

Abstract
Self-amplifying mRNAs (SAM®) are a novel class of nucleic acid vaccines, delivered by a non-viral delivery system. They are effective at eliciting potent and protective immune responses and are being developed as a platform technology with potential to be used for a broad range of targets. However, their mechanism of action has not been fully elucidated. To date, no evidence of in vivo transduction of professional antigen-presenting cells (APCs) by SAM vector has been reported, while the antigen expression has been shown to occur mostly in the muscle fibres. Here we show that bone-marrow-derived APCs rather than muscle cells are responsible for induction of MHC class-I restricted CD8 T cells in vivo, but direct transfection of APCs by SAM vectors is not required. Based on all our in vivo and in vitro data we propose that upon SAM vaccination the antigen is expressed within muscle cells and then transferred to APCs, suggesting cross-priming as the prevalent mechanism for priming the CD8 T-cell response by SAM vaccines.
Keywords: antigen-presenting cells, cross-presentation, muscle cells, nucleic-acid-based vaccines, self-amplifying mRNA
Introduction
Nucleic-acid-based vaccines have the potential to combine the benefits of live-attenuated vaccines, viral vectors and subunit vaccines.1,2 Live-attenuated vaccines and viral vectors are very immunogenic but they are inefficient in subjects that have pre-existing immunity and are generally not good boosters because they generate neutralizing antibodies after the first dose. Subunit vaccines are safe but induce a poor CD8 T-cell response. By contrast, DNA-based and RNA-based vaccines do not generate anti-vector immunity, can be used for multiple doses, work in all subjects, are easy to manufacture and are often less reactogenic compared with live attenuated viruses. Moreover, they induce more efficient and stronger CD8 responses compared with subunit vaccines.3
To date, the majority of pre-clinical and clinical studies using nucleic-acid-based vaccines have been conducted with plasmid DNA4,5 and DNA-based viral vectors.6 However, DNA-based vaccines were found to be less effective in humans7–10 compared with models in small animals, in which they are able to induce potent immune responses.11 The use of electroporation has increased the immunogenicity of DNA vaccines in humans. Recently, electroporation of a human papillomavirus therapeutic DNA vaccine induced good antibody and CD8 T-cell responses.12 However, this delivery system for preventing vaccines is difficult to implement.
More recently, RNA-based vaccines, both mRNA and self-amplifying mRNA (SAM®), are becoming an attractive alternative to DNA for gene vaccination.13–16 Unlike DNA, RNA cannot be integrated into the host cell’s genome, which reduces the risk of gene dysregulation.17 Moreover, mRNA is produced using a cell-free enzymatic transcription reaction, increasing production yields and avoiding safety concerns associated with the use of living organisms.18,19 On the other hand, RNA vaccines mimic a live infection by expressing the antigens in situ after immunization, and inducing an immune response against the encoded antigen.20–23 Moreover, mRNA-based vaccines have the additional advantage that their sequences can be easily manipulated to improve their intrinsic capacity to stimulate the innate immune system or even to induce expression of additional molecules which can then stimulate innate immunity or function as co-stimulatory molecule, finally leading to an enhancement of the antigen-specific immune responses.24–28
We have previously described the SAM vaccine technology,16,29–31 based on a synthetic SAM, delivered by a synthetic lipid nanoparticle (LNP), which is currently in pre-clinical development and may soon be evaluated in humans. The use of an LNP, first explored for systemic delivery of small interfering RNA,32,33 constitutes a novel vaccine delivery system that can successfully replace the more common viral delivery of self-amplifying mRNA using viral replicon particles (VRPs).34,35 In fact, it was shown that the delivery of a 9-kb self-amplifying RNA encapsulated within LNP increases the potency of self-amplifying RNA, avoiding the complications of anti-vector immunity associated with the viral delivery but leading to an immune response comparable to that triggered by VRPs.29
This technology uses an SAM based on an alphavirus genome,36 which contains genes encoding the viral replicase complex responsible for the amplification of the RNA, but lacks the genes encoding the viral structural proteins required to produce infectious viral particles. The viral structural proteins are replaced by genes encoding protein antigens, which are expressed from a subgenomic mRNA. In this way, RNA amplification within the cytoplasm of transfected cells produces many copies of the antigen-encoding mRNA, leading to high levels of antigen expression. In addition, double-stranded RNAs (dsRNAs), that are produced during RNA replication, may act as potent stimulators of innate immunity resulting in the induction of an enhanced immune response.37–39 Hence, SAM vaccines have the potential to be more effective than corresponding mRNA vaccines.1
The SAM vaccines are effective at eliciting broad, potent and functional immune responses against different infectious targets in multiple animal models.29,30,40,41 However, the mechanism by which SAM vectors activate the immune system has not been fully elucidated. In particular, while the cell uptake of small conventional non-amplifying mRNA is known,42 and numerous studies have described that locally administered naked mRNA is taken up by cells in target tissues,43–45 it is not known how larger self-amplifying mRNA are acquired by cells.
Preliminary evidence suggests that muscle cells may play a role in this process. Wolff et al.46 first demonstrated that mRNA could directly transfect muscle cells when injected in vivo, resulting in the expression of the encoded protein. Following Wolff’s results, many studies have shown that intramuscular injection of mRNA-encoding reporter genes results in protein expression in myocytes.29,47,48 In fact, it has been reported that, when mRNA is delivered naked (formulated in buffer without a delivery system), encapsulated, or through a gene gun delivery tool, expression is mostly observed in somatic cells.21,49
To date, no evidence of in vivo transfection of antigen-presenting cells (APCs) by the SAM vectors has been reported, while the antigen expression has been shown to occur mostly in the muscle fibres after administration with a lipid-based delivery system,41 leading to the question of whether somatic muscle cells are able to prime CD8 T cells.
The present study was designed to investigate the respective contribution of muscle cells and bone marrow (BM) -derived professional APCs to CD8 T-cell priming, following SAM vaccine immunization. To address this question, we used chimeric mice that express different MHC class I alleles on BM-derived APCs and muscle cells and the influenza intracellular antigen nucleoprotein (NP) as model antigen. Then we studied CD8 T-cell priming following immunization with a self-amplifying mRNA encoding NP antigen encapsulated in an LNP non-viral delivery system [SAM (NP/LNP)] or delivered with a viral replicon particle, produced using a packaging cell line [VRP (NP)], or formulated in buffer without a delivery system [Naked SAM (NP)].
Materials and methods
Mice
Animals were housed in the Novartis Vaccines Animal Facility and experiments were approved and conducted according to the Institutional Animal Care and Use Committee guidelines. Female, C57BL/6, C3H and B6C3F1 mice, 7–8 weeks of age, used for the generation of BM chimeras were purchased from Charles River Laboratory (Calco, Italy). CD11c.DOG mice, expressing diphtheria toxin receptor under the control of the long CD11c promoter, were kindly provided by N. Garbi (Institute of Molecular Medicine and Experimental Immunology, Bonn, Germany). In these mice, treatment with diphtheria toxin (DT) results in dendritic cell (DC) depletion.50
Generation of BM chimeric mice
Chimeras were prepared as follows. One week before irradiation, recipient mice were given antibiotic drinking water. Water was first autoclaved and then supplemented with 10 μg/ml neomycin (Sigma, St Louis, MO) and 10 μg/ml polymyxin B sulphate (Sigma). Recipient mice were irradiated with approximately 1000 Rad from an X-ray machine source, delivered in two equal doses of 500 Rad given 1 hr apart. Mice were maintained with antibiotic water for 2 weeks after irradiation and then switched to drinking water for the rest of the experimental period. Bone marrow was harvested from the femurs of donor mice and processed to generate a single-cell suspension. After red-cell lysis using RBC lysis buffer (Biolegend, San Diego, CA), T and natural killer cells were depleted by incubation with CD3 and NKp46 biotin antibodies (Miltenyi Biotec, Bergisch Gladbach, Germany) followed by microbeads incubation and magnetic cell separation (Miltenyi Biotec). Five hours after the irradiation, about 2·5 × 106 BM cells were injected by tail vein into each of the recently irradiated recipient mice. The extent of chimerism in each mouse was confirmed by staining peripheral blood cells with FITC-labelled anti-H-2b and phycoerythrin (PE) -labelled anti-H-2k antibodies (BD Pharmingen, San Diego, CA).
RNA synthesis
Influenza NP gene was amplified from the cDNA of influenza A/Puerto Rico/34/2007 (H1N1), using forward primer 5′-GTG AGC GTC GAC GCC ACC ATG GCG TCC CAA GGC ACC AAA C-3′ and reverse primer 5′-GAT TGC GGC CGC TTA ATT GTC GTA CTC CTC TGC ATT GTC TCC G-3′. Amplicon was cloned as a SalI and NotI fragment into an optimized replicon construct.29,36 Plasmid DNA encoding the NP replicon clone was amplified in Escherichia coli and purified using the QiagenPlasmid Maxi kit (Qiagen, Hilden, Germany). DNA was linearized immediately downstream from the 3′ end of the replicon by endonuclease digestion and purified by phenol/chloroform extraction and ethanol precipitation. DNA template was transcribed into RNA using the MEGAscript T7 transcription kit (Life Technologies, Grand Island, NY) and purified by LiCl precipitation. RNA was then capped using the ScriptCap m7G Capping System (CellScript, Madison, WI) and purified again by LiCl precipitation. RNA was resuspended in RNAse-free water and its integrity was evaluated on 1% denaturing agarose-LE gel (Ambion-Life Technologies, Grand Island, NY).
BHK cell transfection and analysis of expressed NP
Eighty per cent of confluent baby hamster kidney (BHK) cells were transiently transfected in a six-well plate with 3 µg of RNA using Lipofectamine 2000 (Life Technologies). RNA and Lipofectamine were first added to 250 μl each of OptiMem Reduced Serum Media (Life Technologies) and incubated for 5 min at room temperature. Two hundred and fifty microlitres of Lipofectamine was then added and the mix was incubated for 20 min at room temperature. Culture media (Dulbecco’s modified Eagle’s medium (DMEM; Gibco-Life Technologies, Grand Island, NY) containing 5% fetal bovine serum (FBS; EuroClone, Pero MI, Italy) and 1× Pen/Strep/Glut (Gibco-Life Technologies) was removed from the cells and 500 μl of Lipofectamine complexed RNA was added to the cells. After 18 hr, cells were trypsinized and stained with the Live/Dead Fixable Yellow viability marker (Molecular Probes-Life Technologies, Eugene, OR). Cells were then fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences, San Jose, CA), washed with Perm-wash buffer (BD Biosciences) and stained with an FITC-labelled anti-NP antibody (Thermo Fisher Scientific, Waltham, MA). Stained cells were acquired on an LSR II flow cytometer (BD Biosciences) and analysed with flowjo software (TreeStar, Ashland, OR).
Transfected cells were also lysed using RIPA buffer (Sigma) supplemented with complete protease inhibitor cocktail (Roche, Mannheim, Germany). Lysates were separated under reducing conditions on a 4–12% Bis–Tris polyacrylamide gel in MOPS electrophoresis buffer (Life Technologies) and blotted to nitrocellulose membrane (Life Technologies). NP was detected with anti-NP antibody (HB65 purified Hybridoma; ATCC Clone H16-R10-4R5).
Production of VRPs
Viral replicon particles were produced in BHK cells as previously described.36 In this system, the antigen expressing alphavirus chimeric replicon vector derived from the genome of Venezuelan equine encephalitis virus engineered to contain the 3′ untranslated region and the packaging signal of Sindbis virus (SV), was electroporated into BHK cells along with defective helper RNAs encoding the Sindbis virus capsid and glycoprotein genes. After 20–24 hr, replicon particles expressing NP were harvested from the culture supernatants and titrated by standard methods. For vaccination, 107 infectious VRPs were delivered bilaterally intramuscularly in mouse quadriceps.
LNP/RNA formulation
Encapsulation of RNA within LNP was performed as previously described.29 Briefly, the lipids 1,2-dilinoleyloxy-3-dimethylaminopropane (DLinDMA),51 1,2-diastearoyl-sn-glycero-3-phosphocholine (DSPC; Genzyme, Cambridge, MA), 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (PEG-DMG 2000; Avanti Polar Lipids, Alabaster, AL) and cholesterol (Sigma-Aldrich, St Louis, MO) were combined at a molar ratio of 40 : 10 : 2 : 48 molar per cent (DLinDMA : DSPC : PEG–DMG 2000 : cholesterol) by an ethanol dilution process to produce the LNP. An 8 : 1 N : P (nitrogen on DLinDMA : phosphate on RNA) molar ratio was used. Equal volumes of lipid in ethanol (Sigma-Aldrich) and RNA in 100 mm citrate buffer, pH 6·0 (Teknova, Hollister, CA) were mixed through a T-junction via a KDS-220 syringe pump (kdScientific, Holliston, MA), and a third syringe with equal volume of buffer was added simultaneously to the lipid/RNA mixture. The resulting LNPs were dialysed overnight at 4° against 1× PBS (Ambion-Life Technologies) pH 7·4 using Pierce slide-A-Lyzer® G2 dialysis cassettes (Thermo Fisher Scientific) with a 3·5K membrane molecular weight cut off. For in vitro and in vivo experiments, formulations were diluted to the required RNA concentration with 1× PBS (Ambion-Life Technologies). Formulations were characterized for particle size, RNA concentration, encapsulation efficiency and RNA integrity (using gel electrophoresis) after the formulation process as previously described29 with slight modifications. Particularly, RNA was extracted from the LNPs by addition of a 400-μl sample to 1·5 ml ethanol (Sigma-Aldrich) and 50 μl of 3 m sodium acetate pH 5·2 (Sigma-Aldrich). After centrifugation the pellet was suspended in nuclease-free water (Ambion-Life Technologies) and used for RNA analysis by gel electrophoresis on mini-gel TAE (Tris–acetate EDTA buffer) 1% agarose gels (Bio-Rad, Hercules CA) and run at 100 V. Gel was stained using 0·1% (vol/vol) SYBR gold according to the manufacturer’s instructions (Molecular Probes-Life Technologies). Images were taken on a Gel Doc EZ imaging system (Bio-Rad).
Immunizations and MHC I Dextramers staining
Chimeric mice were immunized bilaterally intramuscularly (50 μl per quadriceps) on days 0 and 28. One hundred microlitres of heparinized blood was collected and stained with 10 μl of allophycocyanin-labelled H-2Db/ASNENMETM and PE-labelled H-2Kk/SDYEGRLI MHC I Dextramers (Immudex, Copenhagen, Denmark). Ten microlitres of the following antibodies mix was added: peridinin chlorophyll protein Cy5.5-labelled anti-CD3, V500-labelled anti-CD4 and V450-labelled anti-CD44 (all from BD Biosciences) and PE Texas Red-labelled anti-CD8 (Molecular Probes-Life Technologies). Red blood cells were lysed using RBC Lysis buffer (eBioscience, San Diego, CA). Cells were then washed once with PBS (Gibco-Life Technologies), fixed with Cytofix (BD Biosciences) and acquired using an LSR II SOS1 flow cytometer (BD Bioscience). flowjo software (TreeStar) was used to analyse the acquired data. Double positive cells for MHC-I Dextramers and CD44 were gated on the CD3+, CD8+ T-cell population. For experiments with CD11c.DOG mice, long-term depletion of DCs was obtained by DT (Sigma) treatment as follows. CD11c.DOG mice were injected with DT (16 ng/g of body weight in PBS) intraperitoneally 2 days before immunization and then intraperitoneally and subcutaneously at days −1, +1, +3, +5 and +7 with respect to immunization, and CD11c+ cell depletion on the day of immunization was confirmed in peripheral blood using flow cytometry.
In vitro restimulation of antigen-specific CD8 T cells and intracellular cytokines staining
To measure CD8 T-cell responses, spleens were harvested and single-cell suspensions were prepared. Splenocytes were plated at 2 × 106 cells/well in 96-well U-bottom plates in RPMI-1640 medium (Gibco-Life Technologies) supplemented with 25 mm HEPES (Gibco-Life Technologies), 10% heat inactivated FBS (low endotoxin; HyClone, Logan, UT), 1× Pen/Strep/Glut (100×; Gibco-Life Technologies) and 50 μm β-mercaptoethanol (Sigma), and stimulated with 5 μg/ml of H-2Kk restricted CD8 T-cell-specific NP peptide (SDYEGRLI) or H-2Db restricted CD8 T-cell-specific NP peptide (ASNENMETM) in the presence of 5 μg/ml of Brefeldin A (Sigma). At the same time, FITC-labelled anti-lamp1 antibody (BD Biosciences) was added to each stimulus as a CD8 T-cell cytotoxicity marker. After culturing for 4 hr at 37°, cells were washed and stained with Live/Dead Fixable Near-IR viability marker (Molecular Probes-Life Technologies). Cells were then fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences), washed with Perm-wash buffer (BD Biosciences) and stained with the following antibodies: peridinin chlorophyll protein Cy5.5-labelled anti-CD3, V450-labelled anti-CD44, V500-labelled anti-CD4 (all from BD Bioscience), PE Texas red-labelled anti-CD8 (Molecular Probes-Life Technologies), PE-labelled anti-interferon-γ (BD Bioscience). Cells were acquired on an LSR II SOS1 flow cytometer (BD Biosciences) and analysed with flowjo software (TreeStar). Double-positive cells for IFN-γ and CD44 and Lamp1+ cells were gated on the CD3+, CD8+ T-cell population.
C2C12 infection
C2C12 (H-2k) mouse myoblasts cells were obtained from ATCC (Rockville, MD) and maintained in high-glucose DMEM (Gibco-Life Technologies) supplemented with 10% FBS (EuroClone) and 1× Pen/Strep/Glut (Gibco-Life Technologies). For the in vivo immunization with NP-expressing myoblasts, C2C12 cells were infected using VRPs expressing NP. The day before infection, C2C12 cells were plated in a 175-cm2 cell culture flask in culture media without antibiotics. Culture medium was then removed and 8 ml of DMEM containing only 1% FBS and 108 VRP (NP) infectious units were added to the cells. After incubation at 37° for 4 hr, 8 ml of complete culture medium were added to the cells for an overnight incubation. Cells were then washed with PBS, trypsinized and further analysed by flow cytometry for NP and H-2k expression using an FITC-labelled anti-NP (Thermo Fisher Scientific) and a PE-labelled anti- H-2Kk (BD Biosciences) antibodies. A total of 3 × 106 NP C2C12 myoblasts were injected in five separate sites for each quadriceps muscle in a volume of 50 μl per leg.
Generation of BM-derived DCs and isolation of CD11c+ DCs
The BM-DCs were induced from BM cells obtained from 6- to 8-week-old C3H mice as previously described.52 Briefly, a single-cell suspension was prepared from BM obtained from femurs. After lysing red blood cells, 2 × 106 BM cells were cultured in RPMI-1640 medium (Gibco-Life Technologies) supplemented with 25 mm HEPES (Gibco-Life Technologies), 10% heat inactivated FBS (low endotoxin; HyClone), 1× Pen/Strep/Glut (100×; Gibco-Life Technologies), 50 μm β-mercaptoetanol (Sigma), 10 ng/ml mouse granulocyte–macrophage colony-stimulating factor and 5 ng/ml mouse interleukin-4 (both from Miltenyi Biotec) in 10-cm diameter Petri dishes at 37° in 5% CO2. Supplemented medium was replaced every 3 days. On day 8, non-adherent cells and DCs were collected and analysed for CD11c expression by flow cytometry using allophycocyanin-eFluor780-labelled anti-CD11c antibody (eBioscience). All the prepared BM-DC populations used expressed CD11c on at least 90% of the cells. Primary DCs were obtained as follows. Spleens from 6- to 8-week-old C3H mice were harvested, cut into small fragments and treated with 2 mg/ml of collagenase D (Roche) and 0·5 of mg/ml DNase I (Sigma) in RPMI-1640 medium (Gibco-Life Technologies) for 30 min at 37° under continuous agitation. Cell suspension was then filtered through a 30-μm strainer (BD Biosciences) and DCs were isolated using magnetic CD11c MicroBeads (Miltenyi Biotec).
LNP/RNA in vitro transfection
C2C12, BM-DCs and primary DCs were transfected using serial dilutions of LNP formulated SAM (NP). The formulation was diluted in DMEM (Gibco-Life Technologies) containing only 1% FBS (EuroClone) for BHK and C2C12 cells or RPMI-1640 medium (Gibco-Life Technologies) supplemented with 25 mm HEPES (Gibco-Life Technologies), 10% heat inactivated FBS (low endotoxin; HyClone), 1× Pen/Strep/Glut (100×; Gibco-Life Technologies) and 50 μm β-mercaptoetanol (Sigma) for BM-DCs. Cells were incubated at 37° for 18 hr. The medium was then removed, and cells were trypsinized and stained with the Live/Dead Fixable Yellow viability marker (Molecular Probes-Life Technologies). BM-DCs and primary DCs were stained also with allophycocyanin-eFluor780-labelled anti-CD11c antibody (eBioscience). Cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences), washed with Perm-wash buffer (BD Biosciences) and stained with an FITC-labelled anti-NP antibody (Thermo Fisher Scientific) and an anti-dsRNA antibody (Scicons, Szirák, Hungary), previously conjugated with an Invitrogen Zenon Allophycocyanin Labeling kit. Stained cells were acquired on an LSR II SOS1 flow cytometer (BD Biosciences) and analysed with flowjo software (TreeStar). For analysis of BM-DCs and primary DCs, NP+ and dsRNA+ DCs were gated on CD11c+ cells.
Migration assay
C2C12 cells were infected with 2 × 106 IU VRPs expressing NP or transfected with 100 ng LNP formulated SAM (NP) in a 24-well plate. Cells were trypsinized, washed with PBS (Gibco-Life Technologies) to remove any residual VRP or LNP and plated in new wells. After 5–6 hr, BM-DCs were seeded inside trans-well inserts with a 10-μm thick polyethylene membrane containing 0·4-μm diameter pores (BD-Falcon, Franklin Lakes, NJ) on top of VRP-infected or LNP-transfected C2C12 cells for 2 hr at 37°. The DCs in the upper and lower compartments of the trans-well chamber were collected, washed and stained with Live/Dead Fixable Yellow viability marker (Molecular Probes-Life Technologies), allophycocyanin-eFluor780-labelled anti-CD11c (eBioscience) and Alexa Fluor 700-labelled anti-CD45.2 (eBioscience) antibodies. Cells were then fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences), washed with Perm-wash buffer (BD Biosciences) and stained with an FITC-labelled anti-NP antibody (Thermo Fisher Scientific) and allophycocyanin-labelled anti-dsRNA antibody (Scicons). Stained cells were acquired on LSR II SOS1 flow cytometer (BD Biosciences) and analysed with flowjo software (TreeStar). NP+ and dsRNA+ BM-DCs were gated on CD11c and CD45.2 double-positive cells.
Statistical analysis
All results are representative of at least two independent experiments with similar results. All statistics were performed using graphpad prism 6 software (GraphPad Software, S. Diego, CA). The unpaired two-sample Student’s t-test was used. P-values < 0·05 were considered statistically significant; *P < 0·05; **P < 0·01; ***P < 0·001.
Results
BM-derived professional APCs are responsible for direct priming of CD8 T cells upon SAM vaccination
To assess the respective contribution of muscle cells and BM-derived APCs in priming CD8 T cells after SAM vaccination, we generated chimeric mice transferring BM cells from C57BL/6 mice expressing MHC molecules of the H-2b haplotype or C3H mice expressing MHC molecules of the H-2k haplotype into irradiated B6C3F1 recipient mice with a mixed H-2bxk haplotype expressing MHC molecules from both donors.53 The extent of chimerism in each mouse was confirmed 8 weeks after reconstitution by staining peripheral blood cells with anti-H-2b and anti-H-2k antibodies, showing that almost 100% of the cells were derived from the corresponding donor (Fig.1a). The haematopoietic lineage cells in these chimeras, including professional APCs, express only the MHC molecules of donor haplotype (H-2b or H-2k), whereas the somatic cells, such as myocytes, express both parental strain MHC alleles (H-2bxk). As control, irradiated B6C3F1 recipient mice received homologous BM cells.
Figure 1.

Analysis of chimerism and characterization of Self-amplifying mRNA nucleoprotein [SAM (NP)] vector. (a) Peripheral blood cells from chimeric mice were stained with FITC-labelled anti-H-2b and phycoerythrin-labelled anti-H-2k antibodies and analysed by flow cytometry. Representative dot plots corresponding to the three groups of chimeric mice are shown for CD3+ T cells and CD11c+ dendritic cells. (b–d) SAM (NP) vector was characterized analysing RNA integrity and antigen expression after transfection. (b) Denaturing RNA agarose gel electrophoresis: molecular weight ladder (lane 1), SAM (NP) vector (lane 2). (c) Flow cytometry analysis of mock (grey filled) or SAM (NP) (unbroken line) transfected baby hamster kidney (BHK) cells after intracellular staining with FITC-labelled anti-NP antibody. (d) Western blot analysis with anti-NP of BHK cells mock transfected or transfected with pCMV (NP) vector or SAM (NP) vector.
To study if, upon SAM immunization, CD8 T cells are primed by professional APCs expressing either the H-2b or the H-2k haplotype, or by muscle cells expressing both MHC alleles, we used a SAM vector encoding for the influenza NP antigen, SAM (NP), for which an H-2Db (ASNENMETM) and an H-2Kk (SDYEGRLI)-restricted CD8 T-cell peptide are described.53 The NP gene was amplified from the reverse-transcribed RNA genome of influenza virus A/PR/8/34, and then cloned into the self-amplifying RNA vector. The integrity of the transcribed RNA was evaluated by agarose gel electrophoresis (Fig.1b) and the intracellular antigen expression was demonstrated by intracellular staining and flow cytometry (Fig.1c) or Western blot analysis (Fig.1d) after RNA transfection in BHK cells with Lipofectamine. Groups of six or seven BM chimeric mice were intramuscularly immunized twice with a 4-week interval with SAM (NP) (1 μg) encapsulated in an LNP formulation [SAM (NP/LNP)], a conventional viral delivered replicon [VRP (NP)] [107 infectious units (IU)], or PBS as negative control. 1, 2 and 4 weeks after priming and 1 or 2 weeks after boost, antigen-specific CD8 T cells in the peripheral blood were measured by MHC-I Dextramer staining (Fig.2a,b). We found that, independently from the type of RNA delivery through LNP or VRP, CD8 T-cell priming was restricted by the MHC haplotype of donor BM-derived cells. Indeed, using a C57BL/6-specific Dextramer (H-2Db/ASNENMETM), 2 weeks after priming NP-specific CD8 T cells could be detected only in BL/6 chimera (H-2b → H-2bxk) where BM-derived professional APCs express H-2b MHC molecules, but not in C3H chimera (H-2k → H-2bxk) where only somatic cells express H-2b molecule (Fig.2a, left panel). On the contrary, using a C3H-specific Dextramer (H-2Kk/SDYEGRLI), we detected CD8 T cells only in C3H chimera but not in BL/6 chimera (Fig.2a, right panel), suggesting that T cells were primed in vivo by BM-derived antigen-presenting cells rather than muscle cells. As expected in the control B6C3F1 chimera (H-2bxk → H-2bxk), NP-specific CD8 T cells were detected using both C57BL/6 and C3H-specific Dextramers. The same pattern of MHC haplotype restriction was also observed 1 week after boost. Moreover, the kinetics of NP-specific CD8 T cells in the blood, analysed for each of the chimera groups, revealed an increase in the CD8 response after the second immunization with both VRP- and LNP-delivered SAM (NP). The higher frequency of NP-specific CD8 T cells was observed 2 weeks after priming and 1 week after boost. However, the percentage of antigen-specific CD8 T cells decreased 2 weeks after the boost with VRP, when T-cell response was still persistent upon LNP immunization (Fig.2b). Three weeks after the second immunization, splenocytes from each chimera group were stimulated in vitro with either H-2Db-restricted or H-2kk-restricted NP peptides and the frequency of IFN-γ-producing CD8 T cells was analysed. To investigate if CD8 T cells had a cytotoxic phenotype, we also measured Lamp1 expression on the cell surface (Fig.2c). CD8 T cells from immunized BL/6 Chimera were activated only upon stimulation with H-2Db-restricted NP peptide (ASNENMETM) (Fig.2c, left panel), while CD8 T cells from immunized C3H chimera were activated only upon stimulation with H-2Kk-restricted NP peptide (SDYEGRLI) (Fig.2c, right panel). As expected, we observed CD8 T-cell activation upon in vitro stimulation with both NP peptides in B6C3F1 chimeras. Moreover, we found that most of the in vitro responding antigen-specific CD8 T cells were producing IFN-γ and expressed Lamp1. In summary, these results demonstrate that BM-derived professional APCs are responsible for direct priming of MHC class-I restricted CD8 T cells following SAM (NP/LNP) or VRP (NP) immunization.
Figure 2.

CD8 T-cell priming, induced by self-amplifying mRNA (SAM) vaccine, is restricted to the MHC haplotype of donor and requires CD11c+ dendritic cells (DCs). (a–c) Groups of six or seven chimera mice were immunized intramuscularly on days 0 and 28 with either PBS, viral replicon particle nucleoprotein [VRP (NP)] (107 IU) or SAM [NP/lipid nanoparticle (LNP)] (1 μg). (a) Two weeks after priming (upper panels) and 1 week after boost (lower panels) frequency of NP-specific CD8 T cells in the blood was measured by staining with H-2Db/ASNENMETM (left panels) or H-2Kk/SDYEGRLI (right panels) Dextramers and flow cytometry analysis. Data from individual mice (dots) and the mean (solid lines) are reported for each group. Graphs are divided into three sectors, one for each group of chimeric mice. Specific MHC haplotypes of antigen-presenting cells (APCs) and myocytes are indicated at the bottom of the panels. (b) Mean of % of NP-specific CD8 T cells in each chimeric group in the blood at the indicated time-points after immunization with SAM (NP/LNP), VRP (NP) and PBS. BL/6 and B6C3F1 chimeras were stained with H-2Db/ASNENMETM Dextramer, while C3H chimera was stained with H-2Kk/SDYEGRLI Dextramer. (c) Three weeks after the second immunization splenocytes were stimulated in vitro with 5 μg/ml of H-2Db-restricted CD8 T-cell-specific NP peptide (left panel) or H-2Kk-restricted CD8 T-cell specific NP peptide (right panel) and interferon-γ-positive (IFN-γ+) and Lamp1+ CD8 T cells were detected by intracellular staining and flow cytometry. Data are presented as the average plus standard deviation. Graphs are divided into three sectors, one for each group of chimeric mice. Specific MHC haplotypes of APCs and myocytes are indicated at the bottom of the panels. Statistical analysis was performed using unpaired two-sample Student’s t-test versus PBS control: *P < 0·05; **P < 0·01; ***P < 0·001. Data are representative of three experiments. (d) untreated or diphtheria toxin (DT) -treated CD11c.DOG mice were immunized intramuscularly with SAM(NP/LNP) (1 μg) and 8 days later the frequency of NP-specific CD8 T cells in the blood was measured by staining with H-2Db/ASNENMETM Dextramer and flow cytometry analysis. Data from individual mice (squares) and the mean (solid lines) are reported for each group. Statistical analysis was performed using unpaired two-sample Student’s t-test: *P < 0·05.
To better evaluate the nature of APCs involved in CD8 T-cell priming by SAM vaccines and to verify if DCs are required for such an event, we used CD11c.DOG mice expressing the DT receptor under the control of the long CD11c promoter, in which treatment by DT induces a prolonged in vivo depletion of DCs.50 In agreement with published data treatment of CD11c.DOG mice with DT, as described in the Materials and methods, resulted in 60% depletion of CD11c+ cells (data not shown). DC-depleted and non-depleted CD11c.DOG mice were immunized intramuscularly with SAM (NP/LNP) (1 μg) and 8 days later the frequency of NP-specific CD8 T cells in the peripheral blood was measured by MHC-I Dextramer staining. Untreated CD11c.DOG mice were able to develop antigen-specific CD8 T cells upon SAM vaccination, but priming of antigen-specific CD8 T cells was completely abrogated in DC-depleted mice (Fig.2d), suggesting that CD11c+ DCs are indeed required for initiation of the antigen-specific CD8 T-cell response by SAM vaccines.
CD8 priming by naked SAM is also initiated by BM-derived APCs
To test if the mechanism for CD8 T-cell priming triggered by SAM vectors was affected by the delivery system, we compared the LNP-formulated SAM vector (1 μg) with a 10-fold higher dose of the same RNA vector administered in buffer (naked SAM) for the induction of NP-specific CD8 T cells in the peripheral blood of chimeric mice 2 weeks after priming. We found that CD8 T-cell priming was restricted by the MHC haplotype of donor BM-derived cells after immunization with both naked and LNP-formulated SAM vector (Fig.3), suggesting that the mechanism leading to CD8 T-cell priming upon SAM immunization is independent from the delivery system.
Figure 3.
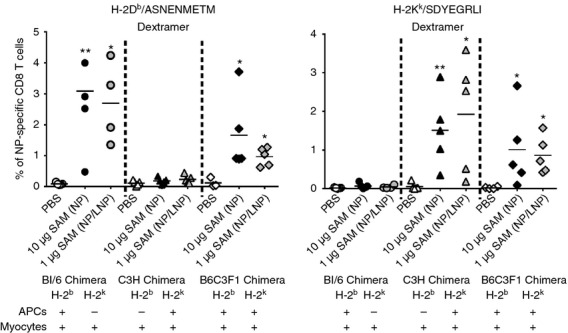
Naked self-amplifying mRNAs (SAM) and lipid nanoparticle (LNP) formulated SAM induce CD8 priming by the same mechanism of action. Groups of four or five chimera mice were immunized intramuscularly with either PBS, naked SAM nucleoprotein (NP) (10 μg) or SAM (NP/LNP) (1 μg). Two weeks after priming, the frequency of NP-specific CD8 T cells in the blood was measured by staining with H-2Db/ASNENMETM (left panel) or H-2Kk/SDYEGRLI (right panel) Dextramers and flow cytometry analysis. Data from individual mice (dots) and the mean (solid lines) are reported. Graphs are divided into three sectors, one for each group of chimeric mice. Specific MHC haplotypes of antigen-presenting cells (APCs) and myocytes are indicated at the bottom of the panels. Statistical analysis was performed using unpaired two-sample Student’s t-test versus PBS control: *P < 0·05; **P < 0·01. Data are representative of at least two experiments.
Direct transfection of professional APCs by SAM vaccine is not required for in vivo CD8 T-cell priming
Having established that professional APCs contribute to SAM-mediated CD8 T-cell priming, we wondered whether this was a result of antigen expression within the transfected APCs or acquisition of antigen by APCs from non-immune transfected cells, or a combination of both. To assess whether APCs need to be directly transfected to prime CD8 T cells, we took advantage of the myoblast cell line C2C12, which is derived from the C3H mouse strain (H-2k MHC haplotype). Using VRP (NP), we infected C2C12 cells to obtain NP-expressing myoblasts (NP C2C12) (Fig.4a), which were injected intramuscularly into each group of chimeric mice on days 0 and 28. In this way, expression of NP was limited only to the injected myoblasts. As negative controls, mice were immunized with PBS and non-infected C2C12 cells. Analysis of the frequency of antigen-specific CD8 T cells in the blood after priming and boost showed that injection of NP C2C12 expressing the H-2Kk molecule, was sufficient to induce a CD8 T-cell response to the antigen expressed by SAM vector not only in C3H chimera, detected by H-2Kk Dextramers (Fig.4b, right panels), but also in BL/6 chimera, detected by H-2Db Dextramers (Fig.4b, left panels), suggesting that BM-derived APCs were able to acquire the antigen from transfected C2C12 myoblasts in vivo. Priming of CD8 T cells in both C3H and BL/6 chimeras following injection of NP C2C12 myoblasts, was demonstrated also by in vitro re-stimulation of splenocytes with either H-2Db-restricted or H-2kk-restricted NP peptides and analysis of IFN-γ-producing and Lamp1-positive CD8 T cells (Fig.4c). In summary, we demonstrated that direct transfection of APCs by SAM vectors is not required for CD8 T-cell priming in vivo.
Figure 4.
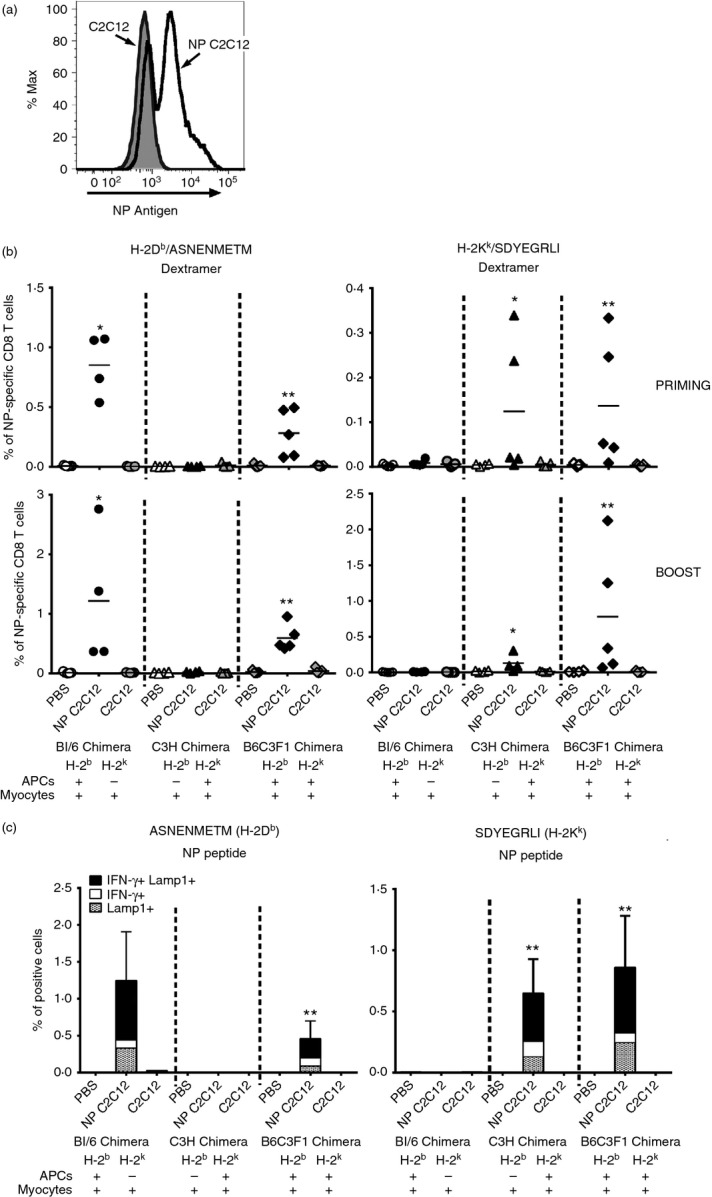
Immunization with antigen-expressing C2C12 myoblasts cell line induces a CD8 T-cell response restricted by the MHC haplotype of donor bone marrow (BM) -derived antigen-presenting cells (APCs). (a) Not infected (grey line) and viral replicon particles (VRPs) nucleoproteins (NP) -infected (black line) C2C12 cells were stained with FITC-labelled anti-NP antibody and analysed by flow cytometry. (b, c) Group of four or five chimera mice were vaccinated intramuscularly on days 0 and 28 with PBS, NP-expressing C2C12 myoblasts (3 × 106) or C2C12 cells (3 × 106). (b) One week after priming (upper panels) and 1 week after boost (lower panels), frequency of NP-specific CD8 T cells in the blood was measured by staining with H-2Db/ASNENMETM (left panels) or H-2Kk/SDYEGRLI (right panels) Dextramers and flow cytometry analysis. Data from individual mice (dots) and the mean (solid lines) are shown. Graphs are divided into three sectors, one for each group of chimeric mice. Specific MHC haplotypes of APCs and myocytes are indicated at the bottom of the panels. (c) Three weeks after boost splenocytes were stimulated in vitro with 5 μg/ml of H-2Db-restricted CD8 T-cell-specific NP peptide (ASNENMETM) or H-2Kk-restricted CD8 T-cell-specific NP peptide (SDYEGRLI) and interferon-γ-positive (IFNγ+) and Lamp1+ CD8 T cells were detected by intracellular staining and flow cytometry. Data are presented as average plus standard deviation. Graphs are divided into three sectors, one for each group of chimeric mice. Specific MHC haplotypes of APCs and myocytes are indicated at the bottom of the panels. Statistical analysis was performed using unpaired two-sample Student’s t-test versus both PBS and C2C12 immunization controls: *P < 0·05; **P < 0·01. Data are representative of at least two experiments.
APCs are not transfected by SAM but can migrate toward transfected myoblasts in vitro and take up their exogenous expressed antigen
To investigate if APCs can be transfected by a SAM vector and express the vector encoded antigen, we treated BM-derived dendritic cells (BM-DCs), primary CD11c+ DCs or C2C12 myoblasts with serial dilution of SAM (NP/LNP), and evaluated both antigen (NP) and dsRNA expression by intracellular staining. We found that C2C12 myoblasts become positive to both stainings (Fig.5a,b upper panels, Fig.5c,d black columns), indicating they were transfected by SAM (NP/LNP), but BM-DCs and primary DCs remained negative (Fig.5a,b middle and lower panels respectively, Fig.5c,d grey and white columns respectively), suggesting they could not be transfected. Similar results were obtained using VRP (NP) (data not shown).
Figure 5.

C2C12 myoblasts but not bone-marrow-derived dendritic cells (BM-DCs) nor primary DCs are transfected in vitro by self-amplifying mRNA (SAM) vectors. C2C12, BM-DCs and primary DCs were treated with serial dilutions of SAM ncleoprotein/lipid nanoparticles (NP/LNP) and stained with FITC-labelled anti-NP and allophycocyanin-labelled anti-dsRNA antibodies, and analysed by flow cytometry. (a, b) Dot plots for anti-NP (a) and anti-dsRNA (b) staining; percentages of cells in the gating region are indicated. (c, d) Histograms report the percentage of NP+ (c) and dsRNA+ (d) C2C12 (black bars), BM-DCs (grey bars) and CD11c+ primary DCs (white bars). Data are representative of at least three experiments.
We then wondered if DCs were able to take up the antigen from transfected myoblasts. To answer this question, BM-DCs were cultured in the upper compartment of a trans-well chamber in which VRP (NP)-infected C2C12 [VRP (NP) C2C12] or SAM (NP/LNP)-transfected C2C12 myoblasts [SAM (NP/LNP) C2C12] were placed in the lower compartment. After 2 hr, DCs in the upper and lower compartments of the trans-well chamber were collected, counted (Fig.6a) and analysed for NP (Fig.6b) and dsRNA (Fig.6c) expression. We found that BM-DCs had the ability to migrate towards the infected or transfected myoblasts and acquire both the antigen and the dsRNA from them. A higher number of BM-DCs migrated toward VRP (NP) C2C12 compared with SAM (NP/LNP) C2C12 myoblasts and a higher frequency of DCs were carrying the antigen and the RNA when migrating to the well with VRP-infected C2C12 cells. To investigate which factors are driving BM-DC migration, we analysed the supernatant of the infected or transfected myoblast cells for the presence of several chemokines [eotaxin, interleukin-8, monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory proteins 1α (MIP-1α) and 1β (MIP-1β), and Regulated upon Activation, Normal T-cell Expressed and Secreted (RANTES)]. Although supernatant from SAM (NP/LNP) C2C12 cells showed the same chemokine expression profile compared with non-transfected cells, VRP (NP) C2C12 supernatant contained higher levels of MCP1, MIP-1α, MIP1β and RANTES, which could account for the increased migration of BM-DCs toward these cells (data not shown).
Figure 6.
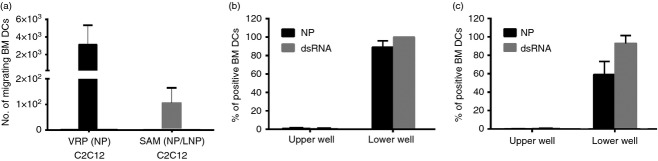
Dendritic cells (DCs) migrate toward transfected C2C12 myoblasts and take up both antigen and RNA. Bone-marrow-derived (BM) -DC were placed in a trans-well insert with 0·4-μm diameter pores above viral replicon particle (nucleoprotein)-infected C2C12 cells [VRP (NP) C2C12] or self-amplifying mRNAs (SAM) (NP/lipid nanoparticle) transfected C2C12 cells [SAM (NP/LNP) C2C12] for 2 hr. (a) Number of BM-DCs recovered in the lower well with VRP (NP) C2C12 or SAM (NP/LNP) C2C12. (b, c) Frequency of NP+ (black bar) and dsRNA+ (grey bar) BM-DCs from both upper and lower wells after culture with VRP (NP) C2C12 (b) or SAM (NP/LNP) C2C12 (c) are shown. Each bar reports the average plus standard deviation from replicate wells. Data are representative of at least three experiments.
Discussion
SAM vaccines are emerging as a novel class of nucleic-acid-based vaccines. They offer the advantage of being fully synthetic and, like other types of nucleic acid vaccine, they have the potential to combine the positive attributes of live attenuated vaccines while avoiding some of their inherent limitations. It has previously been demonstrated that self-amplifying mRNA vaccines are a promising technology platform able to generate potent, versatile and easily produced vaccines to address the health challenges of the twenty-first century.1,29,30,40,41
However, little has been published on the mechanism of action of these vaccines. Providing a better understanding of the mechanism by which SAM vaccines activate the host immune system may enable a rational design of their development and improve their efficacy.
The aim of the current study was to investigate the relative role of muscle cells and professional APCs in the induction of a cellular CD8 T-cell response.
Based on the fact that myocytes appear to be the predominant cell type transfected after SAM immunization,41 we wanted to better understand the nature of the APCs responsible for priming of the immune response. To address this question we generated chimeric mice where BM-derived APCs and muscle cells are discriminated based on the expression of different MHC class I molecules. Influenza NP, an intracellular protein presented via MHC class I molecule to CD8 T lymphocytes, was used as model antigen.
We have demonstrated that in vivo priming of MHC class-I restricted CD8 T cells following SAM immunization involves BM-derived professional APCs. Moreover, we showed that CD11c+ DCs are required for triggering an antigen-specific CD8 T-cell response upon SAM vaccination. Therefore, myocytes can only provide an antigen source for the induction of an MHC class-I-restricted CD8 T-cell response. In addition, we found that direct expression of the antigen in BM-derived APCs is not necessary to prime CD8 T cells. Therefore, we propose that APCs could acquire the antigen from transfected myoblasts, implicating cross-priming as a mechanism for priming the CD8 T-cell response by SAM vaccines.
However, the cellular mechanism by which cross-priming occurs in vivo, still needs to be defined. One hypothesis is that transfected cells at the site of injection undergo apoptosis during RNA amplification, leading to the release of the antigen-associated apoptotic bodies, which are then phagocytosed by APCs and presented via the MHC class I restricted pathway. Our in vitro experiments showed that RNA and antigen are detected in BM-DCs 2 hr after culture with NP-expressing C2C12 myoblasts. The possibility that during this short time-frame the antigen is produced within DCs at detectable levels is very unlikely. Alternatively, we can assume that the antigen expressed by C2C12 or apoptotic bodies from dying cells is taken up by APCs, together with RNA.
When C2C12 myoblasts were transfected with LNP-formulated SAM (NP), they induced less migration of DCs in vitro compared with VRP transfection. However, we know that VRP have a higher transfection efficiency compared with LNP-delivered SAM, suggesting that RNA transfection itself, replication of the transfected RNA or antigen expression by C2C12 are able to induce soluble factors that may promote migration of APCs.
Similar to our data on SAM, it has previously been shown that in vivo CD8 T-cell priming by DNA vaccines requires presentation by professional APCs and that antigen produced by myocytes can be transferred to BM-derived APCs.53 However, in vivo direct APC transfection after DNA vaccination has been demonstrated by the presence of antigens or reporter genes in macrophages or DCs in situ and by the detection of mRNA encoding for the DNA vaccine antigen.54,55 These data suggest that in the case of DNA vaccines, both cross-presentation of myocyte-derived antigens and direct antigen expression on APCs contribute to CD8 responses. Our data strongly suggest that in the case of SAM vaccines, cross-presentation by APCs of myocyte-derived antigens is the primary mechanism for priming CD8 T cells. Indeed, in our work, we were not able to observe direct transfection of APCs by SAM vaccines either in vitro or in vivo. However, we cannot exclude that some APCs are directly transfected by SAM vectors but express the antigen under the limit of detection of the assays used in this study. Indeed, while the majority of transfection events occur in myocytes, we cannot exclude that a small proportion of transfection events into APCs in vivo could account for the major part of the CD8 T-cell responses.
Currently, we are exploring the use of more sensitive technologies to investigate the antigen expression in the site of injection and the draining lymph nodes such as the two photon microscopy,56 which could improve tracking of both RNA and antigen following SAM vaccination and therefore help to better elucidate the mechanism of action of this unique type of vaccine.
In summary, here we demonstrate the central role played by myocyte-expressed antigens in SAM-based vaccines. Our data confirm and extend previous findings obtained with DNA vaccines and suggest that cross-presentation is the principal mechanism of CD8 T-cell priming in all classes of nucleic acid vaccines.
Acknowledgments
The authors would like to thank the FACS facility and the animal care facility of Novartis Vaccines and Diagnostics s.r.l. for their technical support and Prof. C. Baldari (University of Siena) for helpful discussion. This work was supported in part by funding from the Defence Advanced Research Project Agency (DARPA) under agreement HR0011-12-3-001.
Glossary
- APCs
antigen-presenting cells
- BHK
baby hamster kidney
- BM
bone marrow
- DC
dendritic cells
- DMEM
Dulbecco’s modified Eagle’s medium
- DT
diphtheria toxin
- FBS
fetal bovine serum
- IFN-γ
interferon-γ
- LNP
lipid nanoparticle
- mRNA
messenger RNA
- NP
nucleoprotein
- RANTES
Regulated on Activation, Normal T Expressed and Secreted
- SAM
self-amplifying mRNA
- VRP
viral replicon particle
Author contributions
SL and DM performed the experiments; CG prepared constructs and provided molecular biology support; SM prepared the LNP/RNA formulation; DM and BB supervised the molecular biology and formulation work, respectively; SL, UD and CB designed the study and analysed the experiments. AJG and EDG provided the intellectual and strategic support. SL, EDG, UD and CB wrote the paper.
Disclosures
SL, CG, SM, DM, BB, EDG, UD and CB are former or current employees or contractors of Novartis Vaccines and Diagnostics s.r.l (a GSK company). AJG is a Novartis shareholder and was an employee of Novartis Vaccines, Inc. (now GSK Vaccines) during the time this research was conducted. Funding was provided in part from the part from the Defense Advanced Research Project Agency under agreement HROO l 1-12-3-001.
References
- Deering RP, Kommareddy S, Ulmer JB, Brito LA, Geall AJ. Nucleic acid vaccines: prospects for non-viral delivery of mRNA vaccines. Expert Opin Drug Deliv. 2014;11:885–99. doi: 10.1517/17425247.2014.901308. [DOI] [PubMed] [Google Scholar]
- Delany I, Rappuoli R, De Gregorio E. Vaccines for the 21st century. EMBO Mol Med. 2014;6:708–20. doi: 10.1002/emmm.201403876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koup RA, Douek DC. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harb Perspect Med. 2011;1:a007252. doi: 10.1101/cshperspect.a007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro B, Morrow MP, Hutnick NA, Shin TH, Lucke CE, Weiner DB. Clinical applications of DNA vaccines: current progress. Clin Infect Dis. 2011;53:296–302. doi: 10.1093/cid/cir334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011;239:62–84. doi: 10.1111/j.1600-065X.2010.00980.x. [DOI] [PubMed] [Google Scholar]
- Liu MA. Immunologic basis of vaccine vectors. Immunity. 2010;33:504–15. doi: 10.1016/j.immuni.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9:776–88. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ, et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259:1745–9. doi: 10.1126/science.8456302. [DOI] [PubMed] [Google Scholar]
- Davis BS, Chang GJ, Cropp B, Roehrig JT, Martin DA, Mitchell CJ, et al. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J Virol. 2001;75:4040–7. doi: 10.1128/JVI.75.9.4040-4047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver KA, LaPatra SE, Kurath G. Efficacy of an infectious hematopoietic necrosis (IHN) virus DNA vaccine in Chinook Oncorhynchus tshawytscha and sockeye O. nerka salmon. Dis Aquat Organ. 2005;64:13–22. doi: 10.3354/dao064013. [DOI] [PubMed] [Google Scholar]
- Prinz DM, Smithson SL, Kieber-Emmons T, Westerink MA. Induction of a protective capsular polysaccharide antibody response to a multiepitope DNA vaccine encoding a peptide mimic of meningococcal serogroup C capsular polysaccharide. Immunology. 2003;110:242–9. doi: 10.1046/j.1365-2567.2003.01732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagarazzi ML, Yan J, Morrow MP, Shen X, Parker RL, Lee JC, et al. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci Transl Med. 2012;4:155ra38. doi: 10.1126/scitranslmed.3004414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint S, Renmans D, Broos K, Dewitte H, Lentacker I, Heirman C, et al. The ReNAissanCe of mRNA-based cancer therapy. Expert Rev Vaccines. 2015;14:235–51. doi: 10.1586/14760584.2015.957685. [DOI] [PubMed] [Google Scholar]
- Pascolo S. Messenger RNA-based vaccines. Expert Opin Biol Ther. 2004;4:1285–94. doi: 10.1517/14712598.4.8.1285. [DOI] [PubMed] [Google Scholar]
- Ulmer JB, Mason PW, Geall A, Mandl CW. RNA-based vaccines. Vaccine. 2012;30:4414–8. doi: 10.1016/j.vaccine.2012.04.060. [DOI] [PubMed] [Google Scholar]
- Geall AJ, Mandl CW, Ulmer JB. RNA: the new revolution in nucleic acid vaccines. Semin Immunol. 2013;25:152–9. doi: 10.1016/j.smim.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Luo D, Saltzman WM. Synthetic DNA delivery systems. Nat Biotechnol. 2000;18:33–7. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- Mandl CW, Aberle JH, Aberle SW, Holzmann H, Allison SL, Heinz FX. In vitro-synthesized infectious RNA as an attenuated live vaccine in a flavivirus model. Nat Med. 1998;4:1438–40. doi: 10.1038/4031. [DOI] [PubMed] [Google Scholar]
- Petsch B, Schnee M, Vogel AB, Lange E, Hoffmann B, Voss D, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol. 2012;30:1210–6. doi: 10.1038/nbt.2436. [DOI] [PubMed] [Google Scholar]
- Martinon F, Krishnan S, Lenzen G, Magne R, Gomard E, Guillet JG, et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. 1993;23:1719–22. doi: 10.1002/eji.1830230749. [DOI] [PubMed] [Google Scholar]
- Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Kofler RM, Aberle JH, Aberle SW, Allison SL, Heinz FX, Mandl CW. Mimicking live flavivirus immunization with a noninfectious RNA vaccine. Proc Natl Acad Sci USA. 2004;101:1951–6. doi: 10.1073/pnas.0307145101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C, et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70:9031–40. doi: 10.1158/0008-5472.CAN-10-0699. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Duchardt KM, Lorenz C, Pfeiffer R, Ojkic-Zrna S, Probst J, et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother. 2011;34:1–15. doi: 10.1097/CJI.0b013e3181f7dbe8. [DOI] [PubMed] [Google Scholar]
- Sebastian M, Papachristofilou A, Weiss C, Fruh M, Cathomas R, Hilbe W, et al. Phase Ib study evaluating a self-adjuvanted mRNA cancer vaccine (RNActive®) combined with local radiation as consolidation and maintenance treatment for patients with stage IV non-small cell lung cancer. BMC Cancer. 2014;14:748. doi: 10.1186/1471-2407-14-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonehill A, Van Nuffel AM, Corthals J, Tuyaerts S, Heirman C, Francois V, et al. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res. 2009;15:3366–75. doi: 10.1158/1078-0432.CCR-08-2982. [DOI] [PubMed] [Google Scholar]
- Van Lint S, Goyvaerts C, Maenhout S, Goethals L, Disy A, Benteyn D, et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012;72:1661–71. doi: 10.1158/0008-5472.CAN-11-2957. [DOI] [PubMed] [Google Scholar]
- Van Lint S, Wilgenhof S, Heirman C, Corthals J, Breckpot K, Bonehill A, et al. Optimized dendritic cell-based immunotherapy for melanoma: the TriMix-formula. Cancer Immunol Immunother. 2014;63:959–67. doi: 10.1007/s00262-014-1558-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci USA. 2012;109:14604–9. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekele A, Bertholet S, Archer J, Gibson DG, Palladino G, Brito LA, et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerg Microbes Infect. 2013;2:e52. doi: 10.1038/emi.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer JB, Mansoura MK, Geall AJ. Vaccines ‘on demand’: science fiction or a future reality. Expert Opin Drug Discov. 2015;10:101–6. doi: 10.1517/17460441.2015.996128. [DOI] [PubMed] [Google Scholar]
- Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28:172–6. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–38. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundstrom K. Alphavirus-based vaccines. Viruses. 2014;6:2392–415. doi: 10.3390/v6062392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins GJ, Fleeton MN, Sheahan BJ. Therapeutic and prophylactic applications of alphavirus vectors. Expert Rev Mol Med. 2008;10:e33. doi: 10.1017/S1462399408000859. [DOI] [PubMed] [Google Scholar]
- Perri S, Greer CE, Thudium K, Doe B, Legg H, Liu H, et al. An alphavirus replicon particle chimera derived from Venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J Virol. 2003;77:10394–403. doi: 10.1128/JVI.77.19.10394-10403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Wang RY, Qiu Q, Sugauchi F, Grandinetti T, Alter HJ, et al. Induction of potent cellular immune response in mice by hepatitis C virus NS3 protein with double-stranded RNA. Immunology. 2007;122:15–27. doi: 10.1111/j.1365-2567.2007.02607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Sun T, Yu XH, Liu CQ, Yang YX, Lu P, et al. Immunomodulatory effects of dsRNA and its potential as vaccine adjuvant. J Biomed Biotechnol. 2010;2010:690438. doi: 10.1155/2010/690438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. 2011;208:2357–66. doi: 10.1084/jem.20111171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogers WM, Oostermeijer H, Mooij P, Koopman G, Verschoor EJ, Davis D, et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J Infect Dis. 2015;211:947–55. doi: 10.1093/infdis/jiu522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito LA, Chan M, Shaw CA, Hekele A, Carsillo T, Schaefer M, et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol Ther. 2014;22:2118–29. doi: 10.1038/mt.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Fotin-Mleczek M, Roth G, Becker C, Dam TC, Verdurmen WP, et al. Protein expression from exogenous mRNA: uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011;8:627–36. doi: 10.4161/rna.8.4.15394. [DOI] [PubMed] [Google Scholar]
- Probst J, Weide B, Scheel B, Pichler BJ, Hoerr I, Rammensee HG, et al. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 2007;14:1175–80. doi: 10.1038/sj.gt.3302964. [DOI] [PubMed] [Google Scholar]
- Diken M, Kreiter S, Selmi A, Britten CM, Huber C, Tureci O, et al. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 2011;18:702–8. doi: 10.1038/gt.2011.17. [DOI] [PubMed] [Google Scholar]
- Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. 2011;29:154–7. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465–8. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- Johanning FW, Conry RM, LoBuglio AF, Wright M, Sumerel LA, Pike MJ, et al. A Sindbis virus mRNA polynucleotide vector achieves prolonged and high level heterologous gene expression in vivo. Nucleic Acids Res. 1995;23:1495–501. doi: 10.1093/nar/23.9.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggott JM, Sheahan BJ, Soden DM, O’Sullivan GC, Atkins GJ. Electroporation of RNA stimulates immunity to an encoded reporter gene in mice. Mol Med Rep. 2009;2:753–6. doi: 10.3892/mmr_00000168. [DOI] [PubMed] [Google Scholar]
- Conry RM, LoBuglio AF, Wright M, Sumerel L, Pike MJ, Johanning F, et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995;55:1397–400. [PubMed] [Google Scholar]
- Hochweller K, Striegler J, Hammerling GJ, Garbi N. A novel CD11c.DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells. Eur J Immunol. 2008;38:2776–83. doi: 10.1002/eji.200838659. [DOI] [PubMed] [Google Scholar]
- Heyes J, Palmer L, Bremner K, MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release. 2005;107:276–87. doi: 10.1016/j.jconrel.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- Fu TM, Ulmer JB, Caulfield MJ, Deck RR, Friedman A, Wang S, et al. Priming of cytotoxic T lymphocytes by DNA vaccines: requirement for professional antigen presenting cells and evidence for antigen transfer from myocytes. Mol Med. 1997;3:362–71. [PMC free article] [PubMed] [Google Scholar]
- Donnelly JJ, Liu MA, Ulmer JB. Antigen presentation and DNA vaccines. Am J Respir Crit Care Med. 2000;162:S190–3. doi: 10.1164/ajrccm.162.supplement_3.15tac10. [DOI] [PubMed] [Google Scholar]
- Abdulhaqq SA, Weiner DB. DNA vaccines: developing new strategies to enhance immune responses. Immunol Res. 2008;42:219–32. doi: 10.1007/s12026-008-8076-3. [DOI] [PubMed] [Google Scholar]
- Ishii T, Ishii M. Intravital two-photon imaging: a versatile tool for dissecting the immune system. Ann Rheum Dis. 2011;70(Suppl. 1):i113–5. doi: 10.1136/ard.2010.138156. [DOI] [PubMed] [Google Scholar]