represent untreated, etoposide (positive control for caspase-2 activation and Bid truncation, 25 μM), tunicamycin (positive control for ER stress induction, 10 μg/mL), and LPS+ATP (positive control for inflammasome activation, 200 ng/mL and 1mM respectively). Immunoblots in (A–D) are representative of n≥3 independent experiments that were performed and imaged in parallel with identical parameters using a LiCor Odyssey imaging system. Full-length caspase-2 and TOM20 (mitochondrial specific outer membrane protein) serve as loading controls. See also Supplemental Figure S4.
NLRP3 and caspase-2 induce mitochondrial damage via the pore-activating factor Bid
We aimed to elucidate the mechanism by which NLRP3 and caspase-2 could regulate mitochondrial dysfunction. NLRP3 and caspase-2 could lead to truncation and activation of Bid, which damages mitochondria by licensing pore formation of Bax, a pro-apoptotic factor of the Bcl-2 family (Korsmeyer et al., 2000). To determine if Bid was involved in ER stress-induced mitochondrial damage, we infected 4μ8C-treated or Ern1-silenced (IRE1-depleted) macrophages with RB51, and probed for the presence of tBid, using etoposide as a positive control. Total Bid amounts remained constant under all conditions, but Bid truncation was diminished in 4μ8C-treated and Ern1-silenced macrophages (Fig. 5A and Fig. S5A), possibly implicating Bid in ER stress-induced mitochondrial dysfunction. ET induces mitochondrial dysfunction but does not induce ER stress (Hitomi et al., 2004; Wang et al., 1998). Therefore, it was not surprising to see that IRE1 inhibition had no effect on Bid truncation in ET-treated BMDM. To elucidate the role of NLRP3 and caspase-2 in this process, C57BL/6, Nlrp3−/−, and Casp2−/− BMDM were infected with RB51, and the status of Bid was assessed by immunoblot. Bid truncation markedly decreased in the absence of NLRP3 and caspase-2 (Fig. 5B and Fig. S5B). Infected Bid−/− BMDM released less mtDNA into the cytosol than WT controls, confirming that Bid is required for mitochondrial damage, even though Bid-deficiency did not affect bacterial uptake (Fig. 5C and S5C). Bid−/− BMDM infected with RB51 also exhibited diminished secretion of IL-1β and caspase-1 cleavage (Fig. 5D and E). Consistent with our in vitro results, RB51-infected Bid−/− mice showed a decrease in serum IL-1β, as well as an increase in bacterial burden in the spleen, compared to wildtype controls (Fig. 5F and Fig. S5D). Thus, during RB51 infection, NLRP3 and caspase-2 trigger mitochondrial damage through Bid, leading to inflammasome activation.
Figure 5. NLRP3 controls mitochondrial dysfunction by a Bid-dependent mechanism.

Bid truncation in RB51-infected (A) with or without 4μ8c (IRE1 inhibitor, 50 μM) and (B) WT and Nlrp3−/− BMDM – ET╪ identifies duplicate lanes of the same sample. (C) Quantitative PCR of mtDNA in cytosolic extracts from RB51-infected WT and Bid−/− BMDM. (D) ELISA of IL-1β in supernatants from RB51-infected WT and Bid−/− BMDM. (E) Immunoblot analysis of caspase-1 in WT and Bid−/− BMDM infected with RB51. (F) Serum IL-1β in WT (n = 29) and Bid−/− (n = 30) RB51-infected (i.p. CFU 1 × 108) mice. The data in (F) were pooled from 2 independent experiments. Error bars represent mean ± SD of n≥3 independent experiments. * and *** represent p-values of <0.05 and <0.0001 respectively. n.s. = not significant. UNT, ET, TM, and L+A represent untreated, etoposide (positive control for Bid truncation, 25 μM), tunicamycin (positive control for ER stress induction, 10 μg/mL), and LPS+ATP (positive control for inflammasome activation, 200 ng/mL and 1mM) respectively. Immunoblots in (A), (B), and (E) are representative of n≥3 independent experiments that were performed and imaged in parallel with identical parameters using a LiCor Odyssey imaging system. Full length (FL) caspase-1 and Bid serve as loading controls. See also Supplemental Figure S5.
NLRP3 and caspase-2 are required for inflammasome activation in response to chemical ER stress
Our data thus far identified the IRE1-NLRP3-caspase2-Bid axis as a mechanism for relaying infection-induced ER stress signals to the mitochondria, leading to inflammasome activation. We considered the possibility that this pathway might be important for infection-induced inflammasome activation, but not in the general ER stress response. We therefore tested whether IRE1, NLRP3, caspase-2 and Bid were required for inflammasome activation in response to thapsigargin (TG), tunicamycin (TM), and brefeldin A (BFA), three chemical inducers that cause ER stress by distinct mechanisms. Treatment of WT BMDM with TG, TM, or BFA resulted in robust caspase-1 cleavage but low IL-1β production, suggesting that minimal priming was occurring during TG, TM, and BFA treatment in contrast to LPS+ATP (Fig. 6A and B). Since both priming to stimulate Il1b transcription and inflammasome activation are required for IL-1β secretion (Schroder and Tschopp, 2010), we assessed proIL-1β production by immunoblot and found that TM and BFA induced weak proIL-1β production whereas TG did not trigger any detectable proIL-1β production (Fig. S6A). Thus, ER stress is a weak inducer of the first signal (priming) that regulates transcription of Il1b, but can act as a second signal for inflammasome activation. Consistent with our infection model, inhibition of IRE1 abrogated TM- or TG-induced caspase-1 cleavage (Fig. 6B). Although BFA did trigger IRE1 activation as measured by Xbp1 splicing (Fig. S6B), IRE1 was not required for BFA-induced inflammasome activation as previously reported by Tschopp and colleagues (Menu et al., 2012), perhaps due to more extensive perturbations in vesicular trafficking induced by BFA, compared to TM or TG, which act predominantly on the ER. In TM and TG-treated BMDM deficient in NLRP3, caspase-2 or Bid, caspase-1 cleavage was virtually absent (Fig. 6C–E). Although caspase-2 and Bid were critical for chemical ER stress-induced inflammasome activation, neither was required for inflammasome activation by LPS+ATP (Fig. 6D and E). In total, our data support the IRE1-NLRP3-caspase2-Bid axis as a key mechanism by which ER stress drives mitochondrial damage and inflammasome activation.
Figure 6. NLRP3 and caspase-2 are required for caspase-1 activation during general ER stress.

(A) ELISA analysis of IL-1β in supernatants from BMDM treated with 200 ng/mL LPS and 1mM ATP; positive control for inflammasome activation), or the ER stressors BFA (brefeldin A, 20 μM), TM (tunicamycin, 10 μg/mL), and TG (thapsigargin, 10 μM). Error bars represent mean ± SD of n≥3 independent experiments. ** and *** represent p-values of <0.001 and <0.0001 respectively. n.s. = not significant. Immunoblot analysis of caspase-1 in (B) BMDM in the absence or presence of 4μ8c (IRE1 inhibitor, 50 μM), (C) WT and Nlrp3−/− BMDM – L+A╪ identifies duplicate lanes of the same sample, (D) WT and Casp2−/− BMDM, and (E) WT and Bid− /− BMDM. Immunoblots in (B–E) are representative of n≥3 independent experiments that were performed and imaged in parallel with identical parameters using a LiCor Odyssey imaging system. Full-length (FL) caspase-1 serves as a loading control. See also Supplemental Figure S6.
DISCUSSION
ER stress is increasingly implicated in human disease, including infection, Alzheimer’s Disease and diabetes (Wang and Kaufman, 2012). More recent studies have demonstrated a connection between ER stress and the inflammasome, although the mechanisms that control signaling have not been fully elucidated (Lerner et al., 2012; Menu et al., 2012; Oslowski et al., 2012). Our results have revealed that activation of the IRE1 ER stress sensor leads to NLRP3-mediated crosstalk between ER and mitochondria, resulting in release of mitochondrial contents through activation of the caspase-2-Bid signaling axis. Notably, the requirement of NLRP3 in ER stress-induced mitochondrial damage was independent of ASC and caspase-1, suggesting this is not a function of the canonical NLRP3 inflammasome. Our data place NLRP3 upstream of caspase-2 in the ER-mitochondrial signaling pathway, and provide a mechanism by which NLRP3 can facilitate mitochondrial damage to activate the inflammasome. Whether NLRP3 or additional inflammasome regulators act as downstream sensors of mitochondrial damage in ER stress conditions remains to be determined.
The role of mitochondria in activating inflammasomes has been somewhat controversial. Previously, K+ efflux was proposed as the common mechanism by which diverse stresses that increase membrane permeability, e.g., bacterial toxins or LPS+ATP, activate the NLRP3 inflammasome without requiring mitochondrial damage (Allam et al., 2014; Munoz-Planillo et al., 2013). Other studies have reported that mitochondrial damage is critical for inflammasome activation by LPS+ATP or by cytosolic DNA (Nakahira et al., 2011; Shimada et al., 2012). Further increasing the complexity of cellular stress signaling, ROS generated by NADPH oxidase and mitochondrial ROS may stimulate the inflammasome by different pathways (Martinon, 2010; Wynosky-Dolfi et al., 2014). In the conditions we used, NLRP3-dependent inflammasome activation by LPS+ATP was essentially independent of IRE1, caspase-2, Bid, or mitochondrial content release. However, ER stress-induced inflammasome activation required mitochondrial damage. To reconcile our results with previously published reports, we speculate that NLRP3-dependent oligomerization, necessary for inflammasome activation, may exhibit distinct signaling requirements depending on the type or magnitude of stress. A stress that initiates strong K+ efflux, such as pore formation by bacterial toxins or pannexin-1 might allow cytosolic NLRP3 oligomerization, without mitochondrial damage and would therefore not be blocked by cyclosporin A (Kanneganti et al., 2007; Pelegrin and Surprenant, 2006). Stress that triggers weaker K+ efflux might be more dependent on mitochondrial danger signals in order for NLRP3 to use the mitochondria as a platform for oligomerization. Thus, both quality and quantity of a given stress inducer (i.e., infection, chemical stressors, LPS+ATP) may define the critical circuits that are integrated to generate inflammatory output. NLRP3 has previously been shown to bind the mitochondrial phospholipid, cardiolipin, which is exposed on the cytosolic face of mitochondria upon oxidation (Iyer et al., 2013; Korytowski et al., 2011). Cardiolipin-enriched domains have been proposed to act as signaling platforms on the mitochondria, somewhat analogous to the manner in which cholesterol-enriched microdomains act as signaling platforms in the plasma membrane (Schug and Gottlieb, 2009). Notably, protocol-specific conditions may be critical in defining the reliance of inflammasome activation on K+ efflux vs. mitochondrial damage (Horng, 2014; Nakahira et al., 2011; Shimada et al., 2012). In fact, we found that differing times and concentrations of LPS+ATP treatments resulted in substantially different amounts of mtDNA release, perhaps providing an explanation for why conflicting results have been reported for the reliance of LPS+ATP on mitochondrial damage. The molecular context of cellular stress will likely be critical in defining key principles that govern NLRP3 inflammasome activation.
Microbial infection imposes complex stress conditions upon infected cells that are not fully defined for most microbes, but may include K+ efflux, nutrient deprivation, cytoskeletal perturbations and ROS generation. Microbes or microbial ligands activate IRE1 through TLR signaling (Martinon et al., 2010), and in vivo data clearly show that XBP1, a component of the IRE1 signaling pathway, and NLRP3 can mediate resistance to microbial infection (Martinon et al., 2010; von Moltke et al., 2013). These observations are consistent with the idea that activation of ER stress machinery influences the outcome of infection by tuning inflammatory responses. Importantly, a recent study demonstrated that IRE1 is required for optimal secretion of pro-inflammatory cytokines, including IL-1β, in a mouse model of inflammatory arthritis (Qiu et al., 2013). Cellular stress in the form of ER perturbation and mitochondrial dysfunction may provide critical contextual danger signals that together with microbial ligands provoke robust immunity. Because ER stress and mitochondrial dysfunction are also associated with Type 2 diabetes, obesity, Crohn’s disease and cancer (Escames et al., 2012; Garg et al., 2012), it will be of interest to determine whether the NLRP3-caspase-2 regulatory axis is more broadly involved in sterile inflammatory diseases.
NLRP3 has emerged as a critical regulator of the inflammasome in response to ER stress and IRE1 activation. How IRE1 leads to NLRP3-dependent stimulation of caspase-2 is still unclear. NLRP3 binds to the signaling adaptor, TXNIP, whose translation is controlled by activated IRE1 (Lerner et al., 2012; Oslowski et al., 2012; Zhou et al., 2010), suggesting that interaction as a possible interface that may lead to recruitment and cleavage of caspase-2. Alternatively, NLRP3 may interact with an accessory protein similar to ASC that recruits caspase-2 through its CARD domain. IRE1 itself is reported to modulate caspase-2 total protein amounts by controlling degradation of regulatory microRNAs in BFA, TG, or TM-treated murine embryonic fibroblasts (Upton et al., 2012). However, a recent report indicated that overall caspase-2 amounts do not change in response to ER stress induced in human leukemia- and lymphoma-derived cell lines (Sandow et al., 2014), which is consistent with our data in macrophages showing that caspase-2 cleavage, rather than increased caspase-2 protein, is the key parameter induced by ER stressors. Our results emphasize the requirement for NLRP3, caspase-2 and mitochondrial damage in triggering caspase-1 activation specifically in conditions of ER stress, and lay the groundwork for investigating ER stress modulation as a therapeutic strategy for diseases of inflammation and immunity.
EXPERIMENTAL PROCEDURES
Mice
Humane animal care at the University of Michigan is provided by the Unit for Lab Animal Medicine, which is accredited by the American Association for Accreditation of Laboratory Animal Care and the Department of Health and Human Services. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Care and Use of Animals (UCUCA) of the University of Michigan.
Bid WT (n = 29), Bid−/−(Yin et al., 1999) (n = 30), DMSO treated (n = 15),4μ8c treated (n = 15), casp2 WT (n = 15) and Casp2−/− (n = 15) mice (8 -12 weeks) were injected intraperitoneally (i.p.) with Brucella abortus RB51 vaccine strain (1 × 108 CFU) in 200 μl of phosphate-buffered saline (PBS). Mice were matched by sex and age. Mice were treated with DMSO (5% in PBS) or 4μ8c (25mg/kg) daily in 200 μl of PBS. Blood was collected by saphenous vein on day -1, 1, and 3 days pi. Serum was extracted from blood by centrifugation for 3 min at 10,000 rpm and used for assessing IL-1β production by ELISA. At day 3 pi, spleens were removed from euthanized mice, homogenized in 1 ml 0.2% NP-40, and serial dilutions plated onto Brucella agar plates to enumerate CFU.
Cell culture and infection
BMDM were isolated from WT, Casp2−/−, Asc−/−, Nlrp3−/−, Bid−/− and Aim2−/− mice. Casp2−/− with corresponding WT were purchased from Jackson Laboratories (stock #007899) (Bergeron et al., 1998). Nlrp3−/− (Kanneganti et al., 2006) and Asc−/− (Ozoren et al., 2006) were maintained by the Nuñez laboratory. Bid−/− (Yin et al., 1999) and corresponding WT mice were maintained by the Yin laboratory. Aim2−/− (Rathinam et al., 2010) and corresponding WT mice were maintained by the Fitzgerald laboratory (University of Massachusetts, Worcester).
Isolated BMDM were differentiated in DMEM (GIBCO) supplemented with 20% heat-inactivated FBS (Invitrogen), 1% L-glutamine (2 mM), 1% sodium pyruvate (1 mM), 0.1% β-mercaptoethanol (55 μM), and 30% L-929 conditioned medium. BMDM were cultured in non-TC treated plates at 37°C in 5% CO 2, fed fresh media on day 3, and harvested on day 6. Four million RAW264.7 macrophages or BMDM were seeded in 6 well plates 18 hr prior to infection. The LPS+ATP samples were pretreated with LPS (200 ng/mL) overnight, followed by treatment with 1 mM ATP as described below. The following day, where indicated, cells were pretreated with 10 μM cyclosporin A, 300 μM TUDCA, 50 μM 4μ8c or 2 μM Y-VAD-CHO for 1 hr prior to infection. Untreated and pretreated cells were infected with RB51 (MOI 200) for 30 min, after which inoculum was removed and cells were washed with PBS. Medium containing 50 μg/ml gentamicin was added to kill extracellular bacteria. To synchronize infection, cells were spun at 1200 rpm for 3 min after adding inoculum. Cells were treated with 25 μM etoposide, 10 μM thapsigargin, 10 μg/mL tunicamycin, 20 μM brefeldin A or 1 mM ATP for 4 hr. At the indicated times, cells were lysed in buffer containing 1% NP-40 on ice for 15 min and spun at 16,000 x g for 15 min to pellet the insoluble fraction. Soluble fractions were used for immunoblot assays. The insoluble fraction was resuspended in mitochondrial suspension buffer (10mM TrisHCl pH 6.7, 0.15 mM MgCl2, 0.25 sucrose, 1 mM PMSF, 1 mM DTT) and centrifuged at 11,000 x g for 15 minutes at 4°C to pellet the isolated mitochondria. Purity of isolated mitochondria was assessed by immunoblotting for compartment-specific markers: calreticulin (ER), TOM20 (mitochondria), Lamin B1 (nucleus) and Actin (cytosol).
Bacterial strains and reagents
Brucella abortus strain RB51 was obtained from Dr. G. Schurig (Virginia Polytechnic Institute and State University). Reagents were obtained from the following vendors: Sigma-Aldrich (cyclosporin A, tunicamycin, etoposide, brefeldin A, ATP), Fisher (thapsigargin), Calbiochem (TUDCA), Axon (4μ8c), and SCBT (Y-VAD-CHO). Antibodies were obtained from the following vendors: SCBT (anti-p-PERK sc-32577; anti-ATF6 sc-22799; anti-caspase-1 sc-514; anti-TOM20 sc-11415; anti-Lamin-b1 sc-20682), Cell Signaling (anti-PERK 3192S; anti-IRE1 3294S; anti-cytochrome c 4272S; anti-Bid 2003S; anti-Calreticulin 2891), Fisher (anti-NLRP3 MAB7578), BioVision (anti-caspase-2 3027–100) and Thermo Scientific (anti-Actin MS1295P1).
Lentivirus production and silencing of Ern1
HEK293T cells were grown in DMEM with 10% fetal bovine serum (Invitrogen). Lentivirus particles were produced by transfecting the cells with the TRC shRNA encoding plasmid (pLKO.1) along with the packaging plasmids (pVSV-G, pGAG-PAL) obtained from the University of Michigan Vector Core. The medium was changed after 24 hr, and virus particles collected at 48 hr. Virus-containing medium was concentrated 10-fold by centrifugation (24000 rpm) for 2 hr at 4°C. Concentrated virus was used to transduce RA W264.7 cells seeded in 60-mm dishes. The medium was changed 24 hr post transduction, and cells were left to grow for an additional 24 hr. Transduced cells were selected with puromycin (4 μg/mL) using vectors containing sequence from a mouse Ern1-specific shRNA plasmid (Open Biosystems: antisense sequence 5′-TTTCTCTATCAATTCACGAGC-3′) or a non-targeted control shRNA plasmid (Sigma-Aldrich).
Transient silencing of Txnip and Xbp1
Immortalized BMDM were transfected with specific Dharmacon siGENOME Txnip siRNA (cat# M-040441-01-0005), Xbp1 (cat.# M-040825-00-0005) or non-targeted siRNA (cat.# D-001206-13-20) using DharmaFECT4 transfection reagent according to the manufacturer’s protocol. Silencing efficiency was assessed via immunoblot using anti-TXNIP (cat.# NBP1-54578, Novus Biologicals) and anti-XBP1 (cat.# ab37152, Abcam) antibodies.
ROS measurements
BMDM were plated in a 96 well plate with black slides and clear bottom. At designated time points, BMDM were washed with PBS and then incubated with CM-H2DCFDA (Invitrogen) at a final concentration of 2.5 μM in Ringer buffer (155 mM NaCl, 5 mM KCl, 1 mM MgCl2 6H2O, 2 mM NaH2PO4 H2O, 10 mM HEPES, 10 mM glucose). Cell were incubated for 30 min at 37°C, washed 3 times with cold PBS, and incubated for an additional 15 min at 37°C in warm medium for recovery. After recovery, cells were washed one more time with PBS. Florescence was measured at excitation/emission 485nm/525nm.
Immunoblot Assay
Cytosolic extracts were separated by SDS-PAGE, transferred to nitrocellulose membranes (Millipore), blocked with 5% nonfat dry milk in TBS-0.1% Tween20 (TBS-T), and incubated overnight at 4°C with primary antibodies specified above. Membranes were washed with TBS-T and incubated with secondary IRDye 680LT Goat anti-rabbit or IRDye 680LT Goat anti-mouse (1:20,000) at room temperature for 1 hr. Bands were visualized using the Li-Cor Odyssey Infrared Imaging System. Immunoblots shown in the figures are representative of n≥3 independent experiments. All immunoblots shown within an individual panel were analyzed in parallel with identical parameters using the Li-Cor System.
Cytokine Analysis
Culture supernatants were collected at indicated time points from macrophages infected as described. IL-1β concentrations were determined by sandwich ELISA per manufacturer’s instructions (BioLegend). A minimum of 3 technical replicates per experiment and 3 experimental replicates were analyzed for each condition.
Xbp1 splicing assay
Total RNA (2 μg) extracted from samples was prepared using the RNeasy Mini Kit (Qiagen) and used for cDNA synthesis. Primers encompassing the spliced sequences in Xbp1 mRNA (F: 5′-GAACCAGGAGTTAAGAACACG-3′ and R: 5′-AGGCAACAGTGTCAGAGTCC-3′) were used for PCR amplification with GoTaq polymerase (Invitrogen), with amplification using 30 cycles at 94°C 1 min, 60°C 1 min, and 72°C 1 min. PCR produc ts were incubated with PstI (Invitrogen) at 37°C overnight, and separated by electrophoresis (2 .5% agarose gel).
Mitochondrial DNA (mtDNA) release assay
DNA was isolated from 200 μL of the cytosolic fraction using a DNeasy Blood & Tissue Kit (Qiagen). Quantitative PCR was employed to measure mtDNA using Brilliant II SYBR Green with Low ROX (Agilent Technologies) on a Stratagene MX300 QPCR System. The copy number of mtDNA encoding cytochrome c oxidase I was normalized to nuclear DNA encoding 18S ribosomal RNA. The following primers were used: cytochrome c oxidase I (F: 5′-GCCCCAGATATAGCATTCCC-3′ and R: 5′-GTTCATCCTGTTCCTGCTCC-3′) and 18S rRNA (F: 5′-TAGAGGGACAAGTGGCGTTC-3′ and R: 5′-CGCTGAGCCAGTCAGTGT-3′).
Lactate dehydrogenase (LDH) Release Assay
Macrophages were seeded in 96-well plates and infected with RB51 as above. Supernatants were analyzed for LDH enzyme using the CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega) per manufacturer’s instructions. Percentage LDH release was calculated as 100 X [(Experimental LDH Release - Culture Medium Background)/(Maximum LDH Release – Culture Medium Background)].
Statistical Analysis
All p values were generated between identified samples using unpaired two-tailed Student’s t-tests and represent analysis of ≥3 replicates per condition. Asterisks denote the following p values: *p<0.05, **p<0.001 and ***p<0.0001.
Supplementary Material
HIGHLIGHTS.
Infection-associated ER stress initiates mitochondrial damage through IRE1α
Mitochondrial damage is required for IRE1α-dependent IL-1β production
IRE1α promotes mitochondrial damage via NLRP3, caspase-2, and Bid
The IRE1α– NLRP3-casp2 axis drives general ER stress-induced inflammasome activation
Acknowledgments
We acknowledge members of the O’Riordan lab for helpful discussions. We thank Drs. D. Monack and V. Carruthers, for critical review of the manuscript. We are grateful to Dr. M. Swanson, Dr. V. Rathinam, Dr. Yuan He, and Dr. D. Ron for providing critical reagents and experimental guidance. We thank the staff of the UM Unit for Laboratory Animal Medicine. The University of Michigan Vector Core provided the TRC shRNA plasmids for Ern1 silencing. We acknowledge financial support from the University of Michigan Rackham Graduate School (Y.H. and D.N.B.), the UM Genetics Training Program (D.N.B., GM007544), the UM Lung Immunopathology Training Program (B.H.A., HL007517) and the American Heart Association (B.H.A.). This research was supported by funding from the NIH to M.X.D.O. (AI101777), K.A.F. (AI083713), G.N. (AI063331, AR059688), X.M.Y. (AA021751), and the University of Michigan Medical School Unit for Laboratory Animal Medicine and Endowment for Basic Science (Y.H.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author Contributions:
D.N.B., Y.H., and M.X.D.O. designed the experiments. D.N.B., B.H.A., and M.X.D.O. performed the experiments. B.H.A., K.A.F., G.N., Y.H., X.C., and X.M.Y. contributed critical reagents, materials, and discussion. D.N.B. and M.X.D.O. analyzed the data and wrote the paper.
The authors have declared that no competing interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allam R, Lawlor KE, Yu EC, Mildenhall AL, Moujalled DM, Lewis RS, Ke F, Mason KD, White MJ, Stacey KJ, et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014 doi: 10.15252/embr.201438463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes & development. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof LJ, Kao CY, Los FC, Gonzalez MR, Shen Z, Briggs SP, van der Goot FG, Aroian RV. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008;4:e1000176. doi: 10.1371/journal.ppat.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner DN, O’Riordan MX, He Y. Caspase-2 mediates a Brucella abortus RB51-induced hybrid cell death having features of apoptosis and pyroptosis. Frontiers in cellular and infection microbiology. 2013;3:83. doi: 10.3389/fcimb.2013.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MK, Naidoo N. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. 2012;3:263. doi: 10.3389/fphys.2012.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, He Y. Caspase-2 mediated apoptotic and necrotic murine macrophage cell death induced by rough Brucella abortus. PLoS One. 2009;4:e6830. doi: 10.1371/journal.pone.0006830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869–878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escames G, Lopez LC, Garcia JA, Garcia-Corzo L, Ortiz F, Acuna-Castroviejo D. Mitochondrial DNA and inflammatory diseases. Hum Genet. 2012;131:161–173. doi: 10.1007/s00439-011-1057-y. [DOI] [PubMed] [Google Scholar]
- Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med. 2012;18:589–598. doi: 10.1016/j.molmed.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Gomes MT, Campos PC, Oliveira FS, Corsetti PP, Bortoluci KR, Cunha LD, Zamboni DS, Oliveira SC. Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J Immunol. 2013;190:3629–3638. doi: 10.4049/jimmunol.1202817. [DOI] [PubMed] [Google Scholar]
- Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- Hao LY, Liu X, Franchi L. Inflammasomes in inflammatory bowel disease pathogenesis. Curr Opin Gastroenterol. 2013;29:363–369. doi: 10.1097/MOG.0b013e32836157a4. [DOI] [PubMed] [Google Scholar]
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. The Journal of cell biology. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014;35:253–261. doi: 10.1016/j.it.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, et al. Mitochondrial cardiolipin is required for nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesenberger V, Procyk KJ, Yuan J, Reipert S, Baccarini M. Salmonella-induced caspase-2 activation in macrophages: a novel mechanism in pathogen-mediated apoptosis. The Journal of experimental medicine. 2000;192:1035–1046. doi: 10.1084/jem.192.7.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, Nunez G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26:433–443. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem. 2011;286:26334–26343. doi: 10.1074/jbc.M110.188516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, He Y. Caspase-2-dependent dendritic cell death, maturation, and priming of T cells in response to Brucella abortus infection. PLoS One. 2012;7:e43512. doi: 10.1371/journal.pone.0043512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–619. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, Tschopp J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012;3:e261. doi: 10.1038/cddis.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olden K, Pratt RM, Yamada KM. Role of carbohydrates in protein secretion and turnover: effects of tunicamycin on the major cell surface glycoprotein of chick embryo fibroblasts. Cell. 1978;13:461–473. doi: 10.1016/0092-8674(78)90320-3. [DOI] [PubMed] [Google Scholar]
- Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozoren N, Masumoto J, Franchi L, Kanneganti TD, Body-Malapel M, Erturk I, Jagirdar R, Zhu L, Inohara N, Bertin J, et al. Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J Immunol. 2006;176:4337–4342. doi: 10.4049/jimmunol.176.7.4337. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillich H, Loose M, Zimmer KP, Chakraborty T. Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol. 2012;14:949–964. doi: 10.1111/j.1462-5822.2012.01769.x. [DOI] [PubMed] [Google Scholar]
- Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, Zhang Z, Lin Z, Gui M, Li X, et al. Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. The EMBO journal. 2013;32:2477–2490. doi: 10.1038/emboj.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;277:29803–29809. doi: 10.1074/jbc.M204185200. [DOI] [PubMed] [Google Scholar]
- Sandow JJ, Dorstyn L, O’Reilly LA, Tailler M, Kumar S, Strasser A, Ekert PG. ER stress does not cause upregulation and activation of caspase-2 to initiate apoptosis. Cell Death Differ. 2014;21:475–480. doi: 10.1038/cdd.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena G, Chen J, Shalev A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem. 2010;285:3997–4005. doi: 10.1074/jbc.M109.034421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788:2022–2031. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Seimon TA, Kim MJ, Blumenthal A, Koo J, Ehrt S, Wainwright H, Bekker LG, Kaplan G, Nathan C, Tabas I, et al. Induction of ER stress in macrophages of tuberculosis granulomas. PloS one. 2010;5:e12772. doi: 10.1371/journal.pone.0012772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MS, Yu JT, Jiang T, Zhu XC, Tan L. The NLRP3 Inflammasome in Alzheimer’s Disease. Mol Neurobiol. 2013 doi: 10.1007/s12035-013-8475-x. [DOI] [PubMed] [Google Scholar]
- Upton JP, Austgen K, Nishino M, Coakley KM, Hagen A, Han D, Papa FR, Oakes SA. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cell Biol. 2008;28:3943–3951. doi: 10.1128/MCB.00013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H, Ron D. Identification of novel stress-induced genes downstream of chop. The EMBO journal. 1998;17:3619–3630. doi: 10.1093/emboj/17.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynosky-Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D, Zwack EE, Riblett AM, Hu B, Strowig T, et al. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J Exp Med. 2014;211:653–668. doi: 10.1084/jem.20130627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Elbein AD. UDP-N-acetylglucosamine:dolichyl-phosphate N-acetylglucosamine-1-phosphate transferase is amplified in tunicamycin-resistant soybean cells. European journal of biochemistry / FEBS. 1995;233:458–466. doi: 10.1111/j.1432-1033.1995.458_2.x. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Kang R, Zeh HJ, 3rd, Lotze MT, Tang D. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9:451–458. doi: 10.4161/auto.23691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.