

Abstract
Although bacillus Calmette–Guérin (BCG) is an established vaccine with excellent efficacy against disseminated Mycobacterium tuberculosis infection in young children, efficacy in adults suffering from respiratory tuberculosis (TB) is suboptimal. Prime-boost viral vectored vaccines have been shown to induce effective immune responses and lentivectors (LV) have been shown to improve mucosal immunity in the lung. A mucosal boost to induce local immunogenicity is also referred to as a ‘pull’ in a prime and pull approach, which has been found to be a promising vaccine strategy. The majority of infants worldwide receive BCG immunization through current vaccine protocols. We therefore aimed to investigate the role of a boost (or pull) immunization with an LV vaccine expressing the promising TB antigen (Ag85A). We immunized BALB/c mice subcutaneously with BCG or an LV vaccine expressing a nuclear factor-κB activator vFLIP together with Ag85A (LV vF/85A), then boosted with intranasal LV vF/85A. Prime and pull immunization with LV85A induced significantly enhanced CD8+ and CD4+ T-cell responses in the lung, but did not protect against intranasal BCG challenge. In contrast, little T-cell response in the lung was seen when the prime vaccine was BCG, and intranasal vF/85A provided no additional protection against mucosal BCG infection. Our study demonstrates that not all LV prime and pull approaches may be successful against TB in man and careful antigen and immune activator selection is therefore required.
Keywords: BCG, lentivector, prime and pull, prime-boost, tuberculosis, vaccine, viral vector
Introduction
Despite widespread immunization with Mycobacterium bovis bacillus Calmette–Guérin (BCG) strain at birth, M. tuberculosis remains a major public health problem. Tuberculosis (TB) was responsible for an estimated 1·5 million deaths and 9 million cases of disease worldwide in 2013.1 The live vaccine BCG is effective in protecting against disseminated TB infection and meningitis in infants and young children with an estimated efficacy of 77% and 73%, respectively2 but efficacy is limited against pulmonary TB in adults.3 In addition to considerable resultant morbidity and mortality, this lack of efficacy leads to a reservoir of infection. The respiratory route is the major route of spread of disease and an effective vaccine against pulmonary TB is therefore urgently needed. Any new vaccine schedule is unlikely to negate the need for BCG immunization, which is commonly given at birth. Improved vaccine schedules are therefore likely to comprise a prime-boost incorporating a BCG prime and a novel vaccine boost. A localized boost at the respiratory mucosa in a prime and pull approach may induce the development of a localized tissue-resident memory T-cell population at the site of potential mycobacterial exposure, as has been effective in induction of localized immunogenicity against Herpes Simplex Virus 2.4
Viral vectored vaccines have shown considerable promise against TB in pre-clinical studies. Recently the first efficacy trial of a new vaccine against TB for four decades was completed in Africa using a viral vectored vaccine, MVA85A.5 Unfortunately immunogenicity in the field was modest, with a median 136 spot-forming cells/million triple-positive CD4+ cells and no CD8+ T-cell responses were detected. This was far lower than the responses seen in volunteers in the UK, and may have been insufficient to provide protection against disease. This demonstrates the complexities of developing an effective vaccine against this common disease. Further development is therefore needed, particularly to target immunization to the induction of mucosal responses in the lung. Intranasally (i.n.) administered lentivectors (LV) can be used to induce T-cell responses in the lung in mice, and effective mucosal immunity, as has been demonstrated by their ability to protect against influenza.6 We therefore tested the ability of i.n. LV encoding Ag85A to boost BCG immunization in mice and protect animals against mucosal BCG challenge.
Materials and methods
Vectors and their preparation
DNA encoding M. tuberculosis antigen 85A (Ag85A) was a gift from Professor Helen McShane (Jenner Institute, Oxford University). Vesicular stomatitis virus G glycoprotein (VSV-G)-pseudotyped LV were produced as described previously.6 Vectors used the spleen focus-forming virus promoter to express vFLIP from Kaposi’s sarcoma-associated herpesvirus7 and the human phosphoglycerate kinase 1 promoter to express either Ag85A, green fluorescent protein (GFP), or IiOVA.8 To produce LV, 293T cells were transfected with p8.91 (gag/pol), pMD-G (VSV-G envelope) and transfer vector, using Fugene 6 (Roche, Basel, Switzerland). Supernatants were collected and concentrated by ultracentrifugation through a 20% sucrose cushion.
Lentivectors were titred by measuring vector integration in 293T cells by quantitative PCR. Three days after transduction, DNA was prepared from 293T cells using the blood and tissue kit (Qiagen, Hilden, Germany). Incorporated LV sequence was measured by quantitative PCR targeting an invariant region of the inserted LV sequence. Primers used were TGTGTGCCCGTCTGTTGTGT (forward) and GCTCTCTCGACGCAGGACTC (reverse). The 6FAM/TAMRA-labelled probe was CAGTGGCGCCCGAACAGGGA. Quantitative PCR was performed using a Realplex cycler (Eppendorf, Hamburg, Germany).
Animals
Six- to eight-week-old female BALB/c mice were obtained from Charles River Laboratories (Wilmington, MA). All mice were maintained in specific pathogen-free conditions and used in accordance with home office license PPL70/5427. Mice expressing human HLA A2 and HLA DR1, null for mouse MHC I and II, were a gift from Professor Francois Lemonnier, Institute Pasteur, France.9 Vaccines and BCG were resuspended in sterile PBS for administration via the subcutaneous (s.c.) or i.n. route. Bronchoalveolar lavage (BAL) was performed as described previously.6
Tissue processing
Lungs were digested with collagenase (Liberase; Roche) and DNase I (Sigma-Aldrich, St Louis, MO), dissociated via a 70-μm cell strainer, and mononuclear cells were purified using histopaque 1083 (Sigma). Splenocytes were treated with red blood cell lysis solution (Sigma) before washing in PBS. All cells were resuspended in RPMI-1640 medium containing 10% fetal calf serum, l-glutamine and penicillin–streptomycin.
Cell stimulation, staining and flow cytometry
Cells were re-stimulated in culture with a mixture of 66 overlapping 15mer peptides covering the entire sequence of Ag85A in equal concentrations, at 1 μg/ml for each peptide. This peptide mixture has previously been used to assess immune responses to Ag85A in vaccinated mice and humans and is referred to as 66p.10,11
Re-stimulation with 66p for 16 hr was in the presence of 1 μm brefeldin A (eBioscience, San Diego, CA). The number and nature of Ag85A-responding cells were determined by flow cytometry. The staining panel consisted of fixable viability stain e780 (eBioscience) and antibodies against the following targets: CD3 peridinin chlorophyll protein Cy5.5 (clone 17A2; Biolegend, Cambridge, UK); CD4 v500 (RM4-5, BD, Franklin Lakes, NJ); CD8 e450 (53-6.7; eBioscience); interferon-γ (IFN-γ) FITC (XMG1.2; eBioscience); tumour necrosis factor-α (TNF-α) phycoerythrin (MP6-XT22; BD). After washing, cells were blocked with anti-CD16/32 and stained for surface markers (fixable viability dye, anti-CD4, anti-CD8). Cells were washed with PBS then fixed, permeabilized and stained for all other (intracellular) targets using the Foxp3/transcription staining buffer kit (eBioscience).
Data were acquired using an LSR Fortessa cytometer (BD) running FACSDiva software (BD) and was analysed using Flowjo software (Treestar, Ashland, OR).
BCG
BCG Pasteur strain was a gift from Helen McShane, Jenner Institute, Oxford University. BCG SSI was obtained from Statens Serum Institut. BCG was grown in Middlebrook 7H9 media (BD) supplemented with 10% ADC (Sigma), which included 0·05% Tween-80. Colony-forming units were counted by plating onto Middlebrook 7H10 agar (BD) supplemented with 10% Oleic Albumin Dextrose Catalase (OADC) (Sigma) and culturing at 37° until colonies were evident (3 weeks).
BCG was released from mouse tissues by homogenizing excised lung or spleen in PBS using M tubes on a Gentlemax (Miltenyi Biotec, Bergisch Gladbach, Germany), following the pre-installed RNA_01 protocol. Mycobacterial suspensions were diluted and plated onto 7H10/OADC plates for colony counting.
Data presentation and statistical analysis
Data from BAL are shown as total cell counts. From the lung homogenates, the processing of lung may have influenced the recovery of cells and therefore total T-cell numbers. Lung homogenate data are therefore reported as antigen-responding cells as a percentage of total cells rather than as absolute numbers. Numbers and percentages were obtained by subtracting the response of cells (or %) stimulated with the Ag85A peptide mix from that of unstimulated cells.
All data were analysed using Graphpad Prism (GraphPad Software, Inc., La Jolla, CA). Statistical tests applied to each data set are indicated in the relevant figure legend. Data are given as mean or mean ± standard error of the mean (SEM) unless otherwise stated. Data were routinely analysed by one-way analysis of variance, with Bonferroni post-tests unless stated. P < 0·05 was considered significant.
Results
Immunization with LV vFLIP/Ag85A (vF/85A) produces Ag85A-responding T cells
We have previously demonstrated that an LV expressing influenza nucleoprotein (NP), together with the nuclear factor-κB activator vFLIP (vF/NP), can protect mice from lethal influenza challenge.6 In this study a subcutaneous (s.c.) vF/NP prime, followed by an i.n.) vF/NP boost, recruited NP-specific T cells to the lung and completely prevented influenza-associated weight loss and mortality.6 We therefore constructed an LV expressing the mycobacterial antigen Ag85A10 together with vFLIP (vF/85A), and control LV with Ag85A without vFLIP (85A), or with vFLIP and the control antigens GFP (vF/GFP) or IiOVA 8 (vF/OVA). These LV were used to immunize mice, using s.c. prime and an i.n. boost. Antigen-specific cellular immune responses in spleen, BAL and lung homogenate were assessed by re-stimulation of cells with a mixture of peptides spanning the Ag85A protein. Cytokine production was measured using intracellular staining for IFN-γ and TNF-α with the gating strategy shown the Supplementary material (Fig. S1), the majority of Ag85A-responding cells were positive for both IFN-γ and TNF-α. The IFN-γ response is therefore shown in Fig.1 with the TNF-α response presented in the Supplementary material (Fig. S2).
Figure 1.

Lentivectors (LV) encoding Ag85A recruit antigen-specific T cells. BALB/c mice were immunized as shown with vehicle control (saline) or LV encoding vFLIP (vF), Ag85A (85A), IiOVA (OVA), singly or in combination. Priming comprised 4 × 106 TU injected subcutaneously (s.c.) and boosting comprised 6 × 106 TU administered intranasally (i.n.) 2 weeks later. Bronchoalveolar lavage (BAL) (a,d), lung (b,e) and spleen (c,f) lymphocytes were retrieved 2 weeks following the final immunization, some were stimulated with Ag85A peptides (filled bars) others remained unstimulated (open bars), then cells were stained to identify IFNγ in CD4+ (d,e,f) and CD8+ (a,b,c) T cells (values are mean ± SEM, n = 6 except negative control n = 2). Antigen-specific responses were calculated for each group and responses were compared to administration of saline alone (*P < 0·05 or **P < 0·01) or LV vF/85A prime followed by saline boost ($P < 0·05; $$P < 0·01).
Figure1 shows that the s.c. priming dose of vF/85A led to production of Ag85A-specific CD8+ IFN-γ T cells, but not CD4+ IFN-γ T cells, in the spleen (Fig.1). Boosting i.n. with LV expressing Ag85A or vF/85A significantly increased Ag85A-specific CD8+ T cells in the spleen and lung, but only vF/85A significantly increased Ag85A-specific CD8+ T cells in BAL (Fig.1). Prime-boost with vF/85A resulted in a mean of 32 000 Ag85A-specific CD8+ T cells in the BAL, 50% of the total BAL CD8+ T cells. Antigen-specific CD4+ T cells were also increased in the lung by vF/85A priming followed by either Ag85A or vF/85A boosting, though CD4+ responses (up to 1% of total CD4+ T cells in lung) were lower than CD8+ responses. From these data we chose to proceed with vF/85A as our LV immunogen.
Maintenance of T cells in the airway after immunization
To determine how long Ag85A-specific T cells were maintained in the airway and lung after immunization, mice were immunized with LV vF/85A prime/pull and T-cell responses were determined at 2, 4 and 7 weeks post boost (see Supplementary material, Fig. S3). Numbers of both CD8+ and CD4+ T cells in the lungs and BAL declined after 2 weeks. The decline in CD8+ and CD4+ T cells seen after 2 weeks persisted even if a higher dose of LV vF/Ag85A was used to boost (data not shown). Equivalent immunization was seen if the time between prime and boost was increased from 2 weeks to 12 or 30 weeks (data not shown), which supports our previous data that following an s.c. prime, the CD8+ T cells reside in the spleen and are therefore available for boost at later intervals.6
LV T-cell responses in mice expressing MHC I and II
Mice null for murine MHC I and II, expressing human HLA-A2 and HLA-DR1, were used to determine the feasibility of using LV vF/85A prime-boost immunization to recruit Ag85A-responding cells to the human lung. An initial experiment showed a weaker response to LV vF/Ag85A immunization in these mice, so an increased dose of LV vF/85A was used in this experiment (1 × 107 Transforming units (TU) prime, 1·3 × 107 TU boost). CD8+ and CD4+ IFN-γ+ cells were recruited to the spleen following LV vF/85A prime, and a boost immunization led to significant recruitment of these antigen-specific CD8+ cells to the airway (BAL) and lung (Fig.2).
Figure 2.
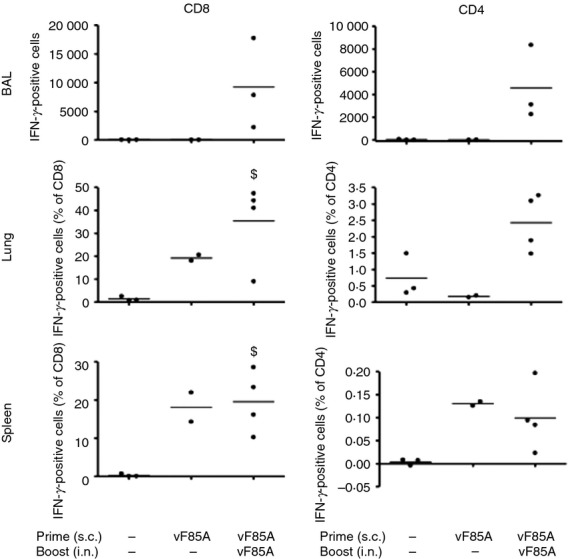
Lentivector (LV) vF/85A stimulates T cells in HLA-A2/DR1 mice. HLA A2 DR1 C57BL/6 mice, expressing human but not mouse MHC molecules, were immunized subcutaneously (s.c., n = 3) with 1 × 107 TU LV vF/85A (LV) or saline as shown. After 2 weeks mice were boosted intranasally (i.n.) with1·3 × 107 TU LV vF/85A or saline. Bronchoalveolar lavage (BAL), lung and spleen T cells were retrieved, stimulated and analysed as in Fig.1. $P < 0·05 versus control (saline alone). This experiment was performed only once.
LV immunization does not does enhance response to a BCG vaccination
New TB vaccines will probably be introduced to a population already immunized with BCG. We therefore examined mice that were immunized s.c. with BCG and then boosted 8 weeks later with LV vF/85A i.n. (Fig.3a). An 8-week interval was selected to optimize the memory response to BCG prime.12 Following BCG prime and LV vF/85A boost, the number of Ag85A-responding cells was not significantly increased when compared with responses to BCG alone in the BAL (Fig.3a). A similar lack of increased response was seen with different strains of BCG and different doses of LV vF/85A (see Supplementary material, Table S1). Hence LV vF/85A prime/pull immunization induced robust Ag85A responses in the lung, whereas BCG prime with LV vF/85A pull did not (Fig.3a and see Supplementary material, Table S1).
Figure 3.
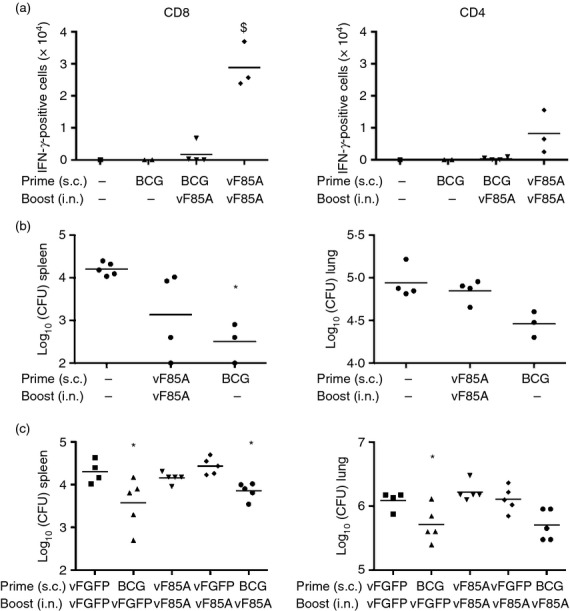
Lentivector (LV) vF/85A does not improve efficacy of bacillus Calmette–Guérin (BCG) vaccination. (a). BALB/c mice (n = 1 to n = 4 as shown) were immunized with saline, BCG [4 × 105 colony-forming units (CFU) subcutaneously (s.c.)] or LV vF/85A (4 × 106 TU s.c.) then boosted after 8 weeks with LV vF/85A [6 × 106 TU intranasally (i.n.)] or saline as shown. T cells were collected after 2 weeks from bronchoalveolar lavage (BAL), stimulated and analysed as Figure1. $P < 0·05 versus BCG. (b) BALB/c mice were immunized with LV vF/85A (4 × 106 TU s.c.) or BCG (4 × 106 CFU s.c.) then some were immunized 3 weeks later with LV vF/85A (2 × 107 i.n.). After a further 3 weeks, mice were challenged with 5 × 106 CFU BCG i.n. Three weeks after BCG challenge lung and spleen homogenates were cultured on 7H10 agar until BCG colonies were visible. Colonies were counted and results are presented as viable BCG remaining in the entire organ. *P < 0·05 versus unimmunized. (c) BALB/c mice (n = 6) were immunized with BCG (4 × 105 CFU s.c.) or LV vF/85A or LV vF/GFP (4 × 106 TU s.c.). Boost immunization after 2 weeks (for LV) or 8 weeks (for BCG) was with LV vF/Ag85A or LV vF/GFP (6 × 106 TU i.n.). After 2 weeks, mice were challenged with 5 × 106 CFU BCG i.n., after a further 3 weeks organs were removed and BCG CFU in lungs and spleens were quantified as above. *P < 0·05 versus unimmunized.
Challenge studies were then performed using mucosal administration of BCG to induce respiratory infection. BCG was not expected to cause manifest illness and did not in these mice. Animals immunized s.c. with BCG were relatively protected against BCG challenge as determined by a lower bacterial load in spleen and lungs than animals that did not receive BCG immunization (Fig.3b, c). Two doses of LV vF/85A in a prime-boost regimen did not significantly protect against BCG challenge, although there was a trend towards lower bacterial loads in spleen but not lungs (Fig.3b, c). Furthermore, boosting BCG s.c. immunization with i.n. LV vF85A did not increase protection compared with that seen with BCG s.c. alone. From these data we conclude that the current formulation of LV vF/85A is not a suitable clinical vaccine candidate to take forward into further development.
Discussion
In this journal Xu et al.13 have recently reported on the efficacy of a BCG prime and a LV boost in protecting against TB in mice. Differences between the study of Xu et al. and our study include their use of a different mycobacterial antigen, Ag85B, fused with a second mycobacterial antigen, Rv3425, and their use of two s.c. LV boost immunizations. We administered a single LV boost via the i.n, route to provide a prime and pull approach and induce local immunogenicity. Although we did induce immunogenicity at the lung and in BAL, suggesting the prime and pull approach was successful; unfortunately this failed to translate into protection against challenge with intranasal BCG.
The inclusion of Rv3425 was one important difference between our approach and that of Xu et al., which should be evaluated further. Xu et al. investigated responses and protection against mycobacteria in C57BL/6 mice whereas we used the BALB/c strain. Mouse strain has a major impact on the T-cell responses induced by BCG and the susceptibility to mycobacterial infection.14,15
We feel that our respiratory BCG challenge is a more stringent mimic of natural processes than the intravenous challenge used by Xu et al.; however, they challenged with M. tuberculosis. Our use of BCG was mainly pragmatic, in an attempt to develop a model for use outside containment level three facilities, as has been investigated previously and found to be possible.12 We also wondered if the lower pathogenicity of this organism might provide better differentiation between vaccine regimens, as in vaccine development sometimes low-level protection is only demonstrated when, for example, the dose of a challenge is reduced to a level at which the difference can be detected.12 It is also possible that BCG would be easier to protect against with a BCG vaccine than M. tuberculosis because of the increase in the similarity of vaccine and challenge organism.
Despite the differences in study design both vaccine regimens induced potent cytotoxic CD8+ T-cell responses, but only Xu et al. induced significant CD4+ T-cell responses. This suggests a major role for CD4+ T-cell responses in protection against mycobacterial infection. However, the CD4+ T-cell response in humans following BCG prime and MVA85A boost was disappointing in the field and the response was insufficient to protect against TB in infants in South Africa.5 This was despite excellent induction of CD4+ and CD8+ T cells using the same regimen in a murine model.16 It has been suggested that the key population of cells required to protect against mycobacterial infection is a CD4+ IL17+ population of Th17 cells and further work is therefore needed to develop vaccines that induce such a population of T cells.17 Alternatively CD4+ IL-2+ T cells may be the key T-cell population.18 Lentivectors expressing cell activators have been used to modify dendritic cell signalling19 and induce subpopulations of CD4+ T cells.20 Intranasal LV immunization is also very effective in recruiting T cells to the lung.6 However, the nuclear factor-κB activator adjuvant recruited CD8+ rather than CD4+ T cells and the CD8+ responses were short-lived. The mycobacterial challenge was administered at the peak of the response so we do not feel that the short-lived duration of response was the explanation for the failure to induce protection. Xu et al. did not observe complete protection against TB challenge. An optimal LV boosting protocol may therefore incorporate the antigens and multiple immunization of Xu et al., but include an intranasal immunization with an LV encoding a dendritic cell activator that generates an appropriate CD4+ T-cell response.
Acknowledgments
GB and ALG designed and performed the experiments, interpreted the results and wrote the paper. GB and DCM performed the experiments. JSB designed the experiments. MKC designed the experiments, interpreted the results and wrote the paper. GB was funded by the UCL/UCLH BRC. DCM was funded by an MRC Clinical Research Fellowship. This work was undertaken at UCLH/UCL, who received a proportion of the funding from the Department of Health’s NIHR Biomedical Research Centre’s funding scheme. ALG was funded by an NIHR Academic Clinical Lectureship and received a BIA project grant, which supported this work. We would like to thank Elspeth Poulton, Mahdad Noursadeghi, Chris Davis, Narjara Gonzalez-Suarez, Jim Periselneris and Ricardo Jose from UCL and E. Stylianou from University of Oxford for their assistance.
Disclosures
ALG (WO/2008/122811) and MKC (WO2004110485) hold patents on vaccine technology but those patents do not refer to the technology used in this manuscript. GB, DCM and JSB have no conflicts of interest to declare.
Supporting Information
Figure S1. Gating strategy for flow cytometry data.
Figure S2. Lentivector vF/85A recruits T cells secreting tumour necrosis factor-α to the lung.
Figure S3. The number of T cells in bronchoalveolar lavage and lung decreases with time.
Table S1. Lentivector vFLIP/Ag85A (vF/85A) boost does not improve immunogenicity of bacillus Calmette–Guérin.
References
- WHO. 2014. Global tuberculosis report.
- Trunz BB, Fine P, Dye C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet. 2006;367:1173–80. doi: 10.1016/S0140-6736(06)68507-3. [DOI] [PubMed] [Google Scholar]
- Andersen P, Doherty TM. The success and failure of BCG – implications for a novel tuberculosis vaccine. Nat Rev Microbiol. 2005;3:656–62. doi: 10.1038/nrmicro1211. [DOI] [PubMed] [Google Scholar]
- Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature. 2012;491:463–7. doi: 10.1038/nature11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet. 2013;381:1021–8. doi: 10.1016/S0140-6736(13)60177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald DC, Singh H, Whelan MA, Escors D, Arce F, Bottoms SE, et al. Harnessing alveolar macrophages for sustained mucosal T-cell recall confers long-term protection to mice against lethal influenza challenge without clinical disease. Mucosal Immunol. 2014;7:89–100. doi: 10.1038/mi.2013.27. [DOI] [PubMed] [Google Scholar]
- Rowe HM, Lopes L, Brown N, Efklidou S, Smallie T, Karrar S, et al. Expression of vFLIP in a lentiviral vaccine vector activates NF-κB, matures dendritic cells, and increases CD8+ T-cell responses. J Virol. 2009;83:1555–62. doi: 10.1128/JVI.00709-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Lopes L, Ikeda Y, Bailey R, Barde I, Zenke M, et al. Immunization with a lentiviral vector stimulates both CD4 and CD8 T cell responses to an ovalbumin transgene. Mol Ther. 2006;13:310–9. doi: 10.1016/j.ymthe.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Pajot A, Michel ML, Fazilleau N, Pancre V, Auriault C, Ojcius DM, et al. A mouse model of human adaptive immune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol. 2004;34:3060–9. doi: 10.1002/eji.200425463. [DOI] [PubMed] [Google Scholar]
- Forbes EK, Sander C, Ronan EO, McShane H, Hill AV, Beverley PC, et al. Multifunctional, high-level cytokine-producing Th1 cells in the lung, but not spleen, correlate with protection against Mycobacterium tuberculosis aerosol challenge in mice. J Immunol. 2008;181:4955–64. doi: 10.4049/jimmunol.181.7.4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkridge T, Scriba TJ, Gelderbloem S, Smit E, Tameris M, Moyo S, et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. J Infect Dis. 2008;198:544–52. doi: 10.1086/590185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian AM, Ronan EO, Poyntz H, Hill AV, McShane H. Preclinical development of an in vivo BCG challenge model for testing candidate TB vaccine efficacy. PLoS ONE. 2011;6:e19840. doi: 10.1371/journal.pone.0019840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Yang E, Wang J, Li R, Li G, Liu G, et al. Prime-boost bacillus Calmette–Guérin vaccination with lentivirus-vectored and DNA-based vaccines expressing antigens Ag85B and Rv3425 improves protective efficacy against Mycobacterium tuberculosis in mice. Immunology. 2014;143:277–86. doi: 10.1111/imm.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugelat S, Ladel CH, Kaufmann SH. Influence of mouse strain and vaccine viability on T-cell responses induced by Mycobacterium bovis bacillus Calmette–Guérin. Infect Immun. 1995;63:2033–40. doi: 10.1128/iai.63.5.2033-2040.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pelayo MC, Bachy VS, Kaveh DA, Hogarth PJ. BALB/c mice display more enhanced BCG vaccine induced Th1 and Th17 response than C57BL/6 mice but have equivalent protection. Tuberculosis. 2015;95:48–53. doi: 10.1016/j.tube.2014.10.012. [DOI] [PubMed] [Google Scholar]
- McShane H, Pathan AA, Sander CR, Keating SM, Gilbert SC, Huygen K, et al. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat Med. 2004;10:1240–4. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- Cruz A, Torrado E, Carmona J, Fraga AG, Costa P, Rodrigues F, et al. BCG vaccination-induced long-lasting control of Mycobacterium tuberculosis correlates with the accumulation of a novel population of CD4+ IL-17+ TNF+ IL-2+ T cells. Vaccine. 2015;33:85–91. doi: 10.1016/j.vaccine.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Kang H, Yuan Q, Ma H, Hu ZD, Han DP, Wu K, et al. Enhanced protective efficacy against Mycobacterium tuberculosis afforded by BCG prime-DNA boost regimen in an early challenge mouse model is associated with increased splenic interleukin-2-producing CD4 T-cell frequency post-vaccination. Immunology. 2014;143:661–9. doi: 10.1111/imm.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D, Lopes L, Lin R, Hiscott J, Akira S, Davis RJ, et al. Targeting dendritic cell signaling to regulate the response to immunization. Blood. 2008;111:3050–61. doi: 10.1182/blood-2007-11-122408. [DOI] [PubMed] [Google Scholar]
- Arce F, Breckpot K, Stephenson H, Karwacz K, Ehrenstein MR, Collins M, et al. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011;63:84–95. doi: 10.1002/art.30099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gating strategy for flow cytometry data.
Figure S2. Lentivector vF/85A recruits T cells secreting tumour necrosis factor-α to the lung.
Figure S3. The number of T cells in bronchoalveolar lavage and lung decreases with time.
Table S1. Lentivector vFLIP/Ag85A (vF/85A) boost does not improve immunogenicity of bacillus Calmette–Guérin.