

Abstract
Shotgun proteomic analysis usually employs multidimensional separations with the first dimension most commonly being strong cation exchange (SCX) liquid chromatography (LC). SCX-LC is necessarily a serial process for preparation of multiple samples. Here we apply a newly available tool, off-gel electrophoresis (OGE), for first dimension separation of peptide mixtures from digests of cerebrospinal fluid (CSF), a complex and low total protein-containing sample. OGE first dimension fractionation enabled identification of a total of 156 unique proteins compared to 115 identified in previous work using first dimension SCX fractionation. OGE can be used to process multiple samples unattended with easy retrieval of the separated fractions. Thus shotgun analysis using OGE as the first dimension separation offers a significant advantage both in terms of sample throughput as well as increased numbers of identified proteins.
Keywords: OGE, cerebrospinal fluid, shotgun proteomics
Introduction
Shotgun analysis is an increasingly applied tool in proteomics.1,2 This method entails digestion of a mixture of intact proteins prior to any form of fractionation, followed by multidimensional separation of the resulting peptide mixture and mass spectrometry (MS) analysis. A variety of separation techniques have been used for shotgun analysis 1 including isoelectric focusing (IEF)3,4, strong cation exchange (SCX) liquid chromatography (LC), capillary electrophoresis (CE) and reversed phase (RP) LC. In the original shotgun proteomic method (dubbed “MudPIT” for “multidimensional protein identification technology”), the multidimensional separation was tandem SCX and RPLC with online electrospray ionization (ESI) MS analysis.5
The online 2D LC-MS/MS approach, although providing an all-in-one setup1,6, results in inefficient use of instrument time. The off-line approach, where peptides are separated in the first dimension before being subjected to second dimension RP-MS analysis, makes more efficient use of mass spectrometer time. On the other hand, it is a tedious process requiring fraction collection of the eluted peptides, and the increased sample handling can increase sample losses. An offline first dimensional separation comparable to SCX, but without some of the disadvantages, would provide a significant improvement in efficiency of both time and sample recovery. We report here the use of Off-Gel electrophoresis (OGE) as the first dimension separation in shotgun analysis of cerebrospinal fluid (CSF).
Another liquid phase IEF separation device used in 2D separations of proteins is the Rotofor, a commercial product available from BioRad. This instrument fractionates proteins in chambers separated by plastic mesh using carrier ampholytes.7,8,9 The ZOOM IEF fractionater, sold by Invitrogen, is a miniaturized system using carrier ampholytes, but with the chambers separated by isoelectric membranes.10 Further miniaturization of this approach in a chip device has also been reported.11 In all of these devices, the separated fractions contain carrier ampholytes, which need to be removed for subsequent steps. A recently available fractionation tool known as the digital Proteome Chip™ (Protein Forest) uses an electric field to fractionate proteins and peptides into a series of immobilized pH gel plugs.12 The OGE system has the advantage of using immobilized pH gradient strips to yield separated fractions without carrier ampholytes. Another advantage of this technique is the commercial availability of IPG strips of various pI ranges.
OGE is a method of isoelectric focusing where sample fractions can be recovered in solution with the option of not using carrier ampholytes13, which may interfere with downstream mass spectrometry analysis. The original design consisted of a protein solution being introduced into a flow chamber containing a specified pH gradient gel strip. Further development of the OGE system included a sample-focusing buffer combination used in a multiwell device that provided a narrow pI range in each well, thereby giving better separation of proteins.8,14 In addition to the separation, OGE has the added advantage of concentrating the focused components. The OGE technique has been used to separate mixtures at the intact protein level, yielding fractions that, in turn, could undergo digestion with the resulting fragments separated at the peptide level using an OGE peptide format.15,16,17 A variety of samples have been analyzed with this OGE approach, including E. coli lysate17, murine macrophage-infected cells18, human plasma 15 and recently, extraocular muscle.19
OGE has been commercialized by the Agilent corporation as the model 3100 OFFGEL Fractionator, which can be used for separation of either protein or peptide mixtures.15 Previous analytical studies show that the OFFGEL device provides a 6-7 mm well width structure that allows the recovery of 90% of peptides in the maximum of two well compartments.17,20 The work reported here applies peptide OGE as the first dimension separation in shotgun analysis of CSF.
Cerebrospinal fluid is a complex biological sample that has been studied in efforts to find protein biomarkers of CNS diseases.21,22 CSF is a clear fluid produced in the choroid plexus of the ventricles that surround the brain and spinal cord containing peptides, proteins, sugars, and salts.23,24,25 Normal human CSF protein concentration is in the range of 0.18-0.8 mg/mL, which is more than two orders of magnitude lower than serum or plasma protein concentration levels.24,25 The low overall concentration and large dynamic range of protein concentration in CSF, as much as twelve orders of magnitude, complicates proteomic analysis.26 Because of this complexity and low total protein concentration, there is a need for not only efficient first dimension separation, but also for concentration of the peptide fragments. In this work, OGE was incorporated into a protocol previously established in our laboratory for shotgun analysis of CSF 27,28, with replacement of the first dimension SCX fractionation with OGE fractionation prior to RP-LC-ESI-MS analysis. This approach yielded an improved method for shotgun proteomics analysis of CSF as reflected by an increased number of identified proteins compared to the conventional method using SCX.
2. Experimental Section
2.1 Materials
Urea, dithiothreitol, iodoacetamide, and proteomics grade trypsin were purchased from Sigma -Aldrich corporation (St. Louis, MO). C-18 Sep-Pak (2.0 mL) vacuum manifold solid-phase extraction (SPE) cartridges were purchased from Waters corporation (Millford, MA). The 3100 OFFGEL Low Resolution Kit, pH 3-10, containing urea, thiourea, dithiothreitol, a glycerol solution, ampholytes, mineral oil, electrode pads, frames, cover seals, and IPG strips, was purchased from Agilent Technologies (Wilmington, DE). Amicon Ultra 5 kDa molecular weight cutoff filters were purchased from the Millipore Corporation (Billerca, MA).
2.2 Human CSF
A pool of human CSF was obtained from excess clinical specimens under IRB approval as previously described.27,28 The total protein concentration of this CSF pool was determined to be 454 μg/mL. The CSF pool was stored at -80° C.
2.3 Buffer Exchange and Removal of Biological Salts
CSF was centrifuged, and the supernatant (2.5 mL, 1.135 mg total) was loaded onto a 15.0 mL capacity 5 kDa Amicon Ultra molecular weight cutoff filter. The volume was then increased to 15.0 mL total by adding 0.2 M NH4HCO3 and cartridges were centrifuged at 3000 × g until the volume was reduced to 200 μL. This buffer exchange process was repeated two more times resulting in the substantial reduction of biological salt concentration. The retentates containing CSF proteins were then removed and aliquotted into 5 equal fractions (40 μL amounts containing 227 μg each), which were dried by vacuum centrifugation.
2.4 Reduction, Alkylation, and Digestion of CSF Proteins
This protocol was as previously described with slight modification.27,28 CSF proteins in each sample were denatured in 9.0 M urea. Disulfide bonds were reduced by 10 mM dithiothreitol for 1 hr at 37° C in the dark. Alkylation was performed via addition of 50 mM iodoacetamide followed by incubation for 30 min at 37° C in the dark. The sample was diluted to 1.85 M urea concentration by addition of 0.2 M NH4HCO3, yielding a pH of 8.5 for protein digestion. Proteomics grade trypsin was added (20 μg, Sigma Aldrich), and the proteins were digested overnight at 37° C in the dark. Tryptic peptides were then isolated via solid-phase extaction with a C-18 Sep-Pak Vac cartridge (Waters Corporation) according to manufacturer’s instructions. Peptides were eluted with 70% acetonitrile/0.1% formic acid and were dried using vacuum centrifugation.
2.5 Off-Gel Electrophoresis
The 3100 OFFGEL Fractionator and the OFFGEL Kit pH 3-10 (Agilent Technologies) were used according to the manufacturer’s protocol. This experiment utilized the 12-well tray. The corresponding 13-cm long IPG gel strip with a linear pH gradient ranging from 3 to10 was rehydrated with the addition of 20 μL of rehydration solution per well. Next, electrode pads were wetted and placed on the anodic and cathodic ends. The strip was allowed to rehydrate for 15 min. The CSF tryptic digest was diluted to 1.8 mL with the addition of OFFGEL peptide fractionation buffer and 150 μL was placed into each well. The cover seal was placed over the well apparatus followed by addition of cover fluid on anodic and cathodic ends of the tray. Electrodes were placed on each side and set to run until 20 kVh was reached (approximately 13 h). Afterwards, the solution was removed from each well, yielding recovery amounts ranging from 100-200 μL per well to give a total of 12 peptide containing fractions. The fractions were then dried via vacuum centrifugation.
The OGE analyses used the same pool of CSF as used for previous experiments employing strong cation exchange (SCX) chromatography as a first-dimension separation.27,28 Offline SCX chromatography was performed using a PolySULFO-ETHYL A column, 200 mm × 2.1 mm (PolyLC, Inc.). The column was equilibrated with buffer A (10 mM KH2PO4 and 25% acetonitrile, pH < 3.0) for 30 min, after which the CSF peptides solubilized in 2.0 mL of buffer A were loaded onto the column. The peptides were eluted with a linear gradient of 0-50% buffer B (10 mM KH2PO4, 1.0 M KCl, and 25% acetonitrile, pH < 3.0) over 60 min. A UV absorption cell (λ = 214 nm) was used to monitor the elution of CSF peptides. A total of 8 peptide-containing fractions were collected in 5 min intervals. Collected fractions were then dried via vacuum centrifugation.
2.6 Reversed-Phase LC/MS/MS
Fractions were analyzed using slight modification of a previously reported procedure regarding SCX fractions.27 OGE fractions were solubilized with 200 μL of 2% acetonitrile/0.1% formic acid (solvent A) and 20 μL was loaded onto a 0.3 mm × 5.0 mm C18 trap column (LC Packings, Dionex), which was then eluted onto an analytical RPC18 column, 15 cm × 75 μm (Microtech Scientfic) and separated with a 100 min linear gradient of 0-50% solvent B (95% acetonitrile/0.1% formic acid) followed by a 20 min linear gradient (50-70% solvent B) at a flow rate of 180 nL/min using a Dionex ULTIMATE system. Eluted peptides were analyzed online with a LTQ mass spectrometer (Thermo Electron) using the Thermo nanospray ion source. The mass spectrometer was set to collect MS/MS spectra on the 5 most intense ions observed in the MS spectrum collected over m/z 400-2000. Other parameters included a nanospray voltage of 2.0 kV, a normalized collision energy of 35%, a default charge state of +5, and an isolation mass window of 2.5 amu. Dynamic exclusion was enabled for all experiments with a duration of 3.0 min, a repeat count of 2, a repeat duration of 0.5 min, and a rejection mass window of 2.0 amu.
The fractions from the SCX separation were analyzed by RP-LC-MS/MS using the same conditions as for the OGE fractions, except that the loading of each SCX fraction on the C18 trap column was followed by a wash for 30 min prior to sample flow onto the RP-C18 analytical column. The wash was unnecessary for the OGE fractions.
2.7 Data Analysis
MS/MS spectra were searched against an indexed Homo sapiens database, which was extracted from the NCBI nonredundant H. sapiens database, using the Turbo SEQUEST algorithm, a component of the Bioworks 3.2 software suite (Thermo Electron). Peptides with up to two missed cleavages were allowed. Dynamic chemical modifications of +16 and +57 mass units corresponding to M-oxidized and C-carboxyamidomethyl modifications, respectively, were included as search parameters. A precursor ion accuracy of 2.0 amu was used. Resulting protein identifications were filtered using two protein and two peptide filters, protein probability P< 0.001, minimum of two unique peptides for a protein identification, peptide probability P< 0.001, and Xcorr (cross correlation) versus charge state of at least 1.5, 2.0, and 2.5 for +1, +2, and +3 ions, respectively. Using Bioworks 3.2, multi-consensus reports were generated for each set of twelve reversed-phase experiments (i.e. LC/MS runs for each of the twelve first-dimension separation fractions) for each sample. All protein identifications resulting from only two unique peptides were further examined. Each peptide sequence was searched using the BLAST algorithm, and when both of the two peptide sequences were found in more than two proteins in the NCBI nonredundant H. sapiens database (e.g. multiple protein variants), the protein identification was deemed inconclusive and eliminated from the identification list. Thus, the identified proteins listed were only those for which at least two peptides were identified that matched to only one protein.
3. Results and Discussion
3.1 LC/MS/MS Analysis of OGE Separated Peptides
The dried CSF protein samples were reduced, alkylated, and digested prior to fractionation using the peptide OFFGEL electrophoresis format. After the separation time was complete, the wells were inspected to make sure that none had gone to dryness (due to osmotic pumping). All wells contained fluid, and a range of ∼100-200 μl of separated peptide solution was recovered from the wells. Each well fraction was analyzed and the proteins identified are shown in Table 1. They are given in the order of relative abundance, which was calculated using the total spectrum count (TSC) method described previously.29 Briefly, this method entails normalizing the TSC (number of peptide MS2 spectra matched to a particular protein) of each protein by dividing by its molecular weight. The LC-MS/MS analyses were repeated two more times. In the repeated analyses additional proteins were identified with the total identification tending toward a plateau after three analyses.29 The same pool of CSF was used as in prior SCX-RP analyses.26 The first run using OGE yielded 97 protein identifications, the second yielded 135, and the third yielded 99, for a total of 156 proteins identified (Table 1). As expected, there were overlapping and unique proteins seen during each run (Fig. 2). Shotgun analysis of the same CSF pool using SCX as the first dimension yielded 86, 94, and 83 proteins in first, second, and third runs, for a total of 115 proteins identified.26 Of these, 101 proteins were observed using either SCX or OGE fractionation (Figure 3). There were a total of 14 unique proteins observed with SCX fractions, while 55 unique proteins were observed in OGE fractions (Table 2). Some of the proteins identified using the OGE separation were ones that were previously seen only after abundant protein depletion of cerebrospinal fluid.28
Table 1.
List of proteins identified using the OGE method in order of abundance as indicated by the total spectrum count (TSC). The percent composition (% comp) was calculated as the percent of the summed normalized TSC’s (see text). Proteins were considered identified if at least two identified peptides were matched to only that protein.
| Protein ID | MW | Accession | TSC | %Comp. |
|---|---|---|---|---|
| Albumin | 69321 | P02768 | 1911 | 22.63 |
| Transthyretin | 15877 | P02766 | 191 | 9.87 |
| Hypothetical protein LOC651928 | 26229 | 946295 | 176 | 5.51 |
| Hypothetical protein LOC649897 | 22059 | Q49AS2 | 102 | 3.80 |
| Cystatin C | 15789 | P01034 | 68 | 3.54 |
| Transferrin | 77000 | P02787 | 303 | 3.23 |
| Anti-RhD monoclonal T125 gamma1 heavy chain | 52253 | Q5EFE5 | 196 | 3.08 |
| Alpha-1 antitrypsin | 46707 | P01009 | 163 | 2.86 |
| Prostaglandin D2-synthase | 21015 | Q5SQ09 | 64 | 2.50 |
| PREDICTED: similar to Ig gamma-3 chain C region | 18069 | 947052 | 51 | 2.32 |
| Apolipoprotein A-II preproprotein | 11167 | P02652 | 27 | 1.98 |
| Apolipoprotein A-I preproprotein | 30758 | P02647 | 70 | 1.87 |
| Apolipoprotein E | 36131 | P02649 | 71 | 1.61 |
| PREDICTED: similar to Ig alpha-1 chain C region isoform 1 | 28490 | Q8NCL6 | 54 | 1.56 |
| Alpha 2 globin | 15247 | Q86YQ5 | 26 | 1.40 |
| Beta globin | 15988 | P68871 | 25 | 1.28 |
| Vitamin D-binding protein | 52883 | P02774 | 80 | 1.24 |
| Apolipoprotein D | 21261 | P05090 | 29 | 1.12 |
| Haptoglobin | 45176 | P00738 | 56 | 1.02 |
| Syntaxin binding protein 2 | 66396 | 15833 | 81 | 1.00 |
| Haptoglobin-related protein | 39004 | P00739 | 44 | 0.93 |
| Hemopexin | 51643 | P02790 | 57 | 0.91 |
| Orosomucoid 1 | 23496 | P02763 | 24 | 0.84 |
| Clusterin isoform 1 | 57795 | Q96AJ1 | 59 | 0.84 |
| Delta globin | 16045 | P02042 | 15 | 0.77 |
| Serpin peptidase inhibitor, clade A, member 3 | 47620 | P36955 | 44 | 0.76 |
| PREDICTED: similar to Ig gamma-4 chain C region . | 47414 | P01861 | 39 | 0.68 |
| PREDICTED: similar to Ig kappa chain V-III region HAH | 16578 | 642113 | 13 | 0.64 |
| Beta-2-microglobulin | 13705 | Q9UM88 | 10 | 0.60 |
| Apolipoprotein A-IV precursor | 45344 | P06727 | 32 | 0.58 |
| Complement component 4A preproprotein | 192663 | Q5INX2 | 117 | 0.50 |
| Complement component 3 | 187045 | Q6LDJ0 | 110 | 0.48 |
| Alpha-2-glycoprotein 1, zinc | 34237 | P02765 | 20 | 0.48 |
| Orosomucoid 2 | 23587 | Q5T538 | 13 | 0.45 |
| Angiotensinogen preproprotein | 53120 | P01019 ACS | 29 | 0.45 |
| Alpha 1B-glycoprotein | 54219 | P04217 | 28 | 0.42 |
| PREDICTED: similar to Ig gamma-1 chain C region | 64280 | 652050 | 32 | 0.41 |
| Serine (or cysteine) proteinase inhibitor, clade F | 46283 | Q4R6H4 | 23 | 0.41 |
| Alpha-2-macroglobulin | 163188 | P01023 | 81 | 0.41 |
| A-gamma globin | 16118 | P69891 | 8 | 0.41 |
| Apolipoprotein H | 38286 | P02749 | 19 | 0.41 |
| Insulin-like growth factor binding protein 6 | 25306 | P24592 | 12 | 0.39 |
| Kininogen 1 | 47852 | P01042 | 22 | 0.38 |
| CD14 antigen precursor | 40050 | P08571 | 18 | 0.37 |
| Retinol-binding protein 4, plasma | 22995 | P02753 | 10 | 0.36 |
| Ceruloplasmin (ferroxidase) | 122127 | P00450 | 53 | 0.36 |
| PREDICTED: similar to Ig kappa chain V-II region Cum | 13912 | P01614 | 6 | 0.35 |
| C-type lectin domain family 3, member B | 22552 | P05452 | 9 | 0.33 |
| Gelsolin isoform B | 80590 | P06396 | 32 | 0.33 |
| Pancreatic ribonuclease | 17632 | P07998 | 7 | 0.33 |
| Alpha-2-HS-glycoprotein | 39299 | P02765 | 15 | 0.31 |
| Family with sequence similarity 3, member C | 24664 | Q5HY75 | 9 | 0.30 |
| Actin, gamma 1 propeptide | 41765 | P63261 | 15 | 0.29 |
| PREDICTED: similar to Ig gamma-2 chain C region | 39968 | P01859 | 14 | 0.29 |
| Kallikrein 6 isoform B | 15045 | Q92876 | 5 | 0.27 |
| EGF-containing fibulin-like extracellular matrix 1 For | 54604 | Q12805 | 18 | 0.27 |
| Complement component 1 inhibitor precursor | 55119 | 060860 | 18 | 0.27 |
| Synovial sarcoma, X breakpoint 4B isoform a | 21844 | O60224 | 7 | 0.26 |
| Complement factor B preproprotein | 85478 | P00751 | 27 | 0.26 |
| Dickkopf homolog 3 | 38365 | Q4R417 | 12 | 0.26 |
| Lysozyme | 16526 | Q8N1E2 | 5 | 0.25 |
| Proprotein convertase subtilisin/kexin type 1 inhibitor | 27355 | P29120 | 8 | 0.24 |
| Galectin 3 binding protein | 65289 | P17931 | 19 | 0.24 |
| Serine (or cysteine) proteinase inhibitor, clade C | 52568 | P32661 | 15 | 0.23 |
| Secreted phosphoprotein 1 isoform A | 35401 | Q4W597 | 10 | 0.23 |
| Alpha-1-microglobulin/bikunin | 38973 | Q5TBD7 | 11 | 0.23 |
| PREDICTED: hypothetical protein XP_939253 | 28963 | 647006 | 8 | 0.23 |
| Chitotriosidase | 51648 | QHVGC6 | 14 | 0.22 |
| Insulin-like growth factor binding protein 7 | 29111 | Q16270 | 7 | 0.20 |
| Complement factor H isoform b | 50974 | P08603 | 11 | 0.18 |
| Amyloid precursor-like protein 1 isoform 2 | 72131 | P51693 | 15 | 0.17 |
| Serine (or cysteine) proteinase inhibitor, clade F | 46283 | Q4R6H4 | 9 | 0.16 |
| Secretogranin III | 52972 | Q8WXD2 | 10 | 0.15 |
| PREDICTED: similar to FXYD domain-containing ion | 10640 | A8K0R4 | 2 | 0.15 |
| Complement component 1, q subcomponent, B chain | 26704 | Q5T960 | 5 | 0.15 |
| PREDICTED: similar to Ig heavy chain V-III region | 16286 | 646057 - | 3 | 0.15 |
| VGF nerve growth factor inducible | 67217 | Q9UDW8 | 12 | 0.15 |
| Osteoglycin preproprotein | 33900 | P20774 | 6 | 0.15 |
| Autotaxin isoform 2 preproprotein | 98929 | 1035181 | 17 | 0.14 |
| PREDICTED: similar to peptidylprolyl isomerase A | 18660 | P62937 | 3 | 0.13 |
| Complement component 7 | 93457 | Q8TCS7 | 14 | 0.12 |
| SPARC-like 1 | 75201 | Q14515 ACS | 11 | 0.12 |
| Phospholipid transfer protein isoform a | 54704 | Q53H91 | 8 | 0.12 |
| Afamin | 69024 | P43652 | 10 | 0.12 |
| PREDICTED: similar to Ig heavy chain V region 102 | 13898 | P01743 | 2 | 0.12 |
| Insulin-like growth factor binding protein 2 | 35114 | P18065 | 5 | 0.12 |
| Carnosinase 1 | 56656 | Q96KN2 | 8 | 0.12 |
| Fibrinogen, gamma chain isoform gamma-B | 51478 | 068656 | 7 | 0.11 |
| Complement factor H isoform A | 138978* | P08603 | 18 | 0.11 |
| Vitronectin | 54271 | P04004 | 7 | 0.11 |
| Peroxiredoxin 2 isoform b | 15979 | Q6P390 | 2 | 0.10 |
| Cell adhesion molecule with homology to L1CAM | 136612 | Q59FY0 | 17 | 0.10 |
| Superoxide dismutase 3, extracellular | 25864 | O14618 | 3 | 0.10 |
| Complement component 9 | 63132 | Q9UG14 | 7 | 0.09 |
| Plasminogen For | 90510 | P06733 | 10 | 0.09 |
| PREDICTED: similar to actin-like protein | 46174 | O96019 | 5 | 0.09 |
| Fibulin 1 isoform B | 65427 | Q59G97 | 7 | 0.09 |
| PREDICTED: similar to Ubiquitin carboxyl-terminal | 113359 | Q9H9C5 | 12 | 0.09 |
| Leucine-rich alpha-2-glycoprotein 1 | 38154 | P02750 | 4 | 0.09 |
| DJ-1 protein | 19878 | Q99497 | 2 | 0.08 |
| Hypothetical protein LOC79441 | 69606 | Q725Q5 | 7 | 0.08 |
| Complement component 1, r subcomponent | 80147 | Q53HT9 | 8 | 0.08 |
| Hypothetical protein LOC160518 | 140512 | Q6NUJ0 | 14 | 0.08 |
| PREDICTED: similar to Prostate, ovary, testis alpha-N- acetylglucosaminidase |
121366 | P54802 | 12 | 0.08 |
| Fibronectin 1 isoform 3 preproprotein | 259061 | P02751 | 25 | 0.08 |
| Alpha 1 actin | 42023 | P68133 | 4 | 0.08 |
| Chitinase 3-like 1 | 42586 | P36222 | 4 | 0.08 |
| Alpha-2-plasmin inhibitor | 54561 | P08697 | 5 | 0.08 |
| Fibrinogen, beta chain preprotein | 55892 | P02765 | 5 | 0.07 |
| Tissue inhibitor of metalloproteinase 1 | 23155 | P01033 | 2 | 0.07 |
| Coagulation factor II | 69992 | Q53H04 | 6 | 0.07 |
| UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 1 | 47088 | Q43505 | 4 | 0.07 |
| Complement component 1, s subcomponent | 76634 | Q53HU9 | 6 | 0.06 |
| Fructose-bisphosphate aldolase C | 39431 | P09972 | 3 | 0.06 |
| Kallikrein 6 isoform A preproprotein | 26838 | Q6T774 | 2 | 0.06 |
| Peptidoglycan recognition protein L | 67927 | Q96PD5 | 5 | 0.06 |
| Heparin cofactor II | 57034 | P05546 | 4 | 0.06 |
| Microfibrillar-associated protein 4 | 28629 | A8KAJ1 | 2 | 0.06 |
| Inter-alpha (globulin) inhibitor H4 | 103293 | Q59FS1 | 7 | 0.06 |
| Histidine-rich glycoprotein | 59540 | P04196 | 4 | 0.06 |
| Inter-alpha globulin inhibitor H2 polypeptide | 106396 | A2RTY6 | 7 | 0.05 |
| Nel-like 2 | 91284 | Q99435 | 6 | 0.05 |
| PREDICTED: similar to Fc fragment of IgG binding | 30792 | 945689 | 2 | 0.05 |
| PREDICTED: similar to Ceruloplasmin (Ferroxidase) | 32078 | 877964 | 2 | 0.05 |
| Neuronal cell adhesion molecule isoform B | 130964 | A4D0S3 | 8 | 0.05 |
| Limbic system-associated membrane protein | 37370 | Q13449 | 2 | 0.04 |
| Fibulin 1 isoform D | 77190 | Q96K89 | 4 | 0.04 |
| Intercellular adhesion molecule 5 | 97185 | Q9UMF0 | 5 | 0.04 |
| Aldolase A | 39395 | P04075 | 2 | 0.04 |
| UNC13 (C. elegans)-like | 180529 | O14795 ACS | 9 | 0.04 |
| ATP-binding cassette, sub-family E, member 1 | 67271 | P61221 | 3 | 0.04 |
| Neogenin homolog 1 | 159859 | Q59FP8 | 7 | 0.04 |
| Chromogranin A | 50657 | P10645 | 2 | 0.03 |
| Inter-alpha (globulin) inhibitor H1 | 101338 | P19827 | 4 | 0.03 |
| Hypothetical protein LOC440307 | 53187 | A6NNM8 | 2 | 0.03 |
| Hypothetical protein LOC51244 | 53924 | Q6P1I3 | 2 | 0.03 |
| Hypothetical protein LOC9816 | 170460 | Q14146 | 6 | 0.03 |
| Amyloid beta A4 protein precursor, isoform A | 86888 | P05067 | 3 | 0.03 |
| MutS homolog 5 isoform c | 92816 | O46196 | 3 | 0.03 |
| Complement component 4 binding protein, alpha chain | 66989 | Q5VVQ8 | 2 | 0.02 |
| Coagulation factor XII | 67774 | P00748 | 2 | 0.02 |
| Integrator complex subunit 8 | 113015 | Q75QN2 | 3 | 0.02 |
| Chromogranin B | 78199 | P05060 | 2 | 0.02 |
| Carbamoylphosphate synthetase 2 | 242827 | P27708 | 6 | 0.02 |
| Complement component 2 | 83214 | A2AAQ4 | 2 | 0.02 |
| Apolipoprotein B | 515209 | P04114 | 12 | 0.02 |
| Tuberous sclerosis 2 isoform 3 | 195776 | P49815 | 4 | 0.02 |
| Trinucleotide repeat containing 15 | 149977 | Q6Y7W6 | 3 | 0.02 |
| Calsyntenin 1 isoform 1 | 109723 | Q94985 | 2 | 0.01 |
| Ephrin receptor EphA4 | 109789 | P54764 | 2 | 0.01 |
| Contactin 1 isoform 1 For | 113249 | Q12860 | 2 | 0.01 |
| Calcium channel, voltage-dependent, alpha 2/delta | 123105 | Q17R45 | 2 | 0.01 |
| Fc fragment of IgG binding protein | 571719 | P01876 | 9 | 0.01 |
| Myosin VIIA | 254242 | Q13402 | 3 | 0.01 |
| Complement component 4B preproprotein | 192629 | Q6U2E9 | 2 | 0.01 |
| Thyroid hormone receptor interactor 11 | 227498 | Q15643 | 2 | 0.01 |
Figure 2.
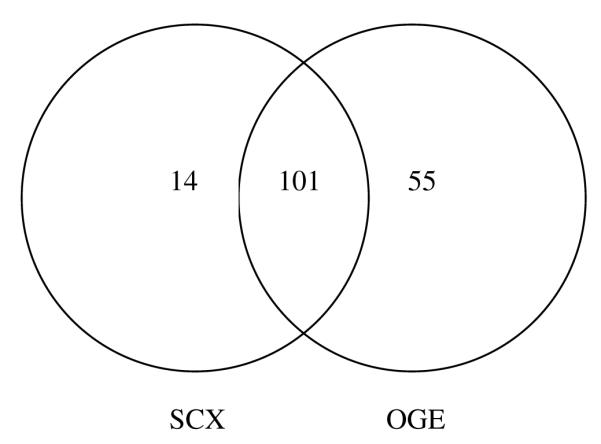
Venn diagram showing the numbers of proteins identified using SCX and using OGE as the first dimension separation in shotgun analysis of undepleted CSF. A total of 101 identified proteins were observed in both of the methods, while 14 additional proteins were unique to the SCX fmethod, and 55 were unique to OGE method.
Table 2.
Lists of uniquely identified proteins observed using the SCX and using the OGE methods (i.e. observed using one method but not the other). Proteins denoted by an asterisk were observed in previous SCX fractionation analysis only after abundant protein depletion.
| Unique Protein Identifications Observed in SCX Fractions | Unique Proteins Identifications Observed in OGE Fractions |
|---|---|
| Antitrypsin | Actin, gamma 1 propeptide* |
| Brevican isoform 1 | Aldolase A* |
| CD59 antigen p18-20 | Alpha 1 actin |
| Complement component 1, q subcomponent | Amyloid beta A4 protein, isoform a |
| Epididymal secretroy protein E1 | Apolipoprotein B* |
| Fibrinogen, alpha polypeptide isoform alpha | ATP-binding cassette, sub-family E, member 1* |
| Fibulin 1 isoform C | Calcium channel, voltage-dependent, alpha 2/delta* |
| Gelsolin isoform A | Carbamoylphosphate synethetase 2 |
| Insulin receptor substrate 1 | Coagulation factor XII* |
| Neural cell adhesion molecule 1 isoform 1 | Complement component 2* |
| Prion protein preproprotein For | Complement component 4 binding protein, alpha chain* |
| Prosaposin | Contactin isoform 1* |
| Thy-1 cell surface antigen | DJ-1 protein |
| Thymosin-like 3 | Ephrin receptor EphA4 |
| Fructose-bisphosphate aldolase C* | |
| Heparin cofactor II* | |
| Hypothetical protein LOC160518 | |
| Hypothetical protein LOC440307 | |
| Hypothetical protein LOC51244 | |
| Hypothetical protein LOC 79441 | |
| Hypothetical protein LOC9816 | |
| Insulin-like growth factor binding protein 6 | |
| Integrator complex subunit 8 | |
| Inter-alpha (globulin) inhibitor H1* | |
| Intercellular adhesion molecule 5 | |
| Kallikrein 6 isoform A preproprotein | |
| Leucine-rich alpha-2-glycoprotein 1* | |
| Microfibrillar-associated protein 4* | |
| MutS homolog 5 isoform c | |
| Myosin VIiA | |
| Nel-like 2* | |
| Neogenin homolog 1* | |
| Peroxiredoxin 2 isoform b | |
| Phospholipid transfer protein isoform a* | |
| PREDICTED: hypothetical protein XP_939253 | |
| PREDICTED: similar to actin-like protein | |
| PREDICTED: similar to ceruloplasmin* | |
| PREDICTED: similar to Fc fragment of IgG binding* | |
| PREDICTED: similar to FXYD domain-containing ion | |
| PREDICTED: similar to Ig heavy chain V region 102 | |
| PREDICTED: similar to Ig heavy chain V-III region | |
| PREDICTED: similar to Ig kappa chain V-II region C | |
| PREDICTED: similar to peptidylprolyl isomerase A | |
| PREDICTED: similar to prostrate, ovary, testis alpha-N-acetylglucosaminidase | |
| PREDICTED: similar to ubiquitin carboxyl-terminal | |
| Serine (or cysteine) proteinase inhibitor, clad | |
| Serine (or cysteine) proteinase inhibitor, clade | |
| Superoxide dismutase 3, extracellular | |
| Synovial sarcoma, X breakpoint 4B isoform a | |
| Syntaxin binding protein 2 | |
| Thyroid hormone receptor interactor 11 | |
| Tissue inhibitor of metalloproteinase 1 | |
| Trinucleotide repeat containing 15 | |
| Tuberous sclerosis 2 isoform 3 | |
| UNC13 (C. elegans)-like |
3.2 Advantages of OGE-LC/MS/MS Method
Because OGE does not require addition of salts or detergents, peptides do not have to undergo an extensive or time consuming cleaning step prior to the second dimension separation and MS analysis. A 30 min wash step in the SCX method is eliminated by use of OGE, significantly reducing the instrument time required for the analysis. Also, OGE provides an automated environment for sample separation and stability of the separation in that after separation, the peptides remain focused until the fractions are removed from the wells. Because OGE concentrates as well as fractionate peptides into their respective pI range, lower abundance proteins can be identified than when the first-dimensional separation only fractionates. OGE also offers the opportunity for increased throughput in that as many as twelve samples can undergo parallel first dimension separation. Table 3 shows a comparison between SCX and OGE workflows. Even though sample run times are longer for OGE analysis, samples can be fractionated in parallel overnight unattended. SCX separation is necessarily a serial process and entails collection of fractions every 5 min for a minimum of 2.5 hrs of instrument time per sample, not including blank gradients normally run between samples to insure no cross contamination.
Table 3.
Comparison of workflows and times for OGE and SCX fractionation. Note the shorter time for instrument setup as well as for the LC-MS/MS runs for the OGE method. A 30 min difference in LC-MS/MS times is due to the absence of a desalting step in the OGE workflow that is normally needed with SCX fractions. The OGE instrument allows the analysis of up to twelve samples at a time, giving a further advantage of parallel sample processing.
| OGE | SCX | |
|---|---|---|
| Instrument Setup Time | 25 min | 3 hrs (includes equilibration time for SCX column) |
| Separation and Fraction Collection Time |
13 hr (fraction collection from wells-5 min) |
3.5 hr (fraction collection every 5 min over a 150 min gradient followed by a 1 hr wash) |
| Sample number | up to 12 samples | 1 sample |
| LC-MS/MS | 180 min per fraction | 210 min per fraction |
4. Conclusions
This work demonstrates the utility of OGE as the first dimension peptide separation for proteomic analysis of CSF. Use of OGE enabled observation of more proteins than with the conventional SCX first dimension separation in CSF that had not been depleted of abundant proteins. OGE also offers increased analytical throughput in that multiple samples can be run in parallel. As such, shotgun analysis using OGE as the first dimension separation offers a significant advance in proteomic analysis technology.
Figure 1.

Venn diagram showing the number of proteins identified in three runs RP-LC-MS/MS analyses of the OGE separated peptides. Each number with no overlap of circles shows the number of proteins uniquely seen in that run, while overlapping circles show the numbers of identifed proteins common to 2 or to 3 of the analyses.
Acknowledgment
L.N.W and K.S. were supported by NIH/NHLBI T32 HL007260 Training to Improve Cardiovascular Drug Therapy. Also supported in part by the MUSC Hollings Cancer Center and the NHLBI Proteomics Initiative via contract NO1-HV 28181.
References
- 1.Fournier ML, Gilmore JM, Martin-Brown SA, Washburn MP. Multidimensional separations-based shotgun proteomics. Chemical Reviews. 2007;107:3654–3686. doi: 10.1021/cr068279a. [DOI] [PubMed] [Google Scholar]
- 2.Chen EI, Hewel J, Felding-Habermann B, Yates JR., III Large scale protein profiling by combination of protein fractionation and multidimensional protein identification technology (MudPIT) Molecular and Cellular Proteomics. 2006;5(1):53–56. doi: 10.1074/mcp.T500013-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Essader AS, Cargile BJ, Bundy JL, Stephenson JL., Jr. A comparison of immobilized pH gradient isoelectric focusing and strong-cation-exchange chromatography as a first dimension in shotgun proteomics. Proteomics. 2005;5:24–34. doi: 10.1002/pmic.200400888. [DOI] [PubMed] [Google Scholar]
- 4.Cargile BJ, Talley DL, Stephenson JL., Jr. Immobilized pH gradients as a first dimension in shotgun proteomics and analysis of the accuracy of pI predictability of peptides. Electrophoresis. 2004;25:936–945. doi: 10.1002/elps.200305722. [DOI] [PubMed] [Google Scholar]
- 5.Wolters DA, Washburn MP, Yates JR., III An automated multidimensional protein identification technology for shotgun proteomics. Analytical Chemistry. 2001;73:5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 6.Winnik WM. Continuous pH/salt gradient and peptide score for strong cation exchange chromatography in 2D-nano-LC/MS/MS peptide identification for proteomics. Analytical Chemistry. 2005;77:4991–4998. doi: 10.1021/ac0503714. [DOI] [PubMed] [Google Scholar]
- 7.Lubman DM, Kachman MT, Wang H, Gong S, Yan F, Hamler RL, O’Neil KA, Zhu K, Buchanan NS, Barder TJ. Two-dimensional liquid separations-mass mapping of proteins from human cancer cell lysates. Journal of Chromatography B. 2002;782:183–196. doi: 10.1016/s1570-0232(02)00551-2. [DOI] [PubMed] [Google Scholar]
- 8.Righetti PG, Castagna A, Herbert B, Reymond F, Rossier JS. Prefractionation techniques in proteome analysis. Proteomics. 2003;3:1397–1407. doi: 10.1002/pmic.200300472. [DOI] [PubMed] [Google Scholar]
- 9.Tran JC, Doucette AA. Rapid and effective focusing in a carrier ampholyte solution isoelectric focusing system: a proteome prefractionation tool. Journal of Proteome Research. 2008;7:1761–1766. doi: 10.1021/pr700677u. [DOI] [PubMed] [Google Scholar]
- 10.Zuo X, Speicher DW. A method for global analysis of complex proteomes using sample prefractionation by solution isoelectrofocusing prior to two-dimensional electrophoresis. Analytical Biochemistry. 2000;284:266–278. doi: 10.1006/abio.2000.4714. [DOI] [PubMed] [Google Scholar]
- 11.Tan A, Pashkova A, Zang L, Foret F, Karger BL. A miniaturized multichamber solution isoelectric focusing device for separation of protein digests. Electrophoresis. 2002;23:3599–3607. doi: 10.1002/1522-2683(200210)23:20<3599::AID-ELPS3599>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 12.Bukshpan S, Zilberstein G. Matrixes, arrays, systems and methods. 7,166,202. US Patent. 2007
- 13.Ros A, Faupel M, Mees H, Oostrum JV, Ferrigno R, Reymond F, Michel P, Rossier JS, Girault HH. Protein purification by Off-Gel electrophoresis. Proteomics. 2002;2:151–156. doi: 10.1002/1615-9861(200202)2:2<151::aid-prot151>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 14.Michel PE, Reymond F, Arnaud IL, Josserand J, Girault HH, Rossier JJ. Protein fractionation in a multicompartment device using Off-Gel isoelectric focusing. Electrophoresis. 2003;24:3–11. doi: 10.1002/elps.200390030. [DOI] [PubMed] [Google Scholar]
- 15.Heller M, Michel PE, Morier P, Crettaz D, Wenz C, Tissot J, Reymond F, Rossier JS. Two-stage Off-Gel isoelectric focusing: protein followed by peptide fractionation and application to proteome analysis of human plasma. Electrophoresis. 2005;26:1174–1188. doi: 10.1002/elps.200410106. [DOI] [PubMed] [Google Scholar]
- 16.Heller M, Ye M, Michel PE, Morier P, Stalder D, Jiinger MA, Aebersold R, Reymond F, Rossier JS. Added value for tandem mass spectrometry shotgun proteomics data validation through isoelectric focusing of peptides. Journal of Proteome Research. 2005;4:2273–2282. doi: 10.1021/pr050193v. [DOI] [PubMed] [Google Scholar]
- 17.Horth P, Miller CA, Preckel T, Wenz C. Efficient fractionation and improved protein identification by peptide OFFGEL electrophoresis. Molecular and Cellular Proteomics. 2006;5(10):1968–1974. doi: 10.1074/mcp.T600037-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Kuhn JF, Hoerth P, Hoehn ST, Preckel T, Tomer KB. Proteomics study of anthrax lethal toxin-treated murine macrophages. Electrophoresis. 2006;27:1584–1597. doi: 10.1002/elps.200500747. [DOI] [PubMed] [Google Scholar]
- 19.Fraterman S, Zeiger U, Khurana TS, Rubinstein NA, Wilm M. Combination of peptide OFFGEL fractionation and label-free quantitation facilitated proteomics profiling of extraocular muscle. Proteomics. 2007;7:3404–3416. doi: 10.1002/pmic.200700382. [DOI] [PubMed] [Google Scholar]
- 20.Lam H, Josserand J, Lion N, Girault HH. Modeling the isoelectric focusing of peptides in an OFFGEL multicompartment cell. Journal of Proteome Research. 2007;6:1666–1676. doi: 10.1021/pr0606023. [DOI] [PubMed] [Google Scholar]
- 21.Burgess JA, Lescuyer P, Halnard A, Burkhard PR, Turck N, Michel P, Rossier JS, Reymond F, Hochstrasser DF, Sanchez J. Identification of brain cell death associated proteins in human post-mortem cerebrospinal fluid. Journal of Proteome Research. 2006;5:1674–1681. doi: 10.1021/pr060160v. [DOI] [PubMed] [Google Scholar]
- 22.Noben J, Dumont D, Kwasnikowska N, Verhaert P, Somers V, Hupperts R, Stinissen P, Robben J. Lumbar cerebrospinal fluid proteome in multiple sclerosis: characterization by ultrafiltration, liquid chromatography, and mass spectrometry. Journal of Proteome Research. 2006;5:1647–1657. doi: 10.1021/pr0504788. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, McKenna T, Hughes C, Leweke FM, Schwarz E, Bahn S. CSF biomarker discovery using label-free nano-LC-MS based proteomic profiling: Technical aspects. Journal of Separation Science. 2007;30:214–225. doi: 10.1002/jssc.200600350. [DOI] [PubMed] [Google Scholar]
- 24.Ogata Y, Charlesworth MC, Muddiman DC. Evaluation of protein depletion methods for the analysis of total-, phosphor- and glycoproteins in lumbar cerebrospinal fluid. Journal of Proteome Research. 2005;4:837–845. doi: 10.1021/pr049750o. [DOI] [PubMed] [Google Scholar]
- 25.Pan S, Wang Y, Quinn JF, Peskind ER, Waichunas D, Wimberger JT, Jin J, Li JG, Zhu D, Pan C, Zhang J. Identification of glycoproteins in human cerebrospinal fluid with a complementary proteomic approach. Journal of Proteome Research. 2006;5:2769–2779. doi: 10.1021/pr060251s. [DOI] [PubMed] [Google Scholar]
- 26.Maccarrone G, Milfay D, Birg I, Rosenhagen M, Holsober F, Grimm R, Bailey J, Zolotarjova N, Turck CW. Mining the human cerebrospinal fluid proteome by immunodepletion and shotgun mass spectrometry. Electrophoresis. 2004;25:2402–2412. doi: 10.1002/elps.200305909. [DOI] [PubMed] [Google Scholar]
- 27.Shores KS, Knapp DR. Assessment approach for evaluating high abundance protein depletion methods for cerebrospinal fluid (CSF) proteomic analysis. Journal of Proteome Research. 2007;6:3739–3751. doi: 10.1021/pr070293w. [DOI] [PubMed] [Google Scholar]
- 28.Shores KS, Udugamasooriya DG, Kodadek T, Knapp DR. Use of peptide analog diversity library beads for increased depth of proteomic analysis: application to cerebrospinal fluid. Journal of Proteome Research. 2007;7:1922–1931. doi: 10.1021/pr7006889. [DOI] [PubMed] [Google Scholar]
- 29.Liu H, Sadygov RG, Yates JR., III A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Analytical Chemistry. 2004;76(14):4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]