

Abstract
Ghrelin is a 28-amino-acid peptide that stimulates the release of pituitary growth hormone. Because of its orexigenic effects, ghrelin is being developed as a therapeutic option for postoperative support and treatment of anorexia-cachexia syndrome of cancer patients. However, ghrelin has a multiplicity of physiological functions, and it also affects cell proliferation. Therefore, the effects of ghrelin administration on carcinogenesis and cancer progression in patients susceptible to cancer should be clarified. In this study, we examined the effects of ghrelin on cancer promotion in vivo using murine intestinal carcinogenesis models. Intestinal tumorigenesis was examined to determine the effects of either exogenous ghrelin administration or ghrelin deficiency following deletion of the Ghrl gene. Two murine intestinal tumorigenesis models were used. The first was the azoxymethane (AOM)/dextran sodium sulfate (DSS)-induced inflammation-associated colon carcinogenesis model and the second was the ApcMin/+ genetic cancer susceptibility model. In AOM/DSS-treated mice, administration of ghrelin significantly suppressed tumor formation in the colon. In contrast, ghrelin administration did not affect the number of intestinal tumors formed in ApcMin/+ mice. The absence of endogenous ghrelin did not affect the incidence of intestinal tumors in either AOM/DSS-treated mice or ApcMin/+ mice, though tumor size tended to be larger in Ghrl−/− colons in the AOM/DSS model. No tumor-promoting effect was observed by ghrelin administration in either tumorigenesis model. In summary, this study provides in vivo experimental evidence for the usefulness of ghrelin administration in the chemoprevention of inflammation-associated colorectal carcinogenesis and may suggest its safety in patients under colitis-associated cancer susceptibility conditions.
Keywords: Carcinogenesis, colon cancer, ghrelin, mouse model, ulcerative colitis
Ghrelin is a novel hormone known to increase appetite and growth hormone secretion. The effect of ghrelin is mediated by its receptor, growth hormone secretagogue receptor (GHSR) 1a.1–3 Recently, its potential usefulness as a therapeutic option for postoperative support and anorexia-cachexia syndrome of cancer patients has come under investigation.4,5 Clinical studies have suggested that administration of ghrelin improves appetite, resulting in increased food intake, weight gain, and better tolerance of chemotherapy.6 This treatment appears to be safe and well tolerated.4–6 However, many in vitro studies have suggested that ghrelin affects the proliferation and survival of cells, including tumor cells.7,8 Ghrelin is a potent regulator of the growth hormone/insulin-like growth factor I axis. Inappropriate regulation of the axis is involved in tumorigenesis.9,10 Therefore, better understanding of ghrelin's effects on carcinogenesis and cancer progression is required for clinical application of ghrelin, particularly for the supportive care of patients who have cancer or are in a cancer-susceptible condition.
Inflammatory bowel diseases (IBD), such as ulcerative colitis (UC) and Crohn's disease, are chronic, relapsing and remitting diseases that can result in significant long-term morbidity, and their incidence is increasing.11 One important concern in IBD is the risk of colorectal cancer.12 It is known that IBD, particularly UC, is associated with a substantial increase in the risk of colitis-associated colorectal cancer (i.e., colitic cancer) and the increased prevalence of colitic cancer in IBD patients depends on disease severity and duration.12 Indeed, UC increases the cumulative risk of colorectal cancer by up to 18–20% after 30 years of disease.12 UC-associated colorectal cancer patients are younger than patients diagnosed with sporadic disease and frequently have a greater number of multiple cancerous lesions and less favorable outcomes.13 Moreover, as inflammation underlies carcinogenesis in UC,12,13 prophylactic surgery (proctocolectomy) does not always eliminate the patients' carcinogenesis risk. Therefore, better understanding of the disease mechanism and the development of chemoprevention therapy for UC are required. To study the molecular mechanism underlying UC, a rodent colitis model induced by dextran sodium sulfate (DSS) has been commonly used.14 Pretreatment with a carcinogen such as azoxymethane (AOM) in the DSS colitis model can be adapted to the study of UC-associated colorectal carcinoma.15
There is accumulating evidence suggesting that ghrelin has significant anti-inflammatory activities in various tissues.16,17 Indeed, ghrelin reduced intestinal inflammation in mouse and rat models of IBD.18–20 Therefore, ghrelin has the potential to significantly contribute to supportive care for patients of colitic cancer. However, its safety and effects on carcinogenesis in patients susceptible to colitis-associated cancer remain to be determined. In this study, we analyzed the effects of ghrelin administration or ghrelin deficiency on intestinal carcinogenesis in vivo, using two distinctive murine carcinogenesis models. Specifically, we used: (i) a UC-associated colorectal carcinogenesis model with combined treatment with AOM and DSS; and (ii) a genetic intestinal tumor susceptibility model based upon mutation of the Apc gene. We found that exogenous administration of ghrelin caused a dramatic decrease in tumor incidence in the AOM/DSS colon carcinogenesis model but not in the mutant Apc model. Loss of ghrelin did not affect the incidence of intestinal tumor formation in either model.
Materials and Methods
Reagents
Acylated ghrelin was obtained from Asubio Pharma (Kobe, Japan). AOM was purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) and DSS with a molecular weight of 36–50 kDa was obtained from MP Biomedicals (Solon, OH, USA). The same lot of DSS was used in all experiments. For immunohistochemistry, following antibodies were used: anti-β-catenin rabbit polyclonal antibody (pAb) (Sigma-Aldrich Chemical Co.), anti-F4/80 rat mAb (Serotec, Oxford, UK) and anti-mouse myeloperoxidase (MPO) rabbit pAb (Thermo Scientific, Waltham, MA, USA), anti-mouse cleaved caspase-3 rabbit pAb (Cell Signaling Technology, Tokyo, Japan), anti-Ki-67 rabbit mAb (Abcam, Cambridge, UK), anti-CD31 rabbit pAb (AnaSpec, San Jose, CA, USA).
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Miyazaki. Mice were housed in plastic cages (four or five mice per cage) in a specific pathogen-free condition with free access to drinking water and a basal diet (Oriental Yeast Co., Ltd., Tokyo, Japan). C57BL/6 mice were obtained from Kyudo (Saga, Japan). Ghrelin knockout (Ghrl−/−) mice were described previously.21 ApcMin/+ mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Ghrelin-deleted Apc mutant mice were established by crossing Ghrl−/− and ApcMin/+ mice.
Murine intestinal tumorigenesis models
The effects of ghrelin deficiency or ghrelin administration on intestinal carcinogenesis were analyzed in genetic and inflammation-associated mouse models. In the inflammation-associated colon carcinogenesis model, the AOM/DSS treatment-induced carcinogenesis protocol was used as previously described.22 Briefly, 8-week-old male C57BL/6 and Ghrl−/− mice were given a single intraperitoneal injection of AOM (10 mg/kg body weight). Starting 1 week after the AOM injection, they received 2% DSS in their drinking water for 7 days followed by a 1-week recovery period with water without DSS. DSS was dissolved in distilled water at a concentration of 2% (w/v) just before use to induce colitis as described.23 This DSS treatment was performed four times. For mice treated with ghrelin, intraperitoneal injection of ghrelin (3 nmol/day, dissolved in saline) was administered during the DSS-treating periods. All mice were killed at week 12 by a lethal dose of sodium pentobarbital and autopsied to determine the effects of ghrelin administration or ghrelin deficiency on colorectal carcinogenesis induced by AOM and DSS. ApcMin/+ mice were used for the genetic intestinal tumorigenesis model. Ghrelin-deficient ApcMin/+ (ApcMin/+/Ghrl−/−) mice were obtained by crossing ApcMin/+ mice with Ghrl−/− mice on a C57BL/6 genetic background. The mice were treated with intraperitoneal administration of saline with or without ghrelin (3 nmol/day) for 4 weeks and were killed 15 weeks after birth. The Apc mutant mice were killed by a lethal dose of sodium pentobarbital at 15 weeks of age and autopsied to evaluate the number and sizes of tumors formed. Scoring of the tumor size was calculated as described previously.22 Histopathological analysis was performed on paraffin-embedded sections after H&E staining.
Murine experimental colitis models
To better understand the effect of ghrelin administration on inflammatory response, we generated an experimental colitis model. Eight-week-old male C57BL/6 mice were treated with 2% DSS for 7 days with intraperitoneal administration of saline with or without ghrelin (3 mmol/day), and then killed. Intestinal tissues were excised and used for RT-PCR and histological analysis.
RT-PCR
Total RNA was prepared with TRIzol (Life Technologies Japan, Tokyo, Japan) followed by DNase I (Takara Bio, Shiga, Japan) treatment. For RT-PCR, 3 μg total RNA was reverse-transcribed with a mixture of Oligo (dT)12-18 (Life Technologies Japan) and random primers (6 mers) (Takara Bio) using 200 units of ReverTra Ace (TOYOBO, Osaka, Japan), and 1/30 of the resulting cDNA was processed for each PCR reaction with 0.2 μM of both forward and reverse primers and AmpliTaq Gold PCR Master Mix (Life Technologies Japan). The primer sequences for β-actin, GHSR1a, GHSR1b, ghrelin, interferon-γ (INF-γ), tumor necrosis factor α (TNFα), interleukin (IL)-1β and IL-6 are described in Table1.
Table 1.
Primers for RT-PCR
| Target | Forward | Reverse | Size |
|---|---|---|---|
| β-actin | 5′TGACAGGATGCAGAAGGAGA3′ | 5′GCTGGAAGGTGGACAGTGAG3′ | 131 |
| GHSR1a | 5′AGCACTTGGGAAGTTGAGGC3′ | 5′TAACCACAACAGCCTGCACC3′ | 234 |
| GHSR1b | 5′ACGCTGTGGGGAGCACGACA3′ | 5′CGCGAGCAGCAGGAAGGATG3′ | 321 |
| Ghrelin | 5′TCTGCAGTTTGCTGCTACTCA3′ | 5′CCTCTTTGACCTCTTCCCAGA3′ | 318 |
| INF-γ | 5′CAGCAACAGCAAGGCGAAA3′ | 5′TCAGCAGCGACTCCTTTTC3′ | 159 |
| TNFα | 5′TCGAGTGACAAGCCTGTAGC3′ | 5′GGAGGTTGACTTTCTCCTGG3′ | 255 |
| IL-1β | 5′GTAATGAAAGACGGCACACCC3′ | 5′GTGCTGATGTACCAGTTGGG3′ | 157 |
| IL-6 | 5′AGCCAGAGTCCTTCAGAGAG3′ | 5′ACTCCTTCTGTGACTCCAGC3′ | 136 |
GHSR, growth hormone secretagogue receptor; IL, interleukin; IFN, interferon; TNF, tumor necrosis factor.
Immunohistochemical analyses
Intestinal tissues of DSS-induced colitis model mice were fixed in 4% paraformaldehyde in PBS overnight and then dehydrated and embedded in paraffin. Immunohistochemistry was performed according to the method described previously.22,23
Statistical analysis
Statistical analysis was done using SPSS 15.0 software (SPSS Japan Inc., Tokyo, Japan). Comparison between two unpaired groups was made with the Mann–Whitney U-test. Significance was set at P < 0.05.
Results
Administration of ghrelin significantly suppressed inflammation-associated colon carcinogenesis
We analyzed the effect of ghrelin on inflammation-associated colon carcinogenesis using an AOM/DSS mouse model. Two independent experiments (Exp. 1 and Exp. 2) were performed. Treatment of the mice with 2% DSS in drinking water resulted in temporary loss of body weight in control mice (Fig.1). Notably, while 25% of mice (1/6 at week 5 and 2/6 at week 9 or 11 in Exp. 1 and Exp. 2, respectively) died before the termination of the experiment in control saline group, none of the ghrelin administration group (0/6 and 0/5 in Exp.1 and Exp.2, respectively) died during the experiments (Table2). Even though these fatal cases were excluded from the control group in a mean body weight analysis, there may be a tendency for the intraperitoneal administration of ghrelin (3 nmol/day) to alleviate the body weight loss after DSS treatment, and a statistically significant difference was observed at week 4 (Fig.1). After 12 weeks of AOM treatment, mice were killed and autopsied. After AOM treatment, ghrelin administration during DSS treatment significantly reduced the multiplicity of adenocarcinomas in the colon (inhibition rates, 94%; P < 0.0001) compared with the control AOM/DSS group (Fig.2). The mean spleen size indicated by spleen/body weight ratio was likely lower (P = 0.08) in the ghrelin group compared to the control group, which may suggest the alleviation of inflammation by ghrelin.
Figure 1.
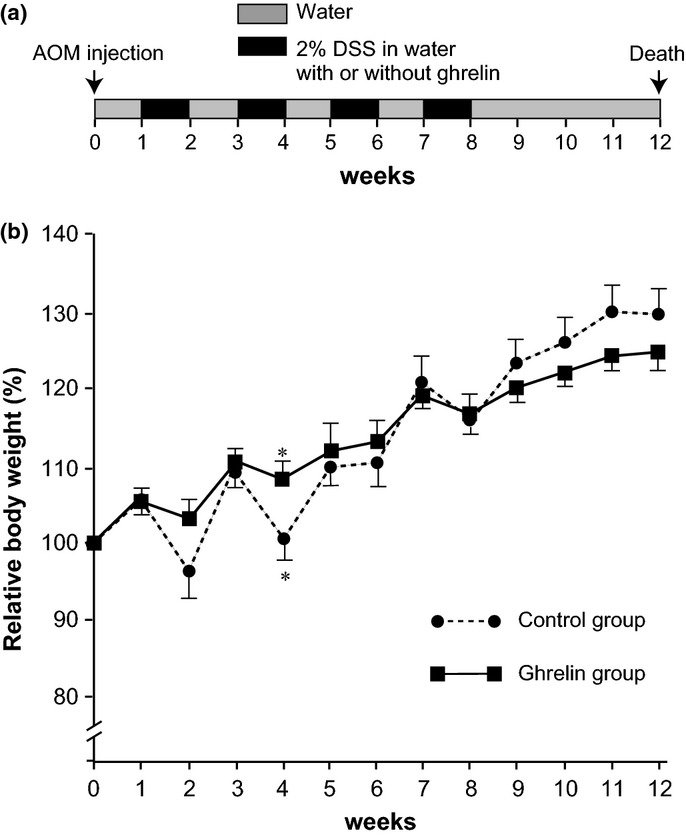
Effect of ghrelin administration on body weight gain in azoxymethane (AOM)/dextran sodium sulfate (DSS) carcinogenesis. (a) The treatment protocol for the experiment. (b) Relative body weight (mean ± SEM) of control (n = 9) and ghrelin (n = 11) groups. Data from three mice that died before the termination of the experiments (Table2) were not included in this graph, which may result in slightly better, though not statistically significant, body weight gain of control mice at week 9–12. *P < 0.001 between two groups.
Table 2.
Effect of ghrelin administration on azoxymethane (AOM)/dextran sodium sulfate (DSS)-induced colon carcinogenesis
| Group (n) | Number of tumors per mouse | Total tumor number of each size | ||
|---|---|---|---|---|
| <2 mm | 2–3 mm | >3 mm | ||
| Exp. 1 | ||||
| Saline (6) | 5, 2, 5, 3, 2, † | 2 | 3 | 12 |
| Ghrelin (6) | 0, 0, 1, 0, 0, 0 | 0 | 1 | 0 |
| Exp. 2 | ||||
| Saline (6) | 3, 3, 3, 2, †, † | 8 | 3 | 0 |
| Ghrelin (5) | 0, 0, 0, 0, 1 | 1 | 0 | 0 |
Died before the termination of the experiment.
Figure 2.
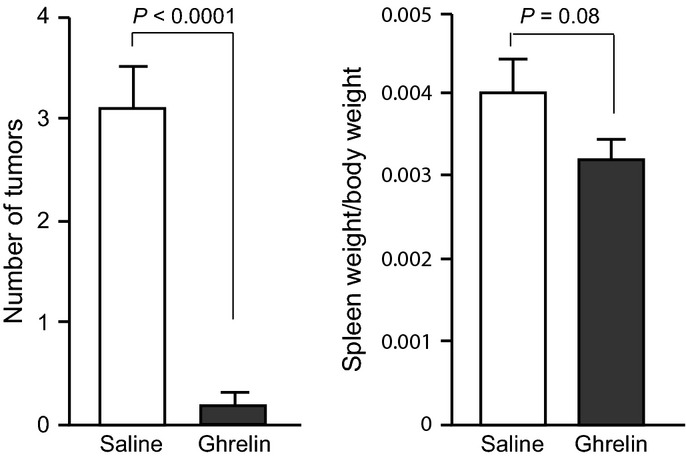
Effect of ghrelin administration on colonic carcinogenesis in the azoxymethane (AOM)/dextran sodium sulfate (DSS) model. (a) Number of tumors (mean ± SEM) was significantly reduced in the ghrelin administration group (ghrelin, n = 11) compared to the control group (saline, n = 9). *P < 0.0001. (b) Spleen weight relative to body weight (mean ± SEM) was modestly reduced by ghrelin treatment (P = 0.08).
To obtain mechanistic insights into the suppressive effects of ghrelin treatment on carcinogenesis, we determined the expression levels of ghrelin receptors in the colons of mice that received 2% DSS in their drinking water for 7 days. The expression levels of endogenous ghrelin, Ghsr1a and Ghsr1b mRNA levels were not altered by ghrelin administration (Fig.3a). In the proximal colon, mRNA levels of pro-inflammatory cytokines tended to be suppressed by ghrelin administration with a statistically significant reduction of the Il1b mRNA level. Interestingly, those differences were not observed in the distal colon, although the distal colon was more susceptible to tumor formation (Fig.3a,b). However, histological and immunohistochemical analysis suggested that the infiltration of F4/80-positive macrophages and MPO-positive neutrophils in the ulcerated portion of distal colon tended to be less in ghrelin-treated mice (Fig.3c).
Figure 3.

Expression of ghrelin, growth hormone secretagogue receptor (GHSR) and pro-inflammatory cytokines in colon tissue of mice after dextran sodium sulfate (DSS) treatment (2% in drinking water, 7 days). (a) RT-PCR analysis of extracts from proximal (pLI) and distal (dLI) colon tissue of mice without (saline) or with ghrelin administration. Data from three mice are indicated for each group. (b) Quantification of pro-inflammatory cytokine mRNA levels by real-time RT-PCR in proximal colon tissue (n = 5 for each group). *P < 0.05. (c) Histological analysis of distal colon on day 7 of DSS treatment. Representative colon tissues with ulcers of similar sizes from control and ghrelin-treated (ghrelin i.p.) mice are shown. Immunostaining photos for macrophage marker (F4/80) and neutrophil marker (MPO) are shown in the lower right panel. Bar, 100 μm.
Deletion of the Ghrl gene did not affect tumor incidence in the inflammation-associated colon carcinogenesis model
Given that exogenous ghrelin administration suppressed AOM/DSS-induced colitis-associated carcinogenesis, we asked whether genetic deletion of endogenous ghrelin enhanced carcinogenesis in this model. We found that the incidence of colon tumor formation was not altered by the deletion of the Ghrl gene (Fig.4a), suggesting that the physiological level of ghrelin may not be functioning as a carcinogenesis suppressor. Alternatively, it is possible that another mechanism compensated for the global absence of ghrelin in Ghrl−/− mice. There was a tendency for tumors formed in Ghrl−/− mice to be larger than those formed in the wild-type mice (Fig.4b,c). Histological findings of the tumors were essentially similar between the two groups, showing a well-differentiated tubular adenocarcinoma (Fig.4d). We also examined Ki-67 labeling index, number of cleaved caspase 3-positive cells, and density of CD31-positive vessels immunohistochemically. However, statistically significant differences were not observed between wild-type and the Ghrl−/− tumors regarding these parameters (data not shown).
Figure 4.
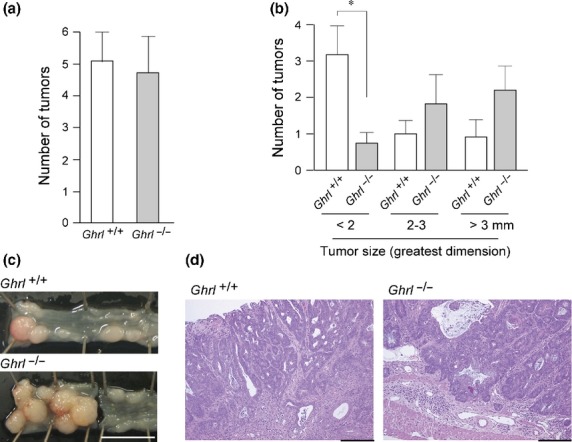
Effect of Ghrl gene deletion on azoxymethane (AOM)/dextran sodium sulfate (DSS)-induced carcinogenesis. (a) Number of tumors (mean ± SE) in the colon of control (Ghrl+/+; n = 5) and ghrelin-deficient (Ghrl−/−; n = 5) mice. (b) Size distribution of the tumors. *P < 0.01. (c) Representative macroscopic images of the colon with tumors. Bar, 1 cm. (d) Histology of tumors (H&E stain). Bar, 200 μm.
Ghrelin did not alter tumor susceptibility in Apc mutant mice
We hypothesized that the suppressing effect of ghrelin on AOM/DSS-induced carcinogenesis could be mediated by either modulation of the cancer-promoting effects of the microenvironment or a direct inhibitory effect on cellular growth. To distinguish between these possibilities, we analyzed the effects of ghrelin administration on intestinal tumor formation in ApcMin/+ mice, a genetic susceptibility model for intestinal tumors. The mice were treated with intraperitoneal administration of saline with or without ghrelin (3 nmol/day) for 4 weeks and were killed 15 weeks after birth (Fig.5a). The intraperitoneal administration of ghrelin did not affect the incidence and size of intestinal tumors (polyps) compared to the saline-treated control mice (Fig.5b,c). Mean body weight of the mice and blood hemoglobin concentration were not altered by the treatment (Fig.5d,e). Nonetheless, nuclear translocation of β-catenin was modestly decreased in ghrelin-treated mice (Fig.5f). Intestinal tumor formation was observed predominantly in the small intestine in both groups, indicating that ghrelin treatment did not alter the distribution of tumors.
Figure 5.
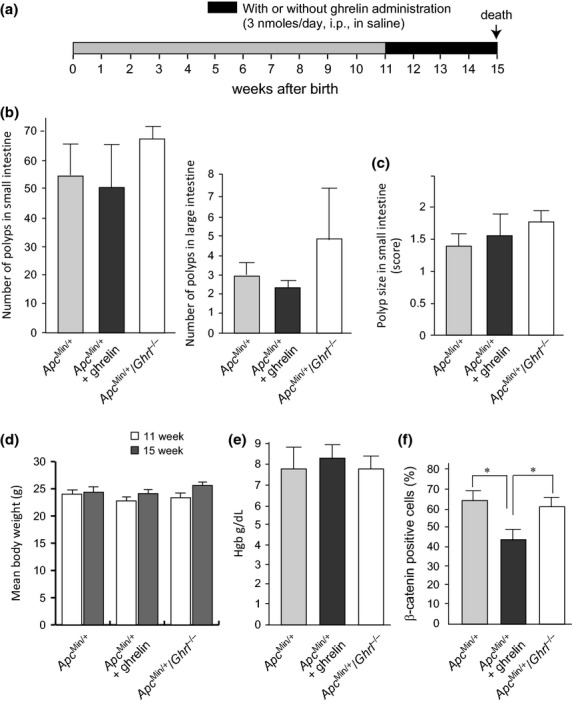
Effect of ghrelin administration or deletion on intestinal tumorigenesis in Apc-mutant mice. (a) Treatment protocol. (b) Number of intestinal polyps (mean ± SEM) of control (APCmin/+; n = 5), ghrelin-treated (APCmin/+ + ghrelin; n = 5) and ghrelin-deficient (APCmin/+/Ghrl−/−; n = 5) APCmin/+ mice. (c) Mean score of polyp size.22 Error bar, SEM. (d) Mean body weight of the mice. Error bar, SEM. (e) Mean hemoglobin concentration of the mice. Error bar, SEM. (f) Mean number of nuclear β-catenin-positive cells in polyp lesions. Error bar, SEM. *P < 0.005.
Finally, we asked whether knocking out ghrelin would affect tumorigenesis in ApcMin/+ mice. While mean intestinal tumor number was modestly increased in ApcMin/+ Ghrl−/− mice compared to control mice, the difference was not statistically significant (P = 0.257) (Fig.5b). Tumor size was not altered by the ghrelin deficiency (Fig.5c). Tumor histologies were similar in all groups, and invasive carcinoma was not observed at the time point of autopsy (15 weeks after birth) (data not shown). We concluded that neither ghrelin administration nor deletion altered intestinal tumorigenesis in Apc mutant mice.
Discussion
To investigate the role of ghrelin in intestinal carcinogenesis, we examined the effect of ghrelin administration or ghrelin loss in the AOM/DSS inflammation-associated colon carcinogenesis model and in an Apc mutant mouse model. The data presented here indicated that administration of ghrelin significantly suppressed tumor incidence in the AOM/DSS carcinogenesis model, but not in the mutant Apc gene-mediated intestinal tumorigenesis model. This study also revealed that ghrelin does not have tumor-promoting effects in either of the intestinal tumor susceptibility models.
The roles of ghrelin in tumor cell biology have been actively investigated for some time. However, most studies analyzed the effects of ghrelin and/or GHSR on cultured cells in vitro or their immunohistochemical expression levels in vivo. These studies have led to controversial results.8 For example, ghrelin may act as either an anti-apoptotic or pro-apoptotic factor in different cancer cells,24,25 suggesting that the effects of ghrelin on cancer cells may be cell type-dependent. Similarly, diverse results have been reported for the growth promoting effect of ghrelin on cancer cells, including colon cancer cells.8,26,27 In the present study, deletion of the Ghrl gene did not affect the number of Ki-67-positive proliferating cells or cleaved caspase 3-positive apoptotic cells in the tumors formed in the AOM/DSS model, though the tumor sizes tended to be larger in the Ghrl−/− mice. This suggests that the onset of inflammation-associated tumor formation may be earlier in the Ghrl−/− mice compared to wild-type mice. Circulating ghrelin levels were also analyzed in various cancers.8 In colorectal cancer patients, total ghrelin levels in serum were increased28 and the serum levels were high in the early stage and low in the late stage of cancer.29 The effects of ghrelin on carcinogenesis have not been studied. In this paper, we provide for the first time direct experimental evidence regarding ghrelin's effects on carcinogenesis in vivo.
The AOM/DSS-induced colon cancer model is commonly used to explore UC-associated colorectal carcinoma.15 In this model, DSS causes oxidative/nitrosative DNA damage in the inflamed colon, and it eventually results in colitis-related colorectal adenocarcinoma (colitic cancer). The control of inflammation can be key for the prevention of colitic cancer. The observed tumor-suppressive effect of ghrelin administration is likely mediated by the anti-inflammatory properties of ghrelin that have been reported in a variety of tissues.6,16,17 In fact, ghrelin suppresses the expression of pro-inflammatory cytokines in colitis models.5,18,21 Furthermore, a recent report suggests that ghrelin prevented the breakdown of intestinal barrier function in DSS-induced colitis, which in turn protects the mucosa from inflammatory and/or carcinogenic stimuli.30 This presumptive mechanism underlying ghrelin's effect in the AOM/DSS model was also supported by the observations made in our Apc mutant mouse model. Ghrelin administration did not show tumor-suppressive properties in ApcMin/+ mice in which an inborn error of the Apc gene already exists and which plays a central role in intestinal tumorigenesis.
In contrast to the ghrelin administration study, homozygous deletion of the Ghrl gene did not enhance the tumor incidence in the AOM/DSS model. This discrepancy may indicate that the physiological level of endogenous ghrelin is not sufficient to exert the anti-inflammatory action necessary to prevent the inflammation-associated colon carcinogenesis. Alternatively, a redundant mechanism may develop that compensates for the anti-inflammatory role of endogenous ghrelin in the case of genetic loss of ghrelin. However, while the tumor incidence was not altered, sizes of the established tumors tended to be larger in Ghrl−/− mice compared to wild-type mice in the AOM/DSS carcinogenesis model. Those results suggest that global loss of endogenous ghrelin may favor the growth of colon cancer cells in vivo. The mechanism underlying this observation remains to be determined.
Ghrelin has the potential to prevent or reverse cancer-associated cachexia, and recent studies have shown improvements in weight stabilization and appetite with its short-term usage.5,6,31 In colorectal cancer patients, postoperative ghrelin administration accelerates gastric emptying and time to first bowel movement.32 However, for further clinical application of ghrelin in cancer patients, more studies are required to fully characterize the role of ghrelin in cancer biology and to establish the safety of ghrelin administration in cancer patients who are frequently susceptible to relapse. For patients with IBD, particularly UC, there is an increased risk of colon cancer in proportion to disease activity and duration.12,13 This study indicates that ghrelin does not promote colitic cancer in the murine UC model and thus, suggests its safety when used as a postoperative supportive medicine in UC-associated colorectal cancer patients. Moreover, this peptide may serve as a significant cancer-preventive compound for IBD-associated colorectal cancer, mechanisms of which should be clarified in future studies.
Acknowledgments
This work was supported in part by Health and Labor Science Research Grants of Japan and Ministry of Education, Science, Sports and Culture, Japan, grants-in-aid for scientific research 24390099.
Glossary
Abbreviations:
- AOM
azoxymethane
- DSS
dextran sodium sulfate
- GHSR
growth hormone secretagogue receptor
- IBD
inflammatory bowel diseases
- IL
interleukin
- INF
interferon
- MPO
myeloperoxidase
- TNF
tumor necrosis factor
- UC
ulcerative colitis
Disclosure Statement
The authors declare no financial or commercial conflicts of interests.
References
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–60. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Nakazato M, Murakami N, Date Y, et al. A role for ghrelin in the central regulation of feeding. Nature. 2001;409:194–8. doi: 10.1038/35051587. [DOI] [PubMed] [Google Scholar]
- Sato T, Nakamura Y, Shiimura Y, Ohgusu H, Kangawa K, Kojima M. Structure, regulation and function of ghrelin. J Biochem. 2012;151:119–28. doi: 10.1093/jb/mvr134. [DOI] [PubMed] [Google Scholar]
- Ali S, Chen J, Garcia JM. Clinical development of ghrelin axis-derived molecules for cancer cachexia treatment. Curr Opin Support Palliat Care. 2013;7:368–75. doi: 10.1097/SPC.0000000000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CK, Wu JC-Y. Role of ghrelin in the pathophysiology of gastrointestinal disease. Gut Liv. 2013;7:505–12. doi: 10.5009/gnl.2013.7.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiguchi S, Murakami K, Yanagimoto Y, et al. Clinical application of ghrelin in the field of surgery. Surg Today. 2015;45:801–7. doi: 10.1007/s00595-014-1040-z. [DOI] [PubMed] [Google Scholar]
- Waseem T, Javaid-Ur R, Ahmad F, Azam M, Qureshi MA. Role of ghrelin axis in colorectal cancer: a novel association. Peptides. 2008;29:1369–76. doi: 10.1016/j.peptides.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos D, Theocharis S, Kouraklis G. Ghrelin: a potential therapeutic target for cancer. Regul Pept. 2010;163:7–17. doi: 10.1016/j.regpep.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Iwakura H, Ariyasu H, Li Y, et al. A mouse model of ghrelinoma exhibited activated growth hormone-insulin-like growth factor I axis and glucose intolerance. Am J Physiol Endocrinol Metab. 2009;297:E802–11. doi: 10.1152/ajpendo.00205.2009. [DOI] [PubMed] [Google Scholar]
- Jeffery PL, Herington AC, Chopin LK. The potential autocrine/paracrine roles of ghrelin and its receptor in hormone-dependent cancer. Cytokine Growth Factor Rev. 2003;14:113–22. doi: 10.1016/s1359-6101(02)00089-8. [DOI] [PubMed] [Google Scholar]
- M'Koma AE. Inflammatory bowel disease: an expanding global health problem. Clin Med Insights Gastroenterol. 2013;6:33–47. doi: 10.4137/CGast.S12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229–44. doi: 10.1007/s00281-012-0352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345:235–41. doi: 10.1016/j.canlet.2013.07.032. [DOI] [PubMed] [Google Scholar]
- Jones-Hall YL, Grisham MB. Immunopathological characterization of selected mouse models of inflammatory bowel disease: comparison to human disease. Pathophysiology. 2014;21:267–88. doi: 10.1016/j.pathophys.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Clapper ML, Cooper HS, Chang W-CL. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007;28:1450–9. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- Takata A, Takiguchi S, Miyazaki Y, et al. Randomized phase II study of the anti-inflammatory effect of ghrelin during the postoperative period of esophagectomy. Ann Surg. 2014 doi: 10.1097/SLA.0000000000000986. ; doi: 10.1097/SLA.0000000000000986. [DOI] [PubMed] [Google Scholar]
- Sun GX, Ding R, Li M, et al. Ghrelin attenuates renal fibrosis and inflammation of obstructive nephropathy. J Urol. 2015;193:2107–115. doi: 10.1016/j.juro.2014.11.098. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Chorny A, Delgado M. Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology. 2006;130:1707–20. doi: 10.1053/j.gastro.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Pamukcu O, Kumral ZN, Ercan F, Yegen BC, Ertem D. Anti-inflammatory effect of obestatin and ghrelin in dextran sulfate sodium-induced colitis in rats. J Pediatr Gastroenterol Nutr. 2013;57:211–8. doi: 10.1097/MPG.0b013e318294711e. [DOI] [PubMed] [Google Scholar]
- Konturek PC, Brzozowski T, Engel M, et al. Ghrelin ameliorates colonic inflammation. Role of nitric oxide and sensory nerves. J Physiol Pharmacol. 2009;60:41–7. [PubMed] [Google Scholar]
- Sato T, Kurokawa M, Nakashima Y, et al. Ghrelin deficiency does not influence feeding performance. Regul Pept. 2008;145:7–11. doi: 10.1016/j.regpep.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Hoshiko S, Kawaguchi M, Fukushima T, et al. Hepatocyte growth factor activator inhibitor type 1 is a suppressor of intestinal tumorigenesis. Cancer Res. 2013;73:2659–70. doi: 10.1158/0008-5472.CAN-12-3337. [DOI] [PubMed] [Google Scholar]
- Kawaguchi M, Takeda N, Hoshiko S, et al. Membrane-bound serine protease inhibitor HAI-1 is required for maintenance of intestinal epithelial integrity. Am J Pathol. 2011;179:1815–26. doi: 10.1016/j.ajpath.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XT, Fan XM, Zha XL. Ghrelin inhibits 5-fluorouracil-induced apoptosis in colonic cancer cells. J Gastroenterol Hepatol. 2011;26:1169–73. doi: 10.1111/j.1440-1746.2011.06715.x. [DOI] [PubMed] [Google Scholar]
- Bonfili L, Cuccioloni M, Cecarini V, et al. Ghrelin induces apoptosis in colon adenocarcinoma cells via proteasome inhibition and autophagy induction. Apoptosis. 2013;18:1188–200. doi: 10.1007/s10495-013-0856-0. [DOI] [PubMed] [Google Scholar]
- Lawnicka H, Mełeń-Mucha G, Motylewska E, Mucha S, Stępień H. Modulation of ghrelin axis influences the growth of colonic and prostatic cancer cells in vitro. Pharmacol Rep. 2012;64:951–9. doi: 10.1016/s1734-1140(12)70890-3. [DOI] [PubMed] [Google Scholar]
- Tian C, Zhang L, Hu D, Ji J. Ghrelin induces gastric cancer cell proliferation, migration, and invasion through GHS-R/NF-κB signaling pathway. Mol Cell Biochem. 2013;382:163–72. doi: 10.1007/s11010-013-1731-6. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos D, Theocharis S, Moutsios-Rentzos A, Kouraklis G, Kostakis A. The role of serum total ghrelin level elevation in colon cancer patients. J BUON. 2014;19:388–93. [PubMed] [Google Scholar]
- Huang Q, Fan YZ, Ge BJ, Zhu Q, Tu ZY. Circulating ghrelin in patients with gastric or colorectal cancer. Dig Dis Sci. 2007;52:803–9. doi: 10.1007/s10620-006-9508-3. [DOI] [PubMed] [Google Scholar]
- Cheng J, Zhang L, Dai W, et al. Ghrelin ameliorates intestinal barrier dysfunction in experimental colitis by inhibiting the activation of nuclear factor-kappa B. Biochem Biophys Res Commun. 2015;458:140–7. doi: 10.1016/j.bbrc.2015.01.083. [DOI] [PubMed] [Google Scholar]
- Tsubouchi H, Yanagi S, Miura A, Matsumoto N, Kangawa K, Nakazato M. Ghrelin relieves cancer cachexia associated with the development of lung adenocarcinoma in mice. Eur J Pharmacol. 2014;743:1–10. doi: 10.1016/j.ejphar.2014.09.025. [DOI] [PubMed] [Google Scholar]
- Falkén Y, Webb DL, Abraham-Nordling M, Kressner U, Hellström PM, Näslund E. Intravenous ghrelin accelerates postoperative gastric emptying and time to first bowel movement in humans. Neurogastroenterol Motil. 2013;25:474–80. doi: 10.1111/nmo.12098. [DOI] [PubMed] [Google Scholar]