

Abstract
The glycophosphatidylinositol-anchored cell surface receptor CD24 (also called heat-stable antigen) promotes the apoptosis of progenitor and precursor B-lymphocytes. However, the immediate proximal events that occur after engagement of CD24 in B cells are not precisely understood. Using a bioinformatics analysis of mouse (Mus musculus) gene expression data from the Immunological Genome Project, we found that known vesicle trafficking and cellular organization genes have similar expression patterns to CD24 during B-cell development in the bone marrow. We therefore hypothesized that CD24 regulates vesicle trafficking. We first validated that antibody-mediated engagement of CD24 induces apoptosis in the mouse WEHI-231 cell line and mouse primary bone marrow-derived B cells. We next found that CD24 surface protein expression is rapidly and dynamically regulated in both WEHI-231 cells and primary immature B cells in response to engagement of CD24. The change in surface expression was not mediated by classical endocytosis or exocytosis. However, we found that CD24-bearing plasma membrane-derived extracellular microvesicles were released in response to CD24 engagement. Furthermore, in response to CD24 engagement we observed a clear exchange of CD24 between different populations of B cells. Hence, we show that engagement of CD24 in immature B cells results in a dynamic regulation of surface CD24 protein and a redistribution of CD24 within the population.
Keywords: apoptosis, B lymphocyte, bioinformatics, CD24, microvesicle
Introduction
B-lymphocyte (B-cell) development is a highly regulated process that occurs in the bone marrow of adult mammals. Bone marrow B-cell development can be divided into discrete stages termed Hardy fractions.1,2 Hardy fractions A through C’, also called progenitor (pro)-B cells, do not express the B-cell receptor. Fraction D are precursor (pre)-B cells that express the pre-B-cell receptor. Fraction E are immature IgM-expressing cells, and Fraction F are mature naive, IgM- and IgD-expressing B cells.1 One of the earliest surface proteins that is expressed during B-cell development is the cell surface receptor CD24, also known as heat-stable antigen.1 CD24 is a small, highly glycosylated, glycophosphatidylinositol-linked protein normally localized to lipid rafts on the plasma membrane.3–5 CD24 expression is detectable by Fraction B cells with the highest expression seen in fraction C/C’ followed by a gradual decline in expression as B cells mature and exit the bone marrow.1
Transgenic mice that over-express CD24 have drastically reduced numbers of both pro-B and pre-B cells, corresponding to Hardy fractions B, C and D,6 due to an increase in B-cell apoptosis.7 Chimeric CD24 knockout mice also showed reduced numbers of cells in Hardy Fraction C, due to a block in the pro-B to pre-B-cell transition.8 The complete CD24 mouse knockout recapitulated this effect, but also showed a major reduction in pre-B cells in Fractions D and E.9 In all cases, near normal numbers of mature peripheral B cells were present suggesting that the cells that pass through the block in development were capable of reconstituting the mature B-cell compartment. Therefore, CD24 function is dependent on both the developmental stage of B cells and the level of CD24 expression, with either increased or reduced expression levels interfering with normal B-cell development.
Several ligands for CD24 have been identified, including P-Selectin, Siglec-G and L1-CAM10–12 with ligand specificity varying by cell type but no ligand has been identified for CD24 on B cells. Antibody-mediated engagement of CD24, which mimics ligand interaction, can induce apoptosis in vitro in mouse bone marrow-derived B-cell cultures and in a number of murine and human pro-B and pre-B cell lines.5,13,14 Enhanced clustering of CD24 by addition of a secondary antibody increased B-cell apoptosis, so demonstrating a dose-dependent regulation of CD24-mediated cell death.13 CD24 engagement also activates several caspase proteins, including Caspase-2, -3, -7 and -8, which are known to be involved in the induction of apoptosis.14 Engagement of CD24 causes the translocation of the Src-family tyrosine kinase, Lyn, into lipid rafts,5 which is presumed to activate downstream signalling. However, other plasma membrane-proximal events that occur in response to CD24 engagement have not been identified.
We used transcriptomics data from the Immunological Genome project15 to identify additional potential functions of CD24. We found that genes with a similar expression profile to CD24 are significantly associated with cytoskeletal organization and vesicle trafficking. In support of the hypothesis that CD24 regulates vesicle trafficking, we found that antibody-mediated engagement of CD24 causes immediate and dramatic changes in its own cell surface expression in both mouse bone marrow-derived primary B cells and in the WEHI-231 immature B-cell line. This dynamic shift is not caused through classical endocytosis or exocytosis events, but is associated with the generation of CD24-bearing extracellular microvesicles (EMV) that can transport CD24 between cells.
Materials and methods
Bioinformatics analysis
Microarray-based expression data were retrieved from the Gene Expression Omnibus (GEO) using accession number GSE15907. RMA normalization of gene expression and identification of differentially expressed genes was performed in R 2.15.016 via TinnR 2.3.7.1.17 using the Bioconductor,18 Biobase,18 Oligo,19 Limma20 and Affycoretools21 packages and the pd.mogene1.1 annotation file. False Discovery Rate was used for multiple testing correction. Unsupervised hierarchical clustering was performed in Genesis 1.7.6.22
The network interaction map was created using the online GeneMANIA tool.23 The Caspase-7 gene was included with the co-expressed genes as it is known to be a target of CD24 signalling14 and was identified in the bioinformatics screen as being expressed during Hardy fractions A through C’, when CD24 expression is highest. Genes lists were annotated automatically for gene ontology (GO) functions by GeneMANIA and AmiGO2,24 and manually annotated using the National Centre for Biotechnology Information Gene database and Vesiclepedia.25
Animal care
The Institutional Animal Care committee at Memorial University of Newfoundland approved all animal procedures. Three-week-old C57BL/6N male mice were obtained from the Quebec facility of Charles River Laboratories (Wilmington, MA).
Cell culture
All materials for cell culture were obtained from Life Technologies (Carlsbad, CA) unless otherwise indicated. Isolated bone-marrow derived immature B cells and the BALB/c × NZB mouse WEHI-231 pre-B-cell lymphoma cell line (ATCC, Manassas, VA) were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 1% antibiotic/antimycotic, 1% sodium pyruvate and 0·1% mercaptoethanol (complete media) at 37° and 5% CO2.
Primary bone marrow B-cell isolation
Femurs were removed from euthanized male C57BL/6N mice (3–6 weeks of age) and bone marrow was flushed out with Quin saline (QS; 25 mm NaHEPES, 125 mm NaCl, 5 mm KCl, 1 mm CaCl2, 1 mm Na2HPO4, 0·5 mm MgSO4, 1 g/l glucose, 2 mm glutamine, 1 mm sodium pyruvate, 50 μm 2-mercaptoethanol, pH 7·2), using a 21-gauge needle. Single-cell suspensions were produced using a 100-µm nylon mesh. The EasySep Mouse B Cell Isolation Kit (cat. no. 19854; StemCell Technologies, Vancouver, BC, Canada) was used to enrich bone marrow isolates following the manufacturer’s protocol. Fc-receptors were blocked on the B cells in this isolation using anti-mouse CD16/CD32 (FcγIII/II receptor) antibodies. All experiments and analyses were performed on total isolated bone marrow-derived B cells.
Flow cytometry
Cells were resuspended in PBS (1·86 mm NaH2PO4·H2O, 8·41 mm Na2HPO4, 150 mM NaCl) containing 1% heat-inactivated fetal bovine serum (FACS buffer) unless stated otherwise. Flow cytometry data were collected on a FACSCalibur flow cytometer using CellQuest Pro v4.0.2 software (BD Biosciences, San Jose, CA) or a FACSAria II cell sorter using FACSDiva v8.0 software (BD Biosciences), at the Medical Education and Laboratory Support Services or the Cold-Ocean Deep-Sea Research Facility, respectively. Analysis was performed using FlowJo v10.0.5 (Tree Star, Ashland, OR). Cell death experiments using Annexin V and propidium iodide were analysed ungated while CD24 expression was analysed after gating on the live cell population based on FSC/SSC.
Apoptosis analysis
Annexin V/propidium iodide staining. Isolated bone marrow B cells or WEHI-231 cells (5 × 105 cells/ml) in complete media were either left untreated, or treated for up to 24 hr with either functional grade 10 μg/ml primary monoclonal M1/69 rat anti-mouse CD24 antibody (cat. no. 16-0242-85) or 10 μg/ml matching primary isotype antibody (cat. no. 16-4031-85) both from eBioscience (San Diego, CA), or with primary antibodies that had been pre-incubated with 5 μg/ml goat anti-rat secondary antibody (secondary; cat. no. 112-005-003; Jackson ImmunoResearch, West Grove, PA). Primary and secondary antibodies were always pre-incubated before addition to cells at a 2 : 1 ratio to ensure efficient cross-linking of primary antibody and to ensure no excess secondary antibody was present. Furthermore, we have confirmed that isotype pre-incubated with secondary antibody did not bind to either WEHI-231 or primary B cells. Cells with treatment times < 24 hr were centrifuged to remove antibody-treated media and then resuspended in complete media and incubated at 37° for a total of 24 hr and then analysed for cell death. For staining, cells were resuspended in Annexin V binding buffer (10 mm HEPES buffer, 140 mm NaCl, 2·5 mm CaCl2) at pH 7·4. Apoptosis was measured using AnnexinV-Alexa488 and propidium iodide using the Dead Cell Apoptosis kit (Life Technologies) following the manufacturer’s instructions. Primary isolated B cells were also stained with 0·125 μg rat anti-mouse CD19-Peridinin chlorophyll protein-Cy5.5 (cat. no. 35-0193-80; eBioscience) antibodies to confirm > 85% of cells were CD19+. Cells were analysed by flow cytometry.
Caspase activation. Cells were treated for up to 3 hr as described above. Thirty minutes before the indicated time, cells were incubated with 5 μm CellEvent™ Caspase-3/7 Green Detection Reagent (Life Technologies) following the manufacturer’s protocol, and then analysed by flow cytometry.
CD24 surface expression and antibody exchange
Isolated bone marrow B cells or WEHI-231 cells (5 × 105 cells/ml in QS) were rested at 37° for 15 min. Cells were then incubated with 10 μg/ml of M1/69 anti-mouse CD24 antibody that had been pre-incubated with 5 μg/ml of biotinylated goat-anti-rat secondary antibody for 1, 5, 15, 40 or 60 min, or left untreated. Cells were stopped in 3 ml of ice-cold FACS buffer and then washed with FACS buffer followed by staining with 0·25 μg streptavidin-linked FITC (cat. no. 11-4317-87; eBioscience) for 30 min in FACS buffer at 4°. Cells were washed three times with FACS buffer and then analysed by flow cytometry.
To determine if CD24 epitopes were saturated by the addition of the pre-incubated primary and secondary antibody, cells were stimulated as above, or left untreated, followed by fixation with 4% paraformaldehyde for 20 min at room temperature, followed by three washes with FACS buffer. Fixed, untreated cells (control) were incubated with 10 μg/ml of M1/69 anti-mouse CD24 antibody that had been pre-incubated with 5 μg/ml of biotinylated goat-anti-rat secondary antibody for 30 min at 4°. All cells were then stained with 0·25 μg streptavidin-linked FITC and with 0·06 μg M1/69-allophycocyanin (APC) (cat. no. 17-0242-80; eBioscience) for 30 min at 4°. Cells were then washed with FACS buffer and analysed by flow cytometry.
For confocal analysis of CD24, cells were stimulated as described above for the indicated times, or were left untreated. Cells were then stained with 0·25 μg streptavidin-linked FITC, washed and fixed in 3·7% paraformaldehyde. Fixed cells were mounted onto glass slides using a Shandon Cytospinner centrifuge (Thermo Scientific, Waltham, MA) for 5 min at 1600 g. Slides were covered with VectaShield hardset mounting media with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA). Confocal imaging was performed using the Olympus FV1000 system and IX81 inverted microscope (Olympus, Shinjuku, Japan) and FV10-ASW (v3.1) software.
For exchange experiments, cells were resuspended as above and then incubated for 15 min at 37° with 10 μg/ml M1/69 anti-CD24 antibody that had been pre-incubated with 5 μg/ml biotinylated secondary antibody and either streptavidin-FITC (0·25 μg) or streptavidin-eFluor660 (0·125 μg, cat. no. 50-4317-80; eBioscience). Cells were centrifuged at 500 g for 5 min to remove unbound antibody and then resuspended in QS. Equal amounts of FITC-labelled and eFluor660-labelled cells were mixed either on ice (control) or at 37° for the indicated times. Cells were washed with FACS buffer and then analysed by flow cytometry.
Inhibition of endocytosis and exocytosis
WEHI-231 cells, resuspended at 5·0 × 105 cells/ml in QS, were pre-incubated in 200 μm Pitstop 2 (Abcam, Cambridge, UK), 50 μm Dynasore (Abcam), 40 μm Exo1 (Abcam), 10 μm Brefreldin A (Life Technologies) or vehicle control (DMSO) at 37° for 30 min and then treated with primary and secondary antibodies, as above, with inhibitor concentrations maintained at half the original concentration, for up to 1 hr.
Transmission electron microscopy
WEHI-231 cells were resuspended and stimulated as described above and then centrifuged and resuspended in Karnovsky fixative for 24 hr. Transmission electron microscopy (TEM) was performed by the Medical Education and Laboratory Support Services facility (Memorial University) according to standard protocols. Briefly, 85-nm resin-embedded sections were mounted on 300-mesh copper grids, and then stained with 3% uranyl acetate in a 30% ethanol. Grids were examined using a JEOL 1200 EX electron microscope (JEOL, Peabody, MA) and images were captured using an SIA-L3C digital camera (SIA, Duluth, GA).
Isolation of extracellular microvesicles
WEHI-231 cells in QS were left untreated, or stimulated as described above for 15 min or 60 min at 37° with either 10 μg/ml of M1/69 anti-mouse CD24 antibody that had been pre-incubated with 5 μg/ml of biotinylated goat-anti-rat secondary antibody. Enrichment of EMV was performed using a similar method to previously published studies.26,27 All steps were performed at 4°. Briefly, cells were centrifuged for 5 min at 400 g to pellet cells then the supernatant was centrifuged for 5 min at 2000 g to pellet cell debris. M1/69 anti-mouse CD24 antibody (10 μg/ml) pre-incubated with 5 μg/ml of biotinylated goat-anti-rat secondary antibody was added to the supernatant from untreated cells, when indicated. The supernatant was centrifuged for 1 hr at 16 800 g to pellet EMV. The pelleted EMV were resuspended in a residual volume of 25 μl QS, and 25 μl of 2 × Annexin V binding buffer was added. EMV were stained with 5 µl of Annexin V Alexa 488 and 0·06 μg streptavidin-APC (eBioscience). EMV were analysed on the FACSAria II with 0·2-μm and 0·4-μm beads (Bangs Laboratories, Inc., Fishers, IN) used to establish a sizing gate for EMV using side scatter following established protocols.28,29
Statistical analysis
Statistical analysis was performed in R v3.0.2.16 Student’s t-test was used to determine differences between two groups. Analysis of variance was used to determine differences between more than two groups followed by either a priori analysis using a generalized linear model30 for more than two groups or Student’s t-test for two groups. Tukey Honest Significant Difference was used for a posteriori analysis of all pairwise comparisons if the anova was found to be significant.
Results
CD24 gene expression is associated with known vesicle trafficking mediators
The co-expression of genes has been previously used to identify regulatory networks and to identify signalling pathway partners.31 To identify genes that may be involved in mediating CD24 function, we made use of global gene expression data generated by the Immunological Genome Project.15 To define a signature profile for genes expressed in a similar manner to CD24 during B-cell development, Hardy fractions A, C’ and F were first analysed as they are known to be CD24-low, CD24-high and CD24-low, respectively. In total, 3759 unique features were differentially expressed across these three stages (False Discovery Rate ≤ 0·005). We then performed unsupervised hierarchical clustering on these 3759 features on microarray data sets representing all stages of B-cell development and activation from the multilineage stem cell, common lymphoid progenitor, Hardy fractions A through F, Splenic transitional B cells and terminally differentiated plasma cells (data not shown). This analysis identified a cluster of 55 features, corresponding to 38 individual genes that clustered with CD24 (Fig.1a and see Supplementary material, Tables S1–S4).
Figure 1.

CD24 is co-expressed with known vesicle trafficking genes during B-cell development. (a) Hierarchical clustering of gene expression based on average linkage clustering. The CD24 co-cluster includes 51 features representing 39 unique genes. n = 3 or n = 4 per array analysed per developmental stage (M: multilineage stem cells, L: common lymphoid progenitor cells, A through F: Hardy Fractions A, B/C, C’,D, E and F, T1-T3: splenic transitional B cells, stages 1 through 3, and PC: terminally differentiated plasma cells). (b) Network analysis of the 37 genes including CD24 in the cluster annotated by GeneMANIA plus caspase-7 (see Supplementary material, Table S4). (c) Summary of gene ontology (GO) enrichment analysis of the 38 genes including CD24 in the cluster as annotated by AmiGO2 (P < 0·05, Bonferroni corrected). The number of genes falling within each GO term is indicated in square brackets. The relationship, if any, between terms is indicated by the relative placement of each bullet where indentations indicate a parent–child relationship in the GO hierarchy. The top-most parent term that encompasses lower children terms is indicated in bold. Parent terms are indicated in italics if the parent term was not significantly enriched. The major groupings are indicated as: lavender, cellular organization; blue, biological regulation of proliferation and apoptosis; and green, response to stimulus.
To generate a network interaction map, we added Caspase 7 to the gene list because we identified Caspase 7 as being expressed during early B-cell development (Fractions A through C’) followed by down-regulation coincident with CD24 expression. Furthermore, Caspase 7 is known to be activated by CD24, as reported previously.14 This network analysis clearly showed that the co-expressed genes share a dense network of known and predicted interactions (Fig.1b), suggesting that they may have common regulators and/or biological functions.
Statistical analysis of GO enrichment showed that genes involved in cellular organization, apoptosis and proliferation, response to stimulus, multicellular organismal aging, and neuromuscular synaptic transmission were significantly enriched (Fig.1c and see Supplementary material, Tables S1–S3). As CD24 has been clearly associated with the regulation of apoptosis, the identification of pro-apoptotic genes as co-expressed with CD24 validated our approach to identify potential partners of CD24. However, the majority of gene ontologies were those associated with cellular organization, in particular vesicle and organelle organization. Manual annotation revealed that 33 genes in this cluster are associated with vesicle formation, transport, or function (see Supplementary material, Table S4).
EHD2,32,33 AP1AR34 and FCHO135 are all involved in regulating the formation and coating of cargo vesicles. MCF2L (also known as ARHGEF14) is a guanine exchange factor that modulates Rho GTPases, which are regulators of cytoskeletal organization and vesicle trafficking.36 The function of the RabL5 gene is not well known, however, it has been shown that RabL5 mRNA is secreted in microvesicles released by tumour cells.37,38 Interestingly, two heat-shock proteins; HSPA2 (from the HSP70 family) and DNAJC21 (a member of the HSP40 family) that are highly enriched in released EMV39–43 were also found to be co-expressed with CD24.
MARCKS is an actin cross-linking protein whose activity is thought to couple actin to the plasma membrane in response to protein kinase C or Ca2+-calmodulin signalling.44,45 Similarly, CSRP2 is also an actin-binding protein whose current function is poorly understood.46 Other cytoskeleton-related genes include SMARCA4, which is an actin-dependent chromatin remodelling protein and is involved in regulating cell adhesion molecule activation in leucocytes,47 and MYL4, a myosin motor protein.48
CD24 is also co-expressed with several calcium-responsive proteins. P2RX3, a calcium-responsive, ATP-dependent cation transporter that has been shown to mediate B- and T-cell recruitment to the spleen.49 A closely related family member, P2X7 has also been shown to directly mediate the release of microvesicles and participate in T-cell fate determination.50,51 PLA2G12A is a calcium-binding lipid kinase52 and is thought to be involved in T-cell activation through calcium-mediated generation of lipid second messengers.53 Lastly, we found the EGFL6 gene, whose protein product contains multiple calcium-binding domains.54 Studies have shown the release of EMV may depend on calcium signalling and the activity of protein kinase C.55,56 Hence, as CD24 stimulation is known to induce increases in intracellular calcium,4 these genes may represent elements that respond to CD24 signalling to cause release of EMV.
As there was a clear over-representation of genes associated with vesicle trafficking, we focused on determining if CD24 is associated with this biological function, and not on the role or regulation of the individual genes.
Antibody-mediated engagement of CD24 causes apoptotic cell death in primary bone marrow-derived B cells and in the WEHI-231 B-cell line
To test the hypothesis that engagement of CD24 may regulate vesicle-mediated transport or associated processes, we first validated that engagement of CD24 would induce apoptosis, a well-established function of CD24, in the WEHI-231 B-cell lymphoma cell line, which expresses high levels of CD24 and in ex vivo primary B cells representing Hardy Fractions B–F. Neither of these cell types have been investigated for their response to engagement of CD24 using this technique, and WEHI-231 cells have previously been anecdotally reported to be resistant to CD24-mediated cell death.13 To mimic ligand engagement, the cells were treated for 24 hr with primary rat anti-mouse CD24 antibody or isotype antibody either with or without additional cross-linking induced by pre-incubating the primary antibody with a secondary anti-rat IgG antibody. The addition of secondary antibody will increase the cross-linking of CD24 as well as increase the avidity of anti-CD24 antibody binding.
We found that WEHI-231 cells did not undergo apoptosis in response to the isotype alone or with secondary antibody, or anti-CD24 primary antibody engagement alone but did undergo significant induction of apoptosis when stimulated with both primary and secondary antibody (Fig.2a–b). In addition, enhanced clustering with primary and secondary antibody, but not with primary antibody alone, significantly increased very late apoptotic or necrotic cell death.
Figure 2.

Enhanced cross-linking of CD24 induces cell death in primary B cells and in the WEHI-231 immature B-cell line. (a, b) WEHI-231 cells or (c, d) primary bone-marrow derived B cells were treated with isotype control or anti-CD24 primary antibody, with or without secondary antibody for 24 hr. Cell death was assessed after 24 hr of antibody stimulation using Annexin V-Alexa488 and propidium iodide staining. (a, c) Representative dot plots are shown, and (b, d) mean ± SEM of per cent Annexin V-positive apoptotic cells or necrotic Annexin V-negative, propidium iodide (PI)-positive cells, n = 3, Student’s paired t-test, *P < 0·05, **P < 0·01.
As with WEHI-231 cells, isotype alone or with secondary antibody did not induce cell death in ex vivo primary cells. In comparison, we found that there was a statistically significant increase in the number of apoptotic cells with anti-CD24 antibody in the presence or absence of secondary cross-linking antibody. However, the amount of apoptosis was substantially increased with the enhanced clustering/binding induced by the secondary antibody (Fig.2c–d). Very late apoptotic or necrotic cell death (propidium iodide-positive, Annexin V-negative) was unaffected by either treatment. This observation is consistent with previous reports showing that stimulation of CD24 with primary antibody alone was sufficient to induce apoptotic cell death and that addition of a secondary antibody potentiates the response.5,13,14
CD24 cross-linking has been shown to activate multiple caspases, including Caspases-2, -3, -7 and -8. Similar to previous reports,14 we found that there was significantly up-regulated caspase-3/7 activity in WEHI-231 cells after 3 hr of antibody stimulation (Fig.3a). Similarly, caspase-3/7 activity was significantly up-regulated in primary cells by the same time point (Fig.3b). These data further confirm that both WEHI-231 cells and ex vivo primary cells can serve as an accurate model for elucidating CD24-mediated functions.
Figure 3.
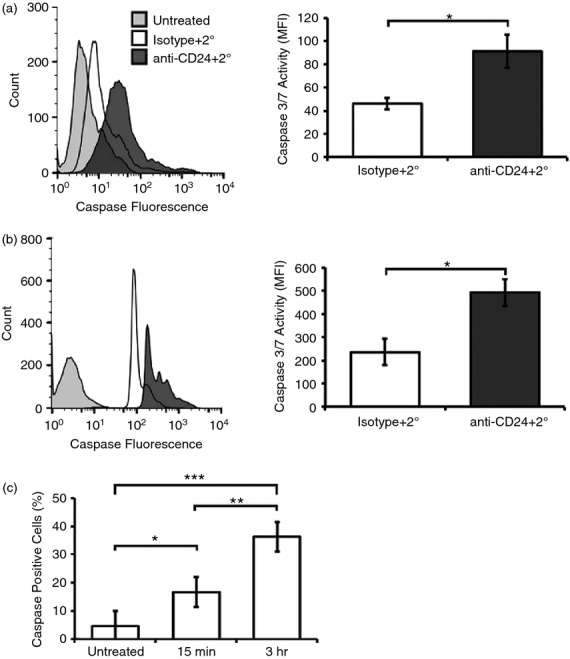
CD24 engagement activates caspase-3/7 in primary B cells and in WEHI-231 cells. (a) WEHI-231 cells or (b) primary cells were treated as for Fig.2. Representative histograms are shown of caspase-3/7 activation (left panel) and the mean ± SEM of the mean fluorescent intensity (right panel), n = 3. Statistical significance was assessed using Students’ paired t-test *P < 0·05, **P ≤ 0·01. (c) WEHI-231 cells were removed from the antibody-treated media after 15 min of stimulation and replaced with untreated media for 2 hr 45 min, or left with antibody for 3 hr. Mean ± SEM of the mean fluorescent intensity, n = 3, are shown; statistical significance was assessed using a one-way analysis of variance followed by a posteriori analysis by Tukey post-hoc, *P < 0·05, **P < 0·01, ***P < 0·001.
We next investigated whether prolonged engagement with antibody was required to induce caspase-3/7 activation by removing unbound antibody following a 15-min incubation of WEHI-231 cells with primary and secondary antibody. We found with only 15 min of stimulation, there was significantly less caspase activity after 3 hr compared with cells exposed continuously to antibody (Fig.3c). These data demonstrate that CD24-mediated cell apoptosis requires prolonged, continuous engagement in conditioned media, rather than a transient engagement, to induce the maximum amount of caspase-3/7 activation.
Thus, we have confirmed that both of these cell types respond in a manner consistent with previous reports on CD24-mediated cell death in other B-cell lines or experimental systems.5,13
Antibody-mediated engagement of CD24 dynamically regulates CD24 surface expression
Engagement of receptors by their ligand can cause changes to their own surface expression, which may be mediated by vesicle formation and/or membrane re-organization, as exemplified by the B-cell receptor.57,58 Therefore, we next examined if expression of CD24 surface protein was altered following antibody-mediated engagement.
We found that both WEHI-231 cells and primary cells responded with a time-dependent, dynamic change in CD24 surface expression. WEHI-231 cells showed an immediate loss of CD24 expression by 1 min before recovering baseline CD24 surface expression by 15 min and then further increasing the amount of cell surface protein over the course of 60 min (Fig.4a–b).
Figure 4.
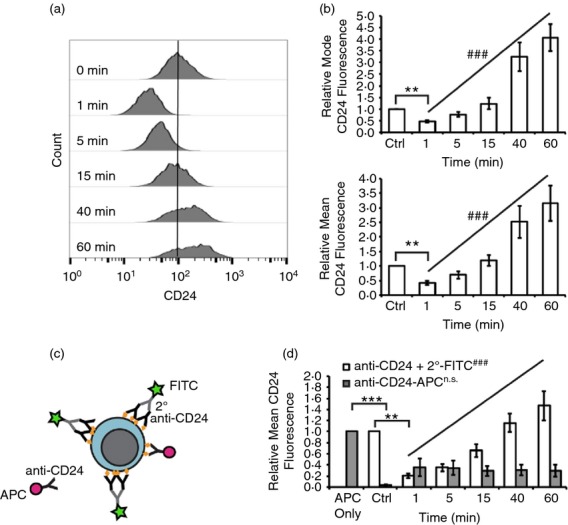
CD24 protein expression is dynamically regulated after CD24 engagement in WEHI-231 cells. (a) WEHI-231 cells were stimulated for the indicated times with anti-CD24 primary antibody and biotinylated secondary antibody. Surface CD24 expression was assessed using streptavidin-FITC. Representative histograms are shown. (b) Mean ± SEM of the relative mean or the modal fluorescence intensity, n = 3, statistical significance was assessed using a one-way analysis of variance followed by a priori analysis of 0 min to 1 min via Student’s one-tailed, uneven variance t-test *P < 0·05, and from 1 min to 60 min via generalized linear model regression analysis. ###P < 0·001. (c) Schematic of CD24 epitope availability assessment. Cells were stimulated for 1 to 60 min with anti-CD24 primary antibody and biotinylated secondary antibody, followed by detection with streptavidin-FITC (green star) as in (a) and (b). Cells were then fixed and available CD24 epitopes (orange diamond) were detected by addition of anti-CD24 antibody directly conjugated to allophycocyanin (red circle). (d) WEHI-231 were stimulated for the indicated times as above and relative surface CD24 expression (white) and free epitopes (grey) detected. The mean ± SEM of the relative mean fluorescence intensity, n = 3, statistical significance was assessed using a one-way analysis of variance followed by a priori analysis of 0 min to 1 min via Student’s one-tailed, uneven variance t-test **P < 0·01, ***P < 0·005, and from 1 min to 60 min via generalized linear model regression analysis. n.s.not significant, ###P < 0·001.
We next confirmed that the changes to CD24 surface expression were not an artefact of the short incubation times by determining if there were any epitopes available for binding of anti-CD24 antibody after stimulation. To do this, cells were fixed after treatment and then incubated with an APC-conjugated anti-CD24 antibody, which will label any unbound epitopes (Fig.4c). CD24 on untreated cells was readily detected by the anti-CD24-APC (Fig.4d, APC only). However, this binding was significantly diminished in cells that had previously been treated with anti-CD24 primary plus secondary antibody (Fig.4d). Furthermore, the degree of binding of the anti-CD24-APC antibody did not vary with the different incubation times with stimulating antibody indicating that the surface CD24 had been saturated by the stimulating antibody at all the times examined.
We next asked if primary cells also showed a change in CD24 surface expression in response to anti-CD24 treatment (Fig.5a). The initial loss of CD24 expression seen in WEHI-231 cells was not observed in primary cells, instead mean CD24 expression was found to significantly increase over the 60-min treatment time (Fig.5b). The increase in expression was due to a majority of the cells having increased levels of CD24 expression (Fig.5b, upper panel). As seen in the representative histogram, a population of cells did not demonstrate a large increase in CD24 expression, resulting in only moderate increases in modal fluorescence intensity that did not reach statistical significance (Fig.5b, upper panel). Clearly, the heterogeneous nature of the isolated primary B cells (Fraction B to F based on CD19 expression) masks some of the changes to CD24 expression at a population level. This heterogeneity may also explain why the early decrease in CD24 expression is not seen in primary cells.
Figure 5.
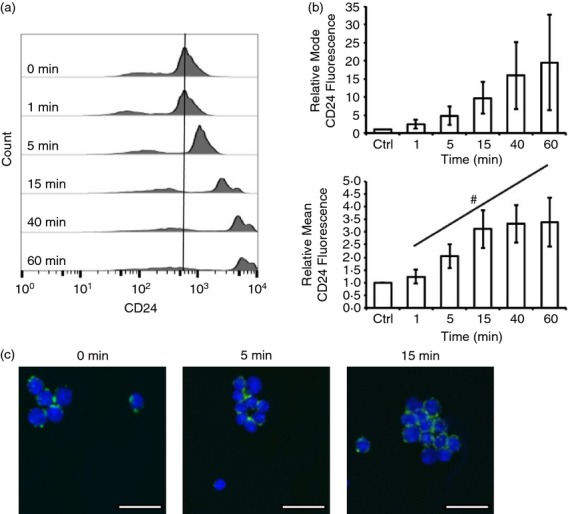
CD24 protein expression is dynamically regulated after CD24 engagement in primary B cells. (a) Primary B cells were stimulated for the indicated times with anti-CD24 primary antibody and biotinylated secondary antibody. Surface CD24 expression was assessed using streptavidin-FITC. Representative histograms are shown. (b) Mean ± SEM of the relative mean or the modal fluorescence intensity, n = 7–8, statistical significance was assessed using a one-way analysis of variance followed by a priori analysis of 0 min to 1 min via Student’s one-tailed, uneven variance t-test and from 1 min to 60 min via generalized linear model regression analysis. #P < 0·05. (c) Primary cells were stimulated as above. Surface CD24 expression was detected as above (green) and nuclei were detected by DAPI (blue). Confocal images represent three biological replicates. Scale bar = 15 μm.
We next assessed if the changes in surface expression were reflected in changes to CD24 localization in primary B cells. We found that in untreated cells, there was punctate staining of surface CD24 that was distributed around the cell membrane (Fig.5c). By 15 min of CD24 engagement, surface CD24 appeared less punctate, occupied the plasma membrane more, and tended to cluster to one side of the cell, predominantly on the surface adjoining a neighbouring cell. Of note, the clusters observed upon stimulation of CD24 are consistent with previous reports that clearly demonstrate that CD24 regulates adhesion of B cells,59 potentially by regulating integrin occupancy of lipid rafts.60 We observed more heterogeneity in surface expression of CD24 in cells imaged by confocal microscopy compared with flow cytometry, probably because of the increased sensitivity of flow cytometry to low-intensity fluorescence signals.61
Hence, the engagement of CD24 on both cell types caused a dynamic change in its own surface expression with both cell types similarly gaining surface expression in a time-dependent manner.
The dynamic regulation of CD24 protein expression does not depend on classical endocytosis or exocytosis processes
The observed changes to CD24 surface expression may be associated with endocytosis and/or exocytosis as seen for other cell surface molecules. For example, endocytosis following stimulation is a common event observed with several other glycophosphatidylinositol-linked cell receptors, such as CD48,62 and CD55.63 To test this hypothesis, we disrupted endocytosis or exocytosis in WEHI-231 cells through chemical inhibition of the respective processes. Primary cells were not able to withstand treatment with the chemical inhibitors without undergoing cell death so these cells were not analysed further. Endocytosis was disrupted by pre-incubation with Pitstop2, which disrupts both clathrin-mediated and non-clathrin-mediated endocytosis, or Dynasore, which prevents dynamin-mediated internalization. Analysis of surface expression showed that Pitstop 2 pre-treatment alone significantly increased CD24 protein expression on the cell surface (Fig.6a). However, Pitstop2 pre-treatment did not prevent the loss or subsequent increase in CD24 surface expression. Treatment with Dynasore had no effect on either the decrease or increase in CD24 surface expression in response to antibody-mediated cross-linking (Fig.6a).
Figure 6.
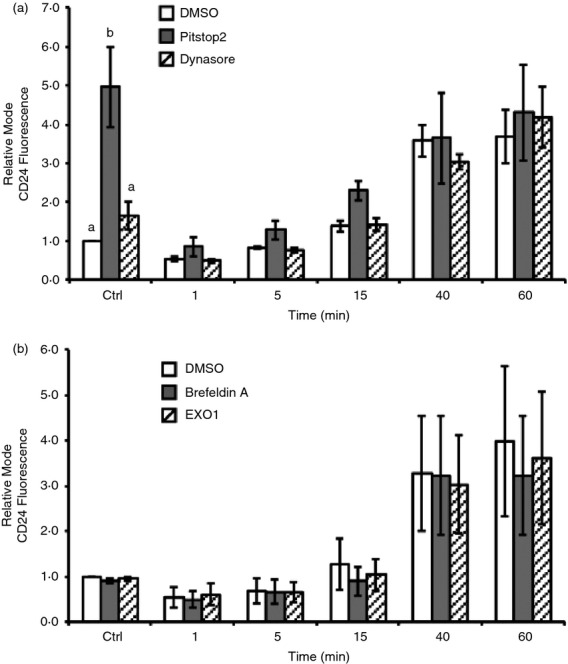
Changes to CD24 cell surface expression are not dependent on classical endocytosis or exocytosis. WEHI-231 cells were pre-treated with (a) Dynasore, or Pitstop2, and (b) Brefeldin A, or Exo1. DMSO was used as a vehicle control in all cases. Cells were treated as described for Fig.4 and CD24 was detected using streptavidin-FITC. Mean ± SEM of the relative modal fluorescence intensity relative to DMSO control treated (Ctrl) is shown, n = 3, statistical significance was determined by a priori analysis of each time-point using one-way analysis of variance, different letters represent different groups at P < 0·05.
To determine if the dynamic changes in CD24 surface expression depend on exocytosis, a process that regulates the formation of exosomes64 and synaptic vesicle release,65 cells were pre-treated with the endoplasmic reticulum to Golgi transport inhibitors Exo1 or Brefeldin A. We found that treatment with either of these inhibitors did not alter CD24 dynamics at any time-point examined, indicating that CD24 surface protein levels are not regulated via classical exocytosis (Fig.6b).
CD24 is released from the cell surface in extracellular microvesicles
As neither endocytosis nor exocytosis was involved in regulating the levels of CD24 surface expression, we next analysed if CD24 could be lost from the cell surface by the generation of plasma membrane-derived EMV released into the extracellular environment. We first analysed changes in size and surface complexity of B cells using flow cytometry following antibody-mediated cross-linking. Both WEHI-231 cells and primary cells showed a large increase in sub-cellular-sized objects within 15 min of CD24 engagement (Fig.7a), a time-point that is too early to be associated with the generation of apoptotic bodies. In addition, the appearance of this sub-cellular fraction was coincident with the time-frame showing dynamic changes in CD24 surface expression. Given the limitations of the FACSCalibur, it is not possible to accurately analyse particles that are in the size range of EMV.66,67
Figure 7.

CD24 antibody-mediated cross-linking induces release of extracellular microvesicles (EMV). (a) WEHI-231 and Primary B cells stimulated with primary and secondary antibody as described for Fig.1, for the indicated times. Representative dot plots are shown with the percentage of sub-cellular sized objects in the upper left corner, n = 7 or n = 8 for primary, n = 3 for WEHI-231. (b) Transmission electron micrographs of WEHI-231 treated with anti-CD24 primary and secondary antibodies for the indicated times. Scale bar = 2 μm for the main image and 1 μm for the inset. (c) Box-and-whisker plot of vesicle sizes observed from the transmission electron microscope. A minimum of three images were analysed with 17, 58 and 46 EMV analysed at 0, 5 and 15 min, respectively. No significant differences in EMV size were found by one-way analysis of variance. (d) Microvesicles isolated from WEHI-231 cells after 15 min of treatment with anti-CD24 primary and eFluor660-labelled secondary antibody analysed by confocal microscopy. CD24 (purple), Annexin V-Alexa488 (green) and DAPI (blue) were detected by confocal microscopy. Note: no DAPI-positive particles were observed. Scale bar = 10 μm. (e) Analysis of isolated microvesicles from WEHI-231 cells. Cells were left unstimulated for 60 min (ctrl) or stimulated with anti-CD24 primary and biotinylated secondary anti-rat antibody for 15 or 60 min. Representative dot plots of isolated vesicles were detected using AnnexinV-Alexa488 and streptavidin-allophycocyanin (APC), after gating on the EMV-sized population, n = 4. (f) Mean ± SEM of the total percentage of AnnexinV+ EMV (upper panel) and the percentage of AnnexinV+ EMV that are CD24+ (lower panel). n = 4, significance was assessed via one-way analysis of variance followed by Tukey post-hoc. Letters represent different groups at P < 0·05.
Therefore, to determine if WEHI-231 cells could produce EMV in response to CD24 engagement, we used TEM to image WEHI-231 cells after treatment with anti-CD24 primary and secondary antibodies (Fig.7b). These images show that engagement of CD24 causes the generation of large numbers of small, plasma membrane-derived EMV within 5 min. By 15 min, the number of EMV closely associated with the cell is reduced, a time-point that is coincident with the appearance of large numbers of sub-cellular sized particles in the bulk population.
Plasma membrane-derived EMV are heterogeneous in both composition and size, ranging from 100 nm to potentially 1000 nm in diameter.55,56 The EMV imaged by TEM ranged in size between 78 nm and 511 nm, with no significant change in size in response to CD24 engagement (Fig.7c). The morphology of these vesicles by TEM indicates that they are formed and released directly by budding from the plasma membrane, a defining characteristic of EMV.68 In contrast, exosomes accumulate in cytosolic multivesicular bodies, which then fuse with the plasma membrane to release the exosomes.68 Apoptotic bodies are much larger than either exosomes or EMV and are associated with fragmentation of the entire cell, including the nucleus and other organelles.68,69
Although a definitive surface marker for EMV has not yet been identified they are unlike exosomes in that they are enriched in surface phosphatidylserine,43,69 which can be detected by Annexin V binding. Hence, to further characterize the plasma-membrane derived vesicles, we viewed EMV that has been isolated from WEHI-231 cells after 15 min of CD24 engagement under confocal microscopy (Fig.7d). The isolated EMV are sub-cellular sized and do not contain DNA as evidenced by their DAPI-negative character. Moreover, CD24 co-localized with Annexin V in a portion of these EMV, demonstrating that the EMV maintained the same orientation of surface proteins as the plasma membrane. When treated with Triton X-100, a non-ionic detergent that disrupts the plasma membrane, we observed an essentially complete loss of Annexin V-positive particles (data not shown), further demonstrating that these are membrane-bound particles.
To quantify the number of EMV that contain CD24, we analysed isolated EMV released after treatment of WEHI-231 cells using the FACSAria II, which is capable of identifying particles < 1 µm in diameter. Using established strategies, we gated on particles in the size range of 0·2–0·4 µm and quantified the number of Annexin V-positive and CD24-positive EMV released after 15 or 60 min of treatment (Fig.7e, f). Overall, we found that supernatant from unstimulated cells contained an average of 6·9 × 105 ± 3·1 × 105 EMV-sized particle after 60 min at 37° (Fig.7e) consistent with a recent report showing that the release of EMV may be an innate biological process in B cells.26 After 15 or 60 min of antibody stimulation, an average of 5·1 × 105 ± 4 × 104 to 5·6 × 105 ± 1·7 × 104 EMV-sized particles were detected, respectively (Fig.7e); however, this was not significantly different from control cells when analysed by one-way analysis of variance (P > 0·05).
In contrast, Annexin V-positive particles, which are generated from and retain the orientation of the plasma membrane, increased significantly following 15 min and 60 min of CD24 stimulation (Fig.7f). Moreover, the percentage of Annexin V-positive EMV that carried CD24 increased significantly from 23·1 ± 1·9% in control cells to 47·8 ± 8·8% and 61·8 ± 3·5% after 15 min and 60 min, respectively (Fig.7f). These findings show that engagement of CD24 promotes the formation of CD24-bearing EMV, in addition to altering the composition of EMV released by B cells.
CD24-bearing EMV can transport CD24 between cells
The EMV can be taken up by other cells via multiple mechanisms to deliver both their intracellular and membrane-bound cargo.68,70 Therefore, we next investigated if the CD24-bearing EMV generated in response to CD24 engagement can be taken up by neighbouring cells and so potentially participate in cell–cell communication. We independently stimulated two different populations of WEHI-231 cells for 15 min with primary and secondary antibody and labelled each with either streptavidin-conjugated FITC or eFluor660. After removal of excess antibody, the differentially labelled cells were mixed together for up to 60 min at 37°. We found that upon mixing, there was a time- and temperature-dependent exchange of CD24 between both WEHI-231 cells and primary cells (Fig.8a, b). When WEHI-231 cells were mixed on ice (i.e. Ctrl), fewer than 5% were double-labelled. There was a statistically significant time-dependent increase in the amount of cells containing both labels over 60 min of co-incubation (Fig.8a, right panel). In contrast, if the cells were paraformaldehyde-fixed before mixing, the exchange of CD24 between WEHI-231 cells was nearly abolished and did not vary with time (Fig.8a). Hence, the exchange of CD24 was dependent on active cellular processes and not due to movement of the anti-CD24 primary antibody, secondary antibody, or streptavidin-conjugated fluorophore from one cell population to the other.
Figure 8.
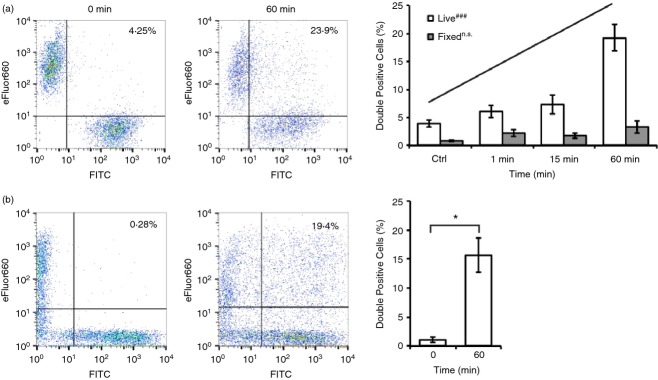
CD24 protein is re-distributed within the B-cell population in response to engagement of CD24. (a) WEHI-231 cells were treated with anti-CD24 primary and secondary anti-rat antibodies conjugated to either eFluor660 or FITC. The two populations of cells (live or fixed) were then mixed and left on ice or incubated at 37° for up to 60 min. Representative dot plots (left panel) and mean ± SEM of the percent of double-positive cells (right panel) are shown (n = 3), statistical significance was determined by one-way analysis of variance followed by generalized linear model regression analysis, n.s.not significant, ###P < 0·001. (b) Primary B cells were treated with anti-CD24 primary and secondary anti-rat antibodies. The two populations were mixed immediately before analysis, or were mixed for 60 min at 37°. Representative dot plots (left panel) and the mean ± SEM of the percent of double-positive cells (right panel) are shown (n = 3), statistical significance was determined by Student’s t-test, *P < 0·05.
We found that primary cells also exchanged CD24 when differentially labelled populations were mixed in a similar manner to WEHI-231 cells (Fig.8b). Hence, engagement of CD24 causes a clear exchange of CD24 protein within a homogeneous population of cells in the case of WEHI-231, or a more heterogeneous population of bone-marrow derived B cells at various stages of development.
Discussion
Using bioinformatics analysis of the transcriptomic profiles of developing B cells, we generated the hypothesis that CD24 engagement may be associated with vesicle trafficking. We then validated this hypothesis at the functional level to show that EMV are generated in response to antibody-mediated CD24 engagement, which promotes its re-distribution throughout a population of cells.
CD24-mediated cell death has been observed in several mouse and human cell lines, as well as in longer-term, in vitro cultures of murine bone marrow extracts co-cultured with bone marrow stromal cells.5,13,14 This study is the first to establish that primary immature bone marrow cells also undergo CD24-mediated apoptosis ex vivo as established by the increase in Annexin V-expressing cells and the activation of Caspase-3/7 (Figs3). We have also clearly established that the WEHI-231 mouse B-cell lymphoma cell line can also undergo CD24-mediated apoptosis, in contrast to a previous anecdotal statement.13 Unlike previous reports, in which engagement of CD24 by primary antibody alone was sufficient to induce apoptosis,13 we found that both primary immature B cells and WEHI-231 cells require more extensive cross-linking of CD24 via primary and secondary antibodies to initiate a substantial increase in apoptosis. Therefore, our findings suggest that CD24 signalling requires a minimum threshold of stimulation that is surpassed upon the additional clustering of CD24 or increased avidity of antibody binding.
During our validation of primary bone marrow-derived B cells and WEHI-231 cells as appropriate models for understanding CD24-mediated signalling, we also refined the timeline of events related to CD24 signalling with respect to the induction of apoptosis. We found that upon engagement of CD24 caspase-3 and -7 activity is substantially and significantly up-regulated within 3 hr in both primary cells and WEHI-231 cells. The caspase family is divided into initiator caspases, such as caspase-8 and executioner caspases, such as caspase-3 and caspase-7.71 Activation of caspases-3 and -8, as detected by Western blot, occurs within 24 hr of anti-CD24 antibody stimulation in Pre-B HPB-Null cells.14 Our data clearly demonstrate that CD24 signalling engages apoptotic machinery in primary B cells and WEHI-213 cells by 3 hr. Furthermore, our results show that removing unbound antibody after a 15-min incubation, which is before the highest levels of CD24 expression are reached in the timeframe examined, reduced the degree of apoptosis within the population. This suggests that the presence of the higher levels of surface CD24 may be necessary to promote maximal apoptosis following CD24 engagement.
Our bioinformatics analysis of CD24 expression has revealed novel associations between CD24 and potential effector partners. It has long been known that CD24 expression is dynamic over the course of B-cell development.1,6–9 We show here that CD24 is closely co-regulated during B-cell development with genes associated with regulation of proliferation and apoptosis, cellular organization, including vesicle trafficking, developmental processes and response to stimuli. The Guilt-by-Association theory31 suggests that these processes could be related to CD24 activity. Identification of the regulation of apoptosis as an enriched GO term validates this strategy for identification of new partners for CD24. However, the most abundant GO terms were associated with the regulation of cellular organization, in particular organelle and vesicle transport, processes that have not been previously shown to be regulated by CD24.
In support of our hypothesis that CD24 is associated with cellular organization, we found that CD24 surface protein expression is complex and dynamic in response to antibody-mediated engagement. In primary B cells, we found that CD24 surface expression increases in response to antibody engagement, whereas WEHI-231 cells show an initial decline, followed by a sustained increase in CD24 surface expression. These data demonstrate that in addition to its regulation during B-cell development, engagement of CD24 can rapidly alter its own expression at the single cell level. Hence, these data demonstrate that there is a positive feedback loop that increases CD24 surface expression in response to its own engagement.
For the first time, we have established that there is a loss of CD24 surface expression that occurs within 1 min in WEHI-231 cells, which is not due to classical exocytosis or endocytosis but via loss of CD24 protein on EMV. Through both TEM and flow cytometry, we have established that EMV appear within 15 min of antibody stimulation and continue to accumulate in the extracellular space through 60 min of stimulation. In addition, we observed an increase in both the total percentage of Annexin V-positive EMV and the percentage of CD24-bearing EMV following CD24 stimulation. There is not a significant increase in the percentage of CD24 + EMV at 60 min compared to 15 min after CD24 engagement. This probably reflects the steady-state equilibrium of EMV where EMV are continually being released and taken up by the cells.
The loss of CD24 due to release of CD24-bearing EMV is consistent with the lack of effect of both endocytosis and exocytosis inhibitors on CD24 surface expression. Previously, CD24 has been associated with exosomes found in urine and amniotic fluid that were secreted by maternal and fetal kidney cells.72 However, as these CD24-bearing vesicles express phosphatidylserine, are not associated with the appearance of secretory vesicles, and have an average size of 165 ± 5 nm, these EMV cannot be classified as exosomes.73 The appearance of CD24 on plasma-membrane-derived EMV is consistent with a general role for CD24 on membrane-bound structures released from cells.
The gain of CD24 surface expression that occurs over the course of 60 min in WEHI-231 cells is not mediated via classical exocytosis pathways, which transport protein from the endoplasmic reticulum via the Golgi to the cell surface. Our data clearly show that isolated EMV can mediate the exchange of CD24 between antibody-stimulated cells. There is a broader distribution of CD24 surface expression at later time-points suggesting that some cells acquire more CD24 than other cells. Therefore, at least some of the increase in CD24 surface protein may be due to the uptake of CD24-bearing EMV. However, as both the mean and mode of CD24 expression increase at the single cell level, there must be additional mechanisms that increase CD24 expression, which are independent of endoplasmic reticulum to Golgi trafficking. For example, it is possible that the increase in CD24 surface expression is due to alterations in epitope availability due to conformational changes in response to CD24 engagement or due to fusion of pre-formed vesicles proximal to the plasma membrane.
Altough the increase in CD24 surface expression in primary B cells occurs within the same timeframe as in WEHI-231 cells, we did not observe the initial loss of expression. Isolated bone marrow-derived primary B cells are at multiple stages of B-cell development (Fraction B to F based on CD19 expression). Therefore, small but biologically significant changes in expression may be masked by the heterogeneity in the starting population leading to variation in the timing of the response. Nonetheless, there is clearly a rapidly activated, positive feedback loop from CD24 engagement leading to increases in CD24 surface expression in primary B cells, similar to that seen in the WEHI-231 cell line.
In addition to the overall increase in CD24 surface expression, we also observed a redistribution in the localization of CD24 surface protein in response to engagement. CD24 changed from a punctate organization to more highly organized and polarized distribution. The polarization of CD24 tended to occur at regions between cells, supporting previous work showing that CD24 can increase cell–cell adhesion.59 However, we cannot conclusively rule out the possibility that the addition of streptavidin-FITC, used to detect CD24, increases the degree of clustering of these cells. Nevertheless, the clustering, in combination with the observation that these cells release CD24-bearing EMV, suggest that engagement of CD24 could preferentially affect neighbouring cells via the directed exchange of CD24 protein.
Previous studies on EMV have shown they can participate in a wide range of cell processes including the regulation of lymphocyte activation, cellular proliferation, the transfer of signalling molecules, the epigenetic modification of cells and the delivery of active second messengers or microRNAs into cells.43,55,56,68,70,74,75 The transfer of EMV in blood transfusions can deliver CD55 and CD59, inhibitors of complement-mediated lysis, to host red blood cells, preventing their complement-mediated destruction.76,77 Although defining the precise function of CD24-bearing EMV in B-cell development requires further investigation, given their broad range of functions and their ubiquitous nature, the production of these CD24-bearing microvesicles from immature B cells may have the potential to affect differentiating B cells as well as the supporting stromal cell environment.
Acknowledgments
We thank Dr Nicholas Fairbridge for critical discussions and Dr David Schneider for statistical advice. DCA and SLC conceived the idea and designed the experiments. DCA, ME, NCS, ESM, and AMH designed, performed, and/or analysed the experiments. DCA and SLC wrote the manuscript. All authors approved the final manuscript. This work was funded by NSERC and a Beatrice Hunter Cancer Research Institute New Investigator Award to SLC. DCA is supported by a trainee award from the Beatrice Hunter Cancer Research Institute with funds provided by The Terry Fox Strategic Health Research Training Program in Cancer Research at CIHR and by Memorial University of Newfoundland.
Glossary
- APC
allophycocyanin
- B cell
B lymphocyte
- DAPI
4′,6-diamidino-2-phenylindole
- EMV
extracellular microvesicles
- GEO
Gene Expression Omnibus
- GO
gene ontology
- Pre-B
precursor B
- Pro-B
progenitor B
- QS
Quin saline
- TEM
transmission electron microscopy
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Table S1. AmiGO2 annotation of CD24 co-expressed genes
Table S2. AmiGO2 gene ontology enrichment analysis
Table S3. GeneMANIA gene ontologyenrichment analysis
Table S4. Gene lists
References
- Hardy RR, Carmack CE, Shinton SA, Kemp JD, Hayakawa K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J Exp Med. 1991;173:1213–25. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- Kay R, Rosten PM, Humphries RK. CD24, a signal transducer modulating B cell activation responses, is a very short peptide with a glycosyl phosphatidylinositol membrane anchor. J Immunol. 1991;147:1412–6. [PubMed] [Google Scholar]
- Fischer GF, Majdic O, Gadd S, Knapp W. Signal transduction in lymphocytic and myeloid cells via CD24, a new member of phosphoinositol-anchored membrane molecules. J Immunol. 1990;144:638–41. [PubMed] [Google Scholar]
- Suzuki T, Kiyokawa N, Taguchi T, Sekino T, Katagiri YU, Fujimoto J. CD24 induces apoptosis in human B cells via the glycolipid-enriched membrane domains/rafts-mediated signaling system. J Immunol. 2001;166:5567–77. doi: 10.4049/jimmunol.166.9.5567. [DOI] [PubMed] [Google Scholar]
- Hough MR, Chappel MS, Sauvageau G, Takei F, Kay R, Humphries RK. Reduction of early B lymphocyte precursors in transgenic mice overexpressing the murine heat-stable antigen. J Immunol. 1996;156:479–88. [PubMed] [Google Scholar]
- Lu L, Chappel MS, Humphries RK, Osmond DG. Regulation of cell survival during B lymphopoiesis: increased pre-B cell apoptosis in CD24-transgenic mouse bone marrow. Eur J Immunol. 2000;30:2686–91. doi: 10.1002/1521-4141(200009)30:9<2686::AID-IMMU2686>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Wenger RH, Kopf M, Nitschke L, Lamers MC, Köhler G, Nielsen PJ. B-cell maturation in chimaeric mice deficient for the heat stable antigen (HSA/mouse CD24) Transgenic Res. 1995;4:173–83. doi: 10.1007/BF01968782. [DOI] [PubMed] [Google Scholar]
- Nielsen PJ, Lorenz B, Muller AM, et al. Altered erythrocytes and a leaky block in B-cell development in CD24/HSA-deficient mice. Blood. 1997;89:1058–67. [PubMed] [Google Scholar]
- Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323:1722–5. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene R, Yang H, Kutsche M, Schachner M. The neural recognition molecule L1 is a sialic acid-binding lectin for CD24, which induces promotion and inhibition of neurite outgrowth. J Biol Chem. 2001;276:21656–63. doi: 10.1074/jbc.M101790200. [DOI] [PubMed] [Google Scholar]
- Sammar M, Aigner S, Hubbe M, Schirrmacher V, Schachner M, Vestweber D, Altevogt P. Heat-stable antigen (CD24) as ligand for mouse P-selectin. Int Immunol. 1994;6:1027–36. doi: 10.1093/intimm/6.7.1027. [DOI] [PubMed] [Google Scholar]
- Chappel MS, Hough MR, Mittel A, Takei F, Kay R, Humphries RK. Cross-linking the murine heat-stable antigen induces apoptosis in B cell precursors and suppresses the anti-CD40-induced proliferation of mature resting B lymphocytes. J Exp Med. 1996;184:1639–49. doi: 10.1084/jem.184.5.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi T, Kiyokawa N, Mimori H, et al. Pre-B cell antigen receptor-mediated signal inhibits CD24-induced apoptosis in human Pre-B Cells. J Immunol. 2003;170:252–60. doi: 10.4049/jimmunol.170.1.252. [DOI] [PubMed] [Google Scholar]
- Heng TS, Painter MW. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–4. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. [Google Scholar]
- Faria JC. Resources of Tinn-R GUI/Editor for R Environment. Ilheus, Bahia, Brasil: Universidade Estadual de Santa Cruz; 2012. [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–7. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:1–25. doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- MacDonald JW. Affycoretools: Functions useful for those doing repetitive analyses with Affymetrix GeneChips. R package version 1.40.5: 2008. [Google Scholar]
- Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–8. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl. 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–9. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra H, Simpson RJ, Ji H, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012;10:e1001450. doi: 10.1371/journal.pbio.1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 2010;115:1755–64. doi: 10.1182/blood-2009-09-242719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescitelli R, Lässer C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzás EI, Lötvall J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles. 2013;2:20677. doi: 10.3402/jev.v2i0.20677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix R, Robert S, Poncelet P, Dignat-George F. Overcoming limitations of microparticle measurement by flow cytometry. Semin Thromb Hemost. 2010;36:807–18. doi: 10.1055/s-0030-1267034. [DOI] [PubMed] [Google Scholar]
- Poncelet P, Robert S, Bouriche T, Bez J, Lacroix R, Dignat-George F. Standardized counting of circulating platelet microparticles using currently available flow cytometers and scatter-based triggering: forward or side scatter? Cytometry A. 2015 doi: 10.1002/cyto.a.22685. ePub ahead of print. DOI: 10.1002/cyto.a.22685. [DOI] [PubMed] [Google Scholar]
- Lindstrom ML, Bates DM. Nonlinear mixed effects models for repeated measures data. Biometrics. 1990;46:673–87. [PubMed] [Google Scholar]
- Wolfe CJ, Kohane IS, Butte AJ. Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinformatics. 2005;6:227. doi: 10.1186/1471-2105-6-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Soriano NA, Bose S, et al. EHD2 and the novel EH domain binding protein EHBP1 couple endocytosis to the actin cytoskeleton. J Biol Chem. 2004;279:10593–605. doi: 10.1074/jbc.M307702200. [DOI] [PubMed] [Google Scholar]
- Moren B, Shah C, Howes MT, Schieber NL, McMahon HT, Parton RG, Daumke O, Lundmark R. EHD2 regulates caveolar dynamics via ATP-driven targeting and oligomerization. Mol Biol Cell. 2012;23:1316–29. doi: 10.1091/mbc.E11-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MR, Maritzen T, Kukhtina V, et al. Regulation of endosomal membrane traffic by a Gadkin/AP-1/kinesin KIF5 complex. Proc Natl Acad Sci U S A. 2009;106:15344–9. doi: 10.1073/pnas.0904268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Boucrot E, Meinecke M, Evergren E, Vallis Y, Mittal R, McMahon HT. FCHo proteins are nucleators of clathrin-mediated endocytosis. Science. 2010;328:1281–4. doi: 10.1126/science.1188462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons M, Rusk N. Control of vesicular trafficking by Rho GTPases. Curr Biol. 2003;13:R409–18. doi: 10.1016/s0960-9822(03)00324-5. [DOI] [PubMed] [Google Scholar]
- Hong BS, Cho JH, Kim H, et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genom. 2009;10:556. doi: 10.1186/1471-2164-10-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, van Rijn S. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood SK, Mambula SS, Gray PJ., Jr Extracellular heat shock proteins in cell signaling and immunity. Ann N Y Acad Sci. 2007;1113:28–39. doi: 10.1196/annals.1391.019. [DOI] [PubMed] [Google Scholar]
- Calderwood SK, Mambula SS, Gray PJ, Jr, Theriault JR. Extracellular heat shock proteins in cell signaling. FEBS Lett. 2007;581:3689–94. doi: 10.1016/j.febslet.2007.04.044. [DOI] [PubMed] [Google Scholar]
- De Maio A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones. 2011;16:235–49. doi: 10.1007/s12192-010-0236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63:2560–70. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–22. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- Nairn AC, Aderem A. Calmodulin and protein kinase C cross-talk: the MARCKS protein is an actin filament and plasma membrane cross-linking protein regulated by protein kinase C phosphorylation and by calmodulin. Ciba Found Symp. 1992;164:145–54. doi: 10.1002/9780470514207.ch10. discussion 54-61. [DOI] [PubMed] [Google Scholar]
- Grubinger M, Gimona M. CRP2 is an autonomous actin-binding protein. FEBS Lett. 2004;557:88–92. doi: 10.1016/s0014-5793(03)01451-0. [DOI] [PubMed] [Google Scholar]
- Chen D, Fang F, Yang Y, Chen J, Xu G, Xu Y, Gao Y. Brahma-related gene 1 (Brg1) epigenetically regulates CAM activation during hypoxic pulmonary hypertension. Cardiovasc Res. 2013;100:363–73. doi: 10.1093/cvr/cvt214. [DOI] [PubMed] [Google Scholar]
- Timson DJ, Trayer HR, Trayer IP. The N-terminus of A1-type myosin essential light chains binds actin and modulates myosin motor function. Eur J Biochem. 1998;255:654–62. doi: 10.1046/j.1432-1327.1998.2550654.x. [DOI] [PubMed] [Google Scholar]
- Coutinho-Silva R, Knight GE, Burnstock G. Impairment of the splenic immune system in P2X(2)/P2X(3) knockout mice. Immunobiology. 2005;209:661–8. doi: 10.1016/j.imbio.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Pizzirani C, Ferrari D, Chiozzi P, Adinolfi E, Sandona D, Savaglio E, Di Virgilio F. Stimulation of P2 receptors causes release of IL-1β-loaded microvesicles from human dendritic cells. Blood. 2007;109:3856–64. doi: 10.1182/blood-2005-06-031377. [DOI] [PubMed] [Google Scholar]
- Frascoli M, Marcandalli J, Schenk U, Grassi F. Purinergic P2X7 receptor drives T cell lineage choice and shapes peripheral γδ cells. J Immunol. 2012;189:174–80. doi: 10.4049/jimmunol.1101582. [DOI] [PubMed] [Google Scholar]
- Gelb MH, Valentin E, Ghomashchi F, Lazdunski M, Lambeau G. Cloning and recombinant expression of a structurally novel human secreted phospholipase A2. J Biol Chem. 2000;275:39823–6. doi: 10.1074/jbc.C000671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho IC, Arm JP, Bingham CO, 3rd, Choi A, Austen KF, Glimcher LH. A novel group of phospholipase A2s preferentially expressed in type 2 helper T cells. J Biol Chem. 2001;276:18321–6. doi: 10.1074/jbc.M008837200. [DOI] [PubMed] [Google Scholar]
- Buchner G, Orfanelli U, Quaderi N, Bassi MT, Andolfi G, Ballabio A, Franco B. Identification of a new EGF-repeat-containing gene from human Xp22: a candidate for developmental disorders. Genomics. 2000;65:16–23. doi: 10.1006/geno.2000.6146. [DOI] [PubMed] [Google Scholar]
- Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20:1487–95. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annu Rev Immunol. 1990;8:773–93. doi: 10.1146/annurev.iy.08.040190.004013. [DOI] [PubMed] [Google Scholar]
- Ketchum C, Miller H, Song W, Upadhyaya A. Ligand mobility regulates B cell receptor clustering and signaling activation. Biophys J. 2014;106:26–36. doi: 10.1016/j.bpj.2013.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmon G, Eckert M, Sammar M, Schachner M, Altevogt P. Nectadrin, the heat-stable antigen, is a cell adhesion molecule. J Cell Biol. 1992;118:1245–58. doi: 10.1083/jcb.118.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz S, Mierke CT, Joumaa S, Behrens J, Fabry B, Altevogt P. CD24 induces localization of β1 integrin to lipid raft domains. Biochem Biophys Res Commun. 2008;365:35–41. doi: 10.1016/j.bbrc.2007.10.139. [DOI] [PubMed] [Google Scholar]
- Basiji DA, Ortyn WE, Liang L, Venkatachalam V, Morrissey P. Cellular image analysis and imaging by flow cytometry. Clin Lab Med. 2007;27:653–70. doi: 10.1016/j.cll.2007.05.008. viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima K, Ikehara Y, Yamashita K. Functional role played by the glycosylphosphatidylinositol anchor glycan of CD48 in interleukin-18-induced interferon-γ production. J Biol Chem. 2005;280:18056–62. doi: 10.1074/jbc.M413297200. [DOI] [PubMed] [Google Scholar]
- Deckert M, Ticchioni M, Bernard A. Endocytosis of GPI-anchored proteins in human lymphocytes: role of glycolipid-based domains, actin cytoskeleton, and protein kinases. J Cell Biol. 1996;133:791–9. doi: 10.1083/jcb.133.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;113(Pt 19):3365–74. doi: 10.1242/jcs.113.19.3365. [DOI] [PubMed] [Google Scholar]
- Lin RC, Scheller RH. Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol. 2000;16:19–49. doi: 10.1146/annurev.cellbio.16.1.19. [DOI] [PubMed] [Google Scholar]
- Ayers L, Kohler M, Harrison P, et al. Measurement of circulating cell-derived microparticles by flow cytometry: sources of variability within the assay. Thromb Res. 2011;127:370–7. doi: 10.1016/j.thromres.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Bode AP, Hickerson DH. Characterization and quantitation by flow cytometry of membranous microparticles formed during activation of platelet suspensions with ionophore or thrombin. Platelets. 2000;11:259–71. doi: 10.1080/09537100050129279. [DOI] [PubMed] [Google Scholar]
- Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78:838–48. doi: 10.1038/ki.2010.278. [DOI] [PubMed] [Google Scholar]
- Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. 2013;113:1–11. doi: 10.1007/s11060-013-1084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as mediators of intercellular communication in cancer–the emerging science of cellular ‘debris’. Semin Immunopathol. 2011;33:455–67. doi: 10.1007/s00281-011-0250-3. [DOI] [PubMed] [Google Scholar]
- McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S, Rupp C, Stoeck A, et al. CD24 is a marker of exosomes secreted into urine and amniotic fluid. Kidney Int. 2007;72:1095–102. doi: 10.1038/sj.ki.5002486. [DOI] [PubMed] [Google Scholar]
- van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64:676–705. doi: 10.1124/pr.112.005983. [DOI] [PubMed] [Google Scholar]
- Camussi G, Deregibus MC, Bruno S, Grange C, Fonsato V, Tetta C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res. 2011;1:98–110. [PMC free article] [PubMed] [Google Scholar]
- Müller G. Release of exosomes and microvesicles harbouring specific RNAs and glycosylphosphatidylinositol-anchored proteins from rat and human adipocytes is controlled by histone methylation. Am J Mol Biol. 2012;02:187–209. [Google Scholar]
- Sloand EM, Maciejewski JP, Dunn D, Moss J, Brewer B, Kirby M, Young NS. Correction of the PNH defect by GPI-anchored protein transfer. Blood. 1998;92:4439–45. [PubMed] [Google Scholar]
- Sloand EM, Mainwaring L, Keyvanfar K, Chen J, Maciejewski J, Klein HG, Young NS. Transfer of glycosylphosphatidylinositol-anchored proteins to deficient cells after erythrocyte transfusion in paroxysmal nocturnal hemoglobinuria. Blood. 2004;104:3782–8. doi: 10.1182/blood-2004-02-0645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. AmiGO2 annotation of CD24 co-expressed genes
Table S2. AmiGO2 gene ontology enrichment analysis
Table S3. GeneMANIA gene ontologyenrichment analysis
Table S4. Gene lists