

Background: Cobra's “three-finger” nonconventional toxin WTX allosterically modulates muscarinic receptors (mAChRs).
Results: Activity of several WTX mutants was analyzed; toxin spatial structure and dynamics were determined; and complexes of toxin with M1 and M3 mAChRs were modeled.
Conclusion: Flexible loop II is the major determinant for toxin binding to different mAChRs.
Significance: Structural framework for rationalization of target-specific positive/negative allosteric regulation of mAChRs is provided.
Keywords: computer modeling, G protein-coupled receptor (GPCR), nuclear magnetic resonance (NMR), protein dynamic, recombinant protein expression, site-directed mutagenesis, snake neurotoxin
Abstract
Weak toxin from Naja kaouthia (WTX) belongs to the group of nonconventional “three-finger” snake neurotoxins. It irreversibly inhibits nicotinic acetylcholine receptors and allosterically interacts with muscarinic acetylcholine receptors (mAChRs). Using site-directed mutagenesis, NMR spectroscopy, and computer modeling, we investigated the recombinant mutant WTX analogue (rWTX) which, compared with the native toxin, has an additional N-terminal methionine residue. In comparison with the wild-type toxin, rWTX demonstrated an altered pharmacological profile, decreased binding of orthosteric antagonist N-methylscopolamine to human M1- and M2-mAChRs, and increased antagonist binding to M3-mAChR. Positively charged arginine residues located in the flexible loop II were found to be crucial for rWTX interactions with all types of mAChR. Computer modeling suggested that the rWTX loop II protrudes to the M1-mAChR allosteric ligand-binding site blocking the entrance to the orthosteric site. In contrast, toxin interacts with M3-mAChR by loop II without penetration into the allosteric site. Data obtained provide new structural insight into the target-specific allosteric regulation of mAChRs by “three-finger” snake neurotoxins.
Introduction
Snake venom “three-finger” toxins are small β-structural proteins (60–75 amino acids) that consist of three loops (“fingers”) protruding from the compact globular core (“head”) stabilized by four invariant disulfide bonds (1). Separate groups of three-finger toxins act on numerous targets (2), including two classes of acetylcholine receptors as follows: nicotinic acetylcholine receptors (nAChRs)2 belonging to ligand-gated ion channels (3) and muscarinic acetylcholine receptors (mAChRs) of the G protein-coupled receptor (GPCR) family (4). Both types of acetylcholine receptors are responsible for cholinergic signaling in central and peripheral nervous systems and could be considered as promising targets for treatment of a number of disorders, including Alzheimer disease, schizophrenia, and Parkinson disease and chronic obstructive pulmonary disease (3, 5, 6).
Curare mimetic toxins or α-neurotoxins (α-NTs, Fig. 1) inhibiting nAChRs are generally subdivided into two subclasses: short-chain type (four disulfide bonds) and long-chain type (additional fifth disulfide bond is in the central loop II) (1, 2). At present, the central loop II is considered as the major site responsible for interaction of short- and long-chain α-NTs with the different subtypes of nAChRs (7–9).
FIGURE 1.

Alignment of amino acid sequences of rWTX, nonconventional toxins from Naja species, α-neurotoxins, muscarinic toxins, and water-soluble domain of human Lynx1 (ws-Lynx1). Cysteines are shown over a dark gray background. Position of the rWTX loops is shown by the light gray background. Additional N-terminal methionine residue in the rWTX sequence appearing due to translation of the starting atg codon (underlined) is shown. Sequence similarity between rWTX and other proteins was calculated by EMBOSS Stretcher (EMBL-EBI). Amino acid residue numbering is given for wild-type WTX.
There is also a large group of nonconventional toxins with fifth disulfide bond in the N-terminal loop I that usually block nAChRs less efficiently as compared with α-NTs. Function and molecular targets of nonconventional toxins are not comprehensively studied (2). Most of these toxins are characterized by a low level of toxicity (LD50, 5–80 mg/kg), which is significantly less than that of short- and long-chain α-NTs (LD50, 0.04–0.3 mg/kg) (10). The phylogenetic analysis revealed that the family of nonconventional toxins is not homogeneous (11). Some of them, mostly from Bungarus spp., resemble long chain α-NTs, but others, mostly from Naja spp., are similar to three-finger mamba muscarinic toxins (MTs) (11).
MTs (four disulfide bonds) interact with different subtypes of mAChRs and could act as allosteric agonists, antagonists, or modulators (12). The muscarinic toxins MT3 and MT7 from Dendroaspis angusticeps have high specificity for M4 and M1 mAChRs, respectively, although others MTs are less selective (12). Recent mutagenesis data and computer modeling revealed that the positively charged Arg-34 residue located in the central loop II of MT7 represents a major determinant associated with the toxin's high affinity against human M1 mAChR (13).
Nonconventional “weak” toxin from Naja kaouthia (WTX) is nonlethal to mice at concentrations up to 20 mg/kg and shares some biological properties with α-NTs and MTs. WTX irreversibly binds to muscle and α7 nAChRs with IC50 values in the micromolar range (14) and could allosterically interact with different subtypes of mAChRs modulating binding of orthosteric ligands, like acetylcholine and N-methylscopolamine (NMS) (15). Computer modeling study proposed that WTX interacts with nAChRs via the mechanism generally resembling the α-NTs binding with the formation of numerous contacts between the positively charged central loop II and the acetylcholine binding pocket of the receptor (16). However, the molecular mechanism of WTX interaction with mAChRs was not previously described.
Based on the similarity with MT7 (sequence identity ∼59%, Fig. 1), we supposed that the loop II could also be involved in the interaction of WTX with mAChRs. To verify this hypothesis and to define the role of distinct WTX structural features in its activity, the recombinant WTX analogue (rWTX), having additional N-terminal Met residue, and seven of its mutant variants were used. The experiments on competitive binding with [3H]NMS revealed that rWTX demonstrates target-specific activity decreasing NMS binding to M1 and M2 mAChRs and increasing NMS binding to M3 receptors. Mutagenesis data confirmed that arginine residues from the toxin's loop II play an essential role in the rWTX interaction with the M1-, M2-, and M3-mAChRs. The established NMR structure made possible computer docking of the toxin to M1 and M3 receptors. The complexes obtained provide insight into the mechanisms of positive and negative allosteric rWTX activity. A significant conformational plasticity of the loop II observed by NMR and molecular dynamics (MD) simulations could explain the WTX activity toward structurally unrelated targets, nAChRs and mAChRs.
Experimental Procedures
Cloning and Bacterial Expression of WTX Mutants
The mutations were inserted into the WTX gene by site-directed mutagenesis using the expression vector pET22b(+)/wtx (17) as a template. The cultivation of Escherichia coli BL21(DE3) cells transformed with appropriate vectors, gene expression, purification, and refolding of rWTX and its mutants were as described previously (17), with the exception that renaturation buffer contained additionally 0.5 m l-Arg. For production of the 15N-labeled rWTX and P33A mutant, transformed cells were grown on TB medium until A600 0.6. The cells were harvested (2000 × g, 20 min) and resuspended in an equal volume of minimal medium M9 containing (15N)H4Cl as a nitrogen source, and after that gene expression was induced.
Binding of rWTX and Its Mutants to Muscarinic Acetylcholine Receptors
Interaction of wild-type WTX purified from N. kaouthia venom as described in Ref. 14, rWTX, and its mutants with human mAChRs was analyzed using membranes from CHO cells expressing individual subtypes of receptor (kindly supplied by Prof. T. I. Bonner) essentially as described earlier (18). Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum in 10-cm Petri dishes supplemented with 5 mm butyrate for the last 24 h of cultivation to increase receptor expression. Medium was removed, and cells were mechanically detached and washed twice in phosphate-buffered saline and 3 min of centrifugation at 250 × g. Washed cells were diluted in ice-cold homogenization medium (100 mm NaCl, 20 mm NaHEPES, 10 mm EDTA, pH 7.4) and homogenized on ice by two 30-s strokes using a Polytron homogenizer (Ultra-Turrax; Janke & Kunkel GmbH & Co. KG, IKA-Labortechnik, Staufen, Germany) set to maximum with a 30-s pause between strokes. Unbroken cell debris and nuclei were removed by low speed centrifugation at 1000 × g for 5 min at 4 °C. Aliquots of cell homogenates were centrifuged for 30 min at 30,000 × g at 4 °C. Supernatants were discarded, and pellets were resuspended in incubation medium (100 mm NaCl, 10 mm MgCl2, 20 mm NaHEPES, pH 7.4), and centrifuged again under the same conditions. Resulting pellets were kept at −20 °C until assayed for 10 weeks at maximum.
All radioligand binding experiments were carried out in 96-well plates at 30 °C in the incubation medium described above at a final volume of 200 μl. Nonspecific binding was determined in the presence of 10 μm atropine and was always smaller that 10% of total binding. In equilibrium binding experiments, membranes (5–20 μg of membrane proteins) were incubated for 60 min at 30 °C in the presence of 150 pm [3H]NMS (PerkinElmer Life Sciences) without (control) or with a single 10 μm concentration of WTX, rWTX, or its mutants. Incubation was terminated by fast filtration through GF/B filters (Whatman). Filters were dried, and then solid scintillator Meltilex A was applied using heating plate at 105 °C for 75 s. After filters cooled, the radioactivity was measured in Microbeta scintillation counter (Wallac, Finland).
To determine the effects of rWTX on [3H]NMS dissociation, membranes expressing the M1–M3 receptor subtypes (5–9 μg of proteins) were first incubated at 30 °C for 1 h with 4 nm [3H]NMS and then for another hour without or with 10 μm rWTX in a final volume of 150 μl. Dissociation was induced by adding 50 μl of 40 μm atropine (final concentration 10 μm) at 5, 10, 20, 30, 40, or 60 min (M1 and M3 receptors) or at 1, 2, 5, 10, 20, or 30 min (M2 receptor) before fast filtration. Filters were processed as described above. Nonspecific binding was measured in samples with 10 μm atropine added before [3H]NMS. One phase decay equation constrained to 100 and zero was fitted (GraphPad software) to data expressed as percentage of specific binding of [3H]NMS without the addition of atropine to induce dissociation.
NMR Study of Recombinant Neurotoxins
NMR investigation was done using 0.5 mm samples of 15N-labeled rWTX (pH 3.0 and 5.2) or P33A mutant (pH 3.0) in 5% D2O or 100% D2O at 40 °C. NMR spectra were acquired on a Bruker Avance 700 spectrometer equipped with a cryoprobe. 1H and 15N resonance assignment was obtained by a standard procedure using a combination of two- and three-dimensional TOCSY and NOESY spectra (19). Spatial structure calculation of rWTX(P33A) was performed in the CYANA program (Table 1) (20). Upper interproton distance constraints were derived from NOESY (τm 100 ms) cross-peaks via a “1/r6” calibration. Torsion angle restraints were obtained from 3JHNHα, 3JNHβ, 3JHαHβ, and 3JHβHγ coupling constants. Amide protons demonstrating deuterium half-exchange time of >24 h (40 °C) and/or temperature coefficients less than 4.5 ppb/K were supposed to participate in hydrogen bonding. In the final rounds of structure calculation, lower distance constraints (3.0 Å) based on the expected cross-peaks but not presented in the NOESY spectra were introduced. The relaxation parameters of 15N nuclei were analyzed using TENSOR software (21). Hydrodynamic calculations were performed using the program HYDRONMR (22).
TABLE 1.
Statistics for the best CYANA structures of WTX(P33A)
| Distance and angle restraints | |
| Total NOE contacts | 535 |
| Intraresidual | 180 |
| Inter-residual | 355 |
| Sequential (|i − j| = 1) | 170 |
| Medium range (1<|i − j|≤4) | 31 |
| Long range (|i − j|>4) | 154 |
| Hydrogen bonds restraints (20 bonds, upper/lower) | 40/40 |
| S--S bond restraints (5 bonds, upper/lower) | 15/15 |
| Lower distance restraints | 6 |
| Torsion angle restraints | 97 |
| Angle ϕ | 54 |
| Angle ψ | 4 |
| Angle χ1 | 39 |
| Total restraints/residue | 748/12 |
| Statistics for calculated structures | |
| Structures calculated/selected | 400/20 |
| CYANA target function (Å2) | 1.78 ± 0.32 |
| Violations of restraints | |
| Distance (>0.2 Å) | 3 |
| Dihedral angles (>1°) | 2 |
| Root mean square deviation (Å) | |
| Overall | |
| Backbone | 1.26 ± 0.36 |
| Heavy atoms | 2.42 ± 0.49 |
| Overall except loop II (1–28, 38–65) | |
| Backbone | 0.41 ± 0.11 |
| Heavy atoms | 1.21 ± 0.15 |
Computer Modeling of rWTX(P33A) Complexes with M1 and M3 mAChRs
The complexes were built using customized protein-protein docking procedure subdivided into eight steps as follows.
(i) The human M3-mAChR structure was taken from PDB (code 4DAJ). The homology model of human M1-mAChR was generated with MODELLER 8.2 (23) using the M3 receptor structure as a template. A disulfide bridge was imposed between residues 98 and 178. Following the structure of the template, an intracellular loop III between TM helices 5 and 6 is absent from the model.
(ii) A fragment of neuronal membrane-mimicking bilayer (palmitoyloleoylphosphatidylcholine/palmitoyloleoylethanolamine/cholesterol, 144:72:72 molecules, respectively) was assembled, solvated inside the rectangular box with dimensions 8 × 8 × 13 nm3, and equilibrated during 50 ns of MD simulation. All simulations were performed with the GROMACS 4.5.2 (24) using the Gromos96 43a2x parameters set. The Ryckaet-Bellemans torsion potential was used for the lipid hydrocarbon chains (25). Nonbonded interactions were described by the parameters from Ref. 26; SPC water model was used. Simulations were carried out with a time step of 2 fs, imposing three-dimensional periodic boundary conditions, in the isothermal-isobaric (NPT) ensemble with semi-isotropic pressure of 1 bar and a constant temperature of 37 °C. All components of the system (water, phospholipids, and protein) were coupled separately in a temperature bath using Nose-Hoover thermostat. The van der Waals interactions were truncated using 1.6 nm spherical cutoff function. Electrostatic interactions were treated with the Particle Mesh Ewald (PME) algorithm (0.12 nm Fourier spacing).
(iii) M1 or M3 receptors were placed inside the equilibrated bilayer; some phospholipid and cholesterol molecules were removed for this purpose. The system was re-solvated using water molecules and required number of Cl− ions to maintain electroneutrality, equilibrated in several stages, and subjected to another 50 ns of MD.
(iv) rWTX(P33A) mutant was subjected to 100 ns of MD simulation in 5 × 5 × 6 nm3 water box.
(v) The conformational clustering was performed on M1-mAChR, M3-mAchR, and P33A mutant MD trajectories. Gromacs utility g_cluster was used with clustering method Gromos and distance cutoff of 0.2 nm that yielded five, six, and five structural clusters in these three trajectories, respectively.
(vi) 5 × 5 = 25 or 6 × 5 = 30 protein-protein docking runs were performed with either ZDOCK (27) or pyRosetta (28) software for rWTX(P33A)·M1-mAChR and rWTX(P33A)·M3-mAChR complexes, respectively. The necessity to use different docking protocols resulted from abnormal work of ZDOCK that trended to generate unrealistic rWTX(P33A)·M3-mAChR complexes. During generation of the rWTX(P33A)·M1-mAChR complex, to avoid unrealistic docking solutions, the receptor residues inaccessible from the outer side of the bilayer were “blocked.” For each run, ZDOCK systematically generated 2000 structures of the complex; 100 top-scoring structures were used for further analysis (in total, 2500). During generation of the rWTX(P33A)·M3-mAChR complex, a D100_Docking.py script from pyRosetta tutorial was adapted to perform docking. Analogously to ZDOCK, 2000 docking solutions were generated in each of 30 docking runs (totally, 60,000 solutions); in distinction from ZDOCK, no blocking of residues was imposed because pyRosetta does not explicitly provide such an option.
(vii) The docking solutions were “filtered” using in-house re-scoring protocol that requires a simultaneous formation of two ionic or π-aromatic stacking interactions between the toxin and the receptor, and also the building up of favorable specific and nonspecific hydrophobic/hydrophilic contacts in the complex (see “Results”). The analysis of hydrophobic/hydrophilic contacts in the complexes was done with the PLATINUM software (29).
(viii) The “final” solution of the each complex was chosen by a visual inspection, placed in the solvated lipid bilayer, and subjected to a 200-ns MD run.
Results
Design, Refolding, and Characterization of rWTX Mutants
The previously observed conformational heterogeneity of wild-type WTX in solution was attributed to the cis-trans-isomerization of the peptide bond Arg-32–Pro-33, but the additional isomerization of Cys-6–Pro-7 bond was not completely ruled out (30). Moreover, it is well documented that the presence of the sterically restricted Pro residues could force specific conformations of the protein backbone (31). To check the role of these factors in the rWTX biological activity, two mutants (P7A and P33A) were designed. We also suggested four mutant forms of the toxin (R31A, R32A, R31A/R32A, and R37A) to probe the importance of the charged residues of the loop II in the interaction with mAChRs. The unique feature of WTX among other nonconventional neurotoxins isolated from Naja spp. is the tryptophan residue in loop II (32). To study the biological role of this residue, the W36A mutant was produced as well.
It has been shown on the examples of rWTX, water-soluble domain of human Lynx1 protein, and secreted Ly-6/uPAR related human proteins SLURP-1 and SLURP-2 that the expression in the form of E. coli inclusion bodies with subsequent refolding represents the optimal strategy for production of three-finger proteins containing an additional fifth disulfide bond in the loop I (17, 33–35). Therefore, to produce rWTX mutants we used a previously developed protocol (17), but to increase the refolding yield, the renaturation buffer was additionally supplemented with 0.5 m l-Arg. The final yield of the refolded toxins was ∼1–5 mg (for different mutants) per 1 liter of bacterial culture.
The purity (no less than 95%) of the refolded rWTX and its mutants was confirmed by SDS-PAGE, analytical HPLC, and mass spectrometry. The measured molecular masses of the proteins within experimental error coincided with the calculated masses of rWTX mutants having five disulfide bridges and an additional N-terminal Met residue appearing due to translation of starting atg codon. CD and one-dimensional 1H NMR spectroscopy confirmed the proper folding of rWTX mutants.
Muscarinic Acetylcholine Receptor Binding Studies
The ability of rWTX and its mutants to interact specifically with the different subtypes of human mAChR was assessed in competition experiments with [3H]NMS at a single toxin concentration (10 μm). It has been reported that wild-type WTX at this concentration increases the [3H]NMS binding to M1- and M3-mAChRs demonstrating positive cooperativity, but it does not influence the [3H]NMS binding to M2, M4, and M5 receptors (15). Similarly to that, rWTX increases the [3H]NMS binding to M3 receptors and does not affect the [3H]NMS binding to M4 and M5 receptors (Fig. 2A). However, contrary to WTX, rWTX decreases the [3H]NMS binding to M1 and M2 receptors by ∼20%, demonstrating negative cooperativity (Fig. 2A). The presence of common and individual features in the interaction of WTX and rWTX with different mAChR subtypes was confirmed by a direct comparison of their activity on M1 and M3 receptors (Fig. 2A).
FIGURE 2.

Influence of rWTX and its mutants on [3H]NMS binding to mAChRs. Results are given as means ± S.E. of 3–7 values expressed in percent of control binding without toxin obtained in the same experiment. Dashed lines indicate 95% confidence interval of control binding that was in pmol/mg protein: M1, 1.81 ± 0.06, n = 12; M2, 0.28 ± 0.01, n = 9; M3, 1.46 ± 0.05, n = 9; M4, 0.49 ± 0.01, n = 9; M5, 0.32 ± 0.01, n = 9. A single concentration (10 μm) of the toxins was tested. A, comparison of effects of wild-type WTX and rWTX on [3H]NMS binding at mAChRs. No effects of wild-type WTX on [3H]NMS binding with M2, M4, and M5 receptors were previously reported (15). Data indicated as follows: #, p < 0.05; ##, p < 0.01, and ###, p < 0.001 are significantly different from each other. B–D, all mutants were compared with rWTX and control by analysis of variance followed by Dunnett's multiple comparisons test. Data indicated as * and ° (p < 0.05), ** and °° (p < 0.01), and *** and °°° (p < 0.001) are significantly different from rWTX and control, respectively.
To verify the allosteric nature of the interaction between the orthosteric antagonist and rWTX, its effect on the rate of [3H]NMS dissociation was measured at M1, M2, and M3 receptor subtypes (Fig. 3, A–C). The dissociation rate of [3H]NMS in the presence of 10 μm rWTX was significantly reduced as compared with the control experiments (Fig. 3D). The dissociation rates (min−1) decreased from 0.118 ± 0.005 to 0.076 ± 0.003 (n = 3, p < 0.01), from 0.81 ± 0.04 to 0.63 ± 0.03 (n = 3, p < 0.05), and from 0.074 ± 0.002 to 0.065 ± 0.002 (n = 3, p < 0.05) at the M1, M2 and M3 subtypes, respectively (Fig. 3).
FIGURE 3.

Influence of rWTX on rate of orthosteric antagonist [3H]NMS dissociation at M1, M2, and M3 mAChRs. A–C, time course of dissociation of [3H]NMS in the presence and absence of 10 μm rWTX was determined as described under “Experimental Procedures.” Specific binding of [3H]NMS was calculated as a difference between total and nonspecific binding measured in the same experiment and is expressed in percent of initial binding (ordinate). Results are given as means ± S.E. (n = 3). D, 10 μm rWTX decreased [3H]NMS dissociation rate constants (Koff, min−1) from 0.118 ± 0.005 to 0.076 ± 0.003, from 0.81 ± 0.04 to 0.63 ± 0.03, and from 0.074 ± 0.002 to 0.065 ± 0.002 at the M1, M2, and M3 subtypes, respectively. Data designated as *, p < 0.05, and **, p < 0.01, are significantly different from control by two-tailed t test.
The set of rWTX mutants was tested on the M1, M2, and M3 mAChRs. The data obtained for M1 and M3 receptors (Fig. 2, B and D) revealed that P7A and P33A mutations have no remarkable effects on both receptors. Except for the R37A mutation, single substitutions in the toxin's loop II did not change the observed interaction between rWTX and [3H]NMS upon binding to M1 and M3 mAChRs. At the same time, substitution of R37A or simultaneous R31A and R32A substitutions resulted in the disappearance of the toxin effects on the [3H]NMS binding. In the case of M2-mAChRs, all rWTX mutants, except P33A, demonstrated diminished influence on [3H]NMS binding to the receptor (Fig. 2C), pointing to the relevance of the mutated residues for the toxin/receptor interactions. Thus, only the P33A mutation did not affect the toxin activity on the all receptors tested.
NMR Analysis of rWTX and rWTX(P33A)
Similarly to wild-type WTX (30), two sets of signals corresponding to the two structural states of the toxin were observed in the NMR spectra of rWTX (Fig. 4A). According to the NMR data, two structural forms of rWTX have equal population in solution, and the characteristic time of exchange between them significantly exceeded 100 ms (so-called “slow conformational exchange”). The largest differences in the chemical shifts between the two states of the toxin were observed for residues situated in the vicinity of Pro-7 and Pro-33 (Fig. 4C). Analysis of one-dimensional 1H NMR spectra of rWTX mutants (Fig. 4D) revealed that the conformational heterogeneity of rWTX is associated with cis-trans-isomerization of the Arg-32–Pro-33 peptide bond. The rWTX mutant with substitution P33A exhibited only one structural state in solution (Fig. 4, B and D). This (together with the data on functional activity, see above) was the crucial factor in selecting the P33A mutant for further structural studies by NMR and computational modeling. P7A mutation increased the population of trans-Arg-32–Pro-33 conformer in solution (Fig. 4D) pointing to the presence of conformational coupling between loops I and II of the toxin.
FIGURE 4.

Conformational heterogeneity in rWTX molecule associated with the cis-trans-isomerization of the Arg-32–Pro-33 peptide bond. A and B, two-dimensional 1H,15N-HSQC spectra of 0.5 mm rWTX and rWTX(P33A) (pH 3.0, 40 °C). Signals of rWTX conformers with trans- and cis-configuration of Arg-32–Pro-33 peptide bond are marked by red and blue lettering, respectively. The signals of the cis form are also marked by asterisks. C, normalized difference of 1HN, 1Hα, and 15NH chemical shifts (√(Δδ1HN)2 + (Δδ1Hα)2 + (Δδ15NH/5)2) between “trans” and “cis” forms of rWTX. The data for proline residues and for Arg-37 and Tyr-38 (crosses) were not calculated. Signals of Arg-37 and Tyr-38 residues from the trans form of rWTX were not assigned. D, analysis of conformational heterogeneity in rWTX mutants. The fragments of one-dimensional 1H NMR spectra containing HNϵ1 signals of the Trp-36 side chain are shown. The relative content (%) of the trans form is shown above each spectrum.
The determined spatial structure of P33A mutant represents the three-finger fold consisting of two antiparallel β-sheets (Fig. 5). The first of them is formed by two β-strands and involves residues from the loop I (β1, Leu-1–Leu-4; β2, Phe-14–Cys-17). The second one consists of three strands and includes fragments of the loop II (β3, Ile-23–Leu-28; β4, Tyr-38–Ala-43) and loop III (β5, Glu-56–Cys-58). These results are in agreement with the earlier data on the secondary structure of wild-type WTX (30). Apart from the backbone-backbone hydrogen bonds associated with canonical elements of the secondary structure, the head and the loops of the toxin are stabilized by additional H-bonds. For instance, hydrogen bonds HN Lys-62-CO Thr-2, HN Leu-4-Oδ1 Asn-64, and HNϵ Arg-40-CO Asn-64 control the spatial arrangement of the C-terminal region relative to β1 and β4 strands.
FIGURE 5.

Spatial structure and backbone dynamics of rWTX(P33A) in aqueous solution. A, overview of NMR data collected for rWTX(P33A). The positive and negative values of Hα chemical shift indices (CSIs) denote β-strand and α-helical propensity, respectively. The large (>8.5 Hz), small (<5 Hz), and medium (others) 3JHNHα coupling constants are designated by the filled triangles, open squares, and stars, respectively. The filled squares denote amide protons with temperature gradients (ΔδHN/ΔT) less than 4.5 ppb/K. The filled, half-open, and open circles denote HN protons with slow (half-exchange time >24 h), slow-intermediate (half-exchange time >1 h), and intermediate (half-exchange time >15 min) H-D exchange rates (H/DEX), respectively. Amide protons that demonstrate fast exchange with water protons (H2OEX) are shown by open triangles. The corresponding peaks were observed in a 20-ms CLEANEX-PM spectrum. The NOE connectivities are denoted as usual. The widths of the bars correspond to the relative intensity of the cross-peak in the 100-ms NOESY spectrum. Elements of secondary structure are shown on a separate line; the β-strands are designated by arrows and tight β/γ-turns by wavy lines. B, set of the best 20 rWTX(P33A) structures, superimposed over the backbone atoms in regions with well defined structure. The three loops and head of the toxin are labeled. C, ribbon representation of rWTX(P33A) spatial structure. The disulfide bonds are in orange. The side chains of mutated residues are shown. The ribbon of rWTX(P33A) is colored according to obtained dynamical NMR data (see supplemental Table). The residues affected by dynamic processes on the picosecond-nanosecond time scale (one with heteronuclear NOE <0.7 or S2 <0.8) or microsecond-millisecond time scale (having REX >2 Hz) are shown in magenta and yellow, respectively. The residues demonstrating mobility on both time scales are in cyan. D, qualitative data describing changes in the WTX conformation upon introduction of N-terminal Met residue (see Fig. 6, B and C) are mapped on the spatial structure of rWTX(P33A). The residues demonstrating large changes in backbone (1HN, 1Hα) chemical shifts or 3JHNHα coupling constants are shown in blue and red, respectively. The residues demonstrating both types of effects are in cyan. The side chains of Met-0 and Ala-33 are shown in red.
Almost the entire rWTX(P33A) structure is well defined, except the central part of the loop II (His-29–Arg-37) (Fig. 5B). Residues from this region exhibit fast exchange of HN protons with water and show averaged 3JHNHα coupling constants with magnitudes of 6–8 Hz (Figs. 5A and 6C). Moreover, intensities of 1H,15N-HSQC cross-peaks for these residues were significantly lower in comparison with signals from other parts of the toxin molecule (Fig. 6A). Together with 15N relaxation data (Fig. 5C), this points to the enhanced intramolecular mobility of this region in the microsecond to millisecond time scale (fast to intermediate conformational exchange). Extensive dynamics on this time scale was observed for Lys-27–Ile-39 residues from loop II and in spatially proximal fragments of the loops I (Glu-8) and III (Tyr-52–Glu-56) (Fig. 5C). Probably, these exchange processes and conformational coupling between loops I and II are responsible for the relatively low structural convergence observed at the tip of the loop I (Pro-7–Cys-11, Fig. 5B). Analysis of 1H,15N-HSQC spectra of rWTX also revealed diminished intensities of resonances from the tip of the loop II (Fig. 6A). Thus, the latter one in the rWTX molecule is involved in microsecond to millisecond motions on the two time scales (slow, associated with cis-trans-isomerization of the Arg-32–Pro-33 peptide bond, and fast-to-intermediate one). In addition, large fragments of loops II and III participate in the high amplitude motions on the picosecond to nanosecond time scale (Fig. 5C). The head of the toxin demonstrates lesser mobility as compared with the fingers, although extensive dynamics on both picosecond to nanosecond and microsecond to millisecond time scales was detected for the Arg-18–Lys-22 loop protruding from the head (Fig. 5C).
FIGURE 6.

Qualitative comparison of backbone conformation and dynamics for wild-type WTX, rWTX, and rWTX(P33A). A, relative intensity of 15N-HSQC signals for trans and cis forms of rWTX and rWTX(P33A) mutant. For clarity, the intensities for two forms of rWTX are shown in joint columns. Residues with unassigned HN resonances and prolines are marked by crosses. B, difference of 1HN and 1Hα chemical shifts between wild-type WTX and rWTX (trans and cis forms) at pH ∼3.0 and 40 °C. The arbitrarily chosen threshold values (0.05 and 0.03 ppm for 1HN and 1Hα, respectively) are shown. C, comparison of 3JHNHα couplings measured for the trans form of native WTX and rWTX(P33A). The values with difference exceeding 2 Hz are connected by vertical lines. Uncertainties in the measured 3JHNHα values do not exceed 1 Hz. Chemical shifts and J-coupling constants for trans and cis forms of wild-type WTX were taken from Ref. 30.
Analysis of 15N relaxation data also permitted us to characterize rotational diffusion of rWTX(P33A) molecule in solution. The overall tumbling of the protein was found to be axial symmetric, with overall τR ∼4.1 ns and with the ratio of the main components of prolate diffusion tensor (D‖/D⊥) ∼1.5. The data obtained are in relatively good agreement with the values predicted by hydrodynamic calculations (DZ = 4.68·107 s−1, DY = 3.02·107 s−1, and DX = 3.24·107 s−1, τR = 4.5 ns) based on the experimentally determined spatial structure of rWTX(P33A). These data indicate that the protein is in a monomeric state in aqueous solution.
As compared with the wild-type toxin, the bacterially expressed rWTX and its mutants have an additional methionine residue at the N terminus. Comparison of 1HN and 1Hα chemical shifts and 3JHNHα coupling constants for WTX (30), the rWTX and P33A mutant (Fig. 6, B and C) revealed a similar overall conformation of the toxin molecules. The minor changes of the backbone conformation apparently caused by the introduction of N-terminal Met were observed in the three regions of rWTX and rWTX(P33A) as follows: Lys-13–Glu-21, Cys-42–Cys-46, and Cys-63–Arg-65. These fragments belong to the toxin's head and C-terminal tail and are spatially close to the β1 strand (Fig. 5D).
Computer Modeling of rWTX(P33A) Interaction with M1 and M3 mAChRs
In an attempt to understand the mechanisms of target-specific positive and negative allosteric rWTX activity, we modeled the complexes of the rWTX(P33A) mutant with M1- and M3-mAChRs. The established NMR structure of P33A mutant made it possible to dock the toxin into the available x-ray structure of M3-mAChR (36) and to a homology model of M1-mAChR.
To estimate the flexibility of the free receptors and toxin molecules, MD simulations were conducted. Analysis of obtained MD trajectories revealed that the most flexible parts of the molecules are the extracellular loop 2 (ECL2) situated between TM helices 4 and 5 of the receptors and the tip of the toxin's loop II (Fig. 7, black and magenta curves).
FIGURE 7.
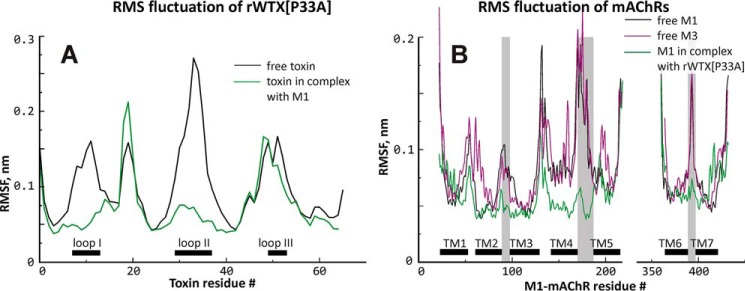
Root-mean-square fluctuation (RMSF) of rWTX(P33A) (A) and mAChRs (B). Free and bound rWTX(P33A) and M1-mAChR are shown with black and green lines, respectively. Free M3-mAChR is shown with purple line. For rWTX(P33A) and mAChRs, location of loops I–III and TM α-helices, respectively, is shown. Intracellular loop 3 of the receptor located between TM helices 5 and 6 is absent from the model. Amino acid residue numbering is provided for rWTX(P33A) and M1 receptor.
Clustering of the conformations extracted from MD trajectories resulted in five representatives for M1-mAChR, six for M3-mAChR, and five for rWTX(P33A). These structures were used in a series of protein-protein docking runs permitting us to take into account the flexibility of toxin and receptors upon binding. The resulting ensembles were filtered (re-scored) using three criteria as follows. (i) Taking into account the obtained mutagenesis data, it was required that the toxin forms at least two ionic bridges or π-cation interactions with the receptor via the positively charged residues Arg-37 and Arg-31 and/or Arg-32. (ii) The match of “hydrophobic/hydrophilic” properties between the toxin and receptor in the complex, which has been proven to be important in molecular recognition (37, 38), was required to exceed a certain threshold (>0.65). (iii) The total number of favorable intermolecular contacts in the complex was required to be above 12. Application of these criteria reduced the initial ensembles of 2500 and 60,000 structures to 24 and 42 models for rWTX(P33A)·M1-mAChR and rWTX(P33A)·M3-mAChR complexes, respectively.
For the P33A mutant complex with the M3 receptor, three clusters of docking solutions containing 23, 8, and 11 structures were obtained. In these clusters, the toxin molecule was located as follows: cluster 1, at the cytoplasmic interface of the receptor (omitted as unrealistic); cluster 2, in the vicinity of the ligand-binding vestibule located between the TM helices at the outer half of the M3-mAChR (so-called allosteric ligand-binding site (36)) (Fig. 8A); and cluster 3, at the receptor/membrane interface (Fig. 8B). The solutions from cluster 2 were analyzed. In these models, the toxin was unable to insert its loops into the allosteric ligand-binding site forming the loose complex with the receptor (Fig. 8A). As a result, the toxin's loop II does not contact with the entrance into orthosteric site situated just below the allosteric site as one may judge by the location of tiotropium molecule in the x-ray structure with M3-mAChR (36). The rWTX(P33A)·M3-mAChR complex is stabilized mainly by ionic interactions between the positively charged loop II residues (Arg-37 and Arg-31 and/or Arg-32) and the negatively charged moieties from ECL2 (Glu-220) and ECL3 (Asp-518) of the receptor (Table 2). The complex was unstable in the course of 200-ns MD simulation in the lipid bilayer that confirms its loose organization. In the cluster 3 of the rWTX(P33A)·M3-mAChR complexes, the toxin also interacts by its positively charged loop II with the negatively charged residues of ECL2 and ECL3 (Glu-228 and Asp-518, respectively), but from the receptor/membrane interface and not from the top of the receptor (Fig. 8B).
FIGURE 8.

Modeled complexes of rWTX(P33A) with M1- and M3-mAChRs. A, close-up view of rWTX(P33A) in complex with M3-mAchR (the structure from cluster 2 of solutions). Toxin molecule is shown in pale green, and the receptor is shown in purple. The M3-mAChR residues from ECL2 and ECL3 are shown. B, close-up view of rWTX(P33A) in complex with M3-mAChR (the structure from cluster 3 of solutions). Toxin molecule is shown in wheat, and the receptor is shown in purple. For comparison, the position of the toxin in cluster 2 of the solutions is shown by a shadow. C, close-up view of rWTX(P33A) (pale green) penetrating by the loop II into M1-mAChR (brown) TM helical bundle; ECL2 of the receptor (dark gray) leaves enough space for this interaction. The M1-mAChR residues that are critical for binding are shown. Negatively or positively charged, polar, and aromatic residues are in red, blue, green and yellow, respectively; toxin's residues are shown over a black background. Disulfide bonds are shown in orange. D, view of rWTX(P33A)·M1-mAChR complex in the POPC/POPE/cholesterol (2:1:1) bilayer after MD relaxation. Hydrophobic atoms of lipids and cholesterol are shown by yellow and orange, respectively. Oxygen and nitrogen atoms of phospholipids are shown by red and blue, respectively. The proximal parts of the membrane, water, and ions are removed for clarity. E, sequence comparison of ECLs in M1-M5 receptors.
TABLE 2.
Contacts between rWTX(P33A) and M1- and M3-mAChR in the modeled complexes
| M1-mAChR residues | rWTX(P33A) residuesa | M3-mAChR residues | rWTX(P33A) residuesa |
|---|---|---|---|
| ECL1 | |||
| His-90, Trp-91 | Arg-31(H) | Asn-132 | Arg-32(H) |
| His-90 | Arg-31(T) | ||
| ECL2 | |||
| Val-168 | Gly-12(H) | Lys-213 | Glu-53(I) |
| Glu-170 | Lys-62, Arg-65(I) | Thr-215 | His-29(H) |
| Glu-170 | Arg-65(H) | Glu-220 | Gln-30(H) |
| Gln-177 | Arg-32(H) | Glu-220 | Arg-32, Arg-37(I) |
| Gln-181 | Arg-32, Ser-35(H) | Gln-224 | Tyr-52(H) |
| Ser-227 | Tyr-52(H) | ||
| ECL3 and top of TM7 | |||
| Asp-393 | Arg-37(I) | Asp-518 | Arg-31(I) |
| Glu-397 | Arg-32, Arg-37(I) | ||
| Glu-397 | Arg-37(H) | ||
| Trp-400 | Arg-32(T) | ||
| Glu-401 | Arg-32(I) | ||
| Tyr-404 | Ala-33(H) | ||
a Types of interactions are as follows: H indicates hydrogen bond; T indicates T-shaped stacking; I indicates ionic.
For the rWTX(P33A) complex with M1-mAChR, all filtered docking solutions were very similar to each other and were significantly different from the solutions described above for the M3 receptor. First of all, the toxin molecule is rotated by ∼180° around the central axis of β-strands (Fig. 8C). Second, contrary to the case of M3-mAChR, the toxin inserts the most protruding loop II into the allosteric ligand-binding vestibule of the M1 receptor just above the orthosteric ligand-binding site. Two positively charged residues (Arg-32 and Arg-37) located in the toxin's loop II form ionic bridges with three negatively charged residues (Asp-393, Glu-397, and Glu-401), located in the ECL3 and the outer part of TM7 of the receptor (Fig. 8C and Table 2). Additionally, two C-terminal positively charged residues of the toxin (Lys-62 and Arg-65) form ionic bridges with Glu-170 from the ECL2 of the receptor. Gly-12 from the toxin loop I forms hydrogen bond with Val-168 from the ECL2 of M1-mAChR (Table 2). Unrestrained MD simulation (200 ns) revealed that the mobility of ECL2 of the receptor and toxin loops I and II dramatically decreased upon the binding (Fig. 7, green curves), pointing to the key role of these regions in the toxin/receptor interaction.
Discussion
The structure-function studies of ligand-receptor interactions require large amounts of the proteins and their mutant variants. In most of the cases, the production of the appropriate protein or peptide samples could be done only via heterologous expression (39). Frequently, the recombinant proteins differ from the native ones, e.g. contain cloning artifacts, stabilizing mutations, purification tags, or non-native glycosylation patterns. Despite that, the recombinant protein production forms the basis for the state-of-the-art structural studies and finally provides most of our current knowledge in the field of membrane receptors and ligand-receptor interactions (40).
Here, we investigated structural and pharmacological properties of rWTX, a recombinant analogue of nonconventional toxin from N. kaouthia, which allosterically modulates the functioning of mAChRs (15). The recombinant toxin was produced by bacterial expression and, as compared with the wild-type toxin, has an additional N-terminal methionine residue. Therefore, rWTX could be considered as a mutant form of wild-type WTX. It turned out that this minor N-terminal modification significantly changes the toxin pharmacological profile. Similar to the native toxin, which was shown to increase the binding of orthosteric ligand NMS to M1- and M3-mAChRs (15), rWTX enhanced the NMS binding, but only to the M3 receptor. At the M1 receptor, the mode of toxin/NMS interaction was switched from “positive” to “negative” and resulted in decreasing of the NMS binding in the presence of rWTX. Moreover, the negative rWTX interaction with NMS was observed at M2-mAChRs, where the wild-type toxin demonstrates a very weak effect (15). The reduction in the rates of [3H]NMS dissociation from M1, M2, and M3 receptors observed in the presence of rWTX (Fig. 3D) confirmed the allosteric mode of the rWTX/mAChRs interaction.
Taking into account the complex nature of the allosteric interactions (6), the rWTX effects described above could not be totally unexpected. First, the negative binding cooperativity between WTX and another orthosteric ligand, acetylcholine, was previously observed at M1, M2, M4, and M5 mAChRs (15). Second, it is well documented that minor changes in the structure of allosteric modulators may lead to a completely different sort of cooperativity (6). For example, the changes in mode of interaction with acetylcholine or NMS binding at different subtypes of mAChR were described for a series of strychnine and brucine analogues (41).
Site-directed mutagenesis indicated that positively charged residues in the rWTX loop II are essential for interaction with M1, M2, and M3 mAChRs. This permits an assumption that the toxin loop II represents a major site of WTX interaction with mAChRs. Our results are in good agreement with studies on other three-finger snake toxins, where a positively charged tip of the loop II was identified as a key determinant of interaction with different targets (7–9, 13, 47), including nAChRs and mAChRs. High conformational mobility of the loop II revealed here (Figs. 5, B and C, and 7A) is not a unique feature of WTX. Recently, large plasticity of loop III was observed for the water-soluble three-finger domain of the human prototoxin Lynx1, which, similarly to WTX, binds with low affinity (of micromolar range) to both nAChRs and mAChRs (42). Thus, we propose that large plasticity of the loops might confer the three-finger proteins, like WTX and Lynx1, the capacity to interact with both types of acetylcholine receptors, while providing rather low affinity for these targets.
To illustrate the structural properties that underlie positive/negative allosteric interaction of rWTX with M1- and M3-mAChRs, we utilized a computer modeling approach based on molecular docking and MD simulations. There are several caveats associated with application of this technique to the membrane receptors and their ligands. (i) Conventional molecular docking protocols do not take into account the dynamics of interacting molecules. This could preclude the finding of correct solution of the complex. (ii) The molecular docking could provide too many different solutions, and choosing of the correct one became a nontrivial task. (iii) The membrane environment of the receptor could influence the ligand binding and should be taken into account. To overcome these obstacles, we used a special customized protein-protein docking procedure (see under “Results”). (i) To increase structural sampling and partially take into account the flexibility of the interacting molecules, we started docking runs not from the single receptor/toxin structures but from the structural ensembles obtained via MD simulations. (ii) In an attempt to filter out “incorrect” or unrealistic docking solutions, we used available mutagenesis data and additional score-function restricting complementarity of the “hydrophobic/hydrophilic” properties of the toxin/receptor molecules in the complex (37, 38). (iii) The resulting complexes were subjected to unrestrained MD runs in the hydrated lipid bilayer to estimate the influence of the environment on their stability.
Wild-type WTX and rWTX molecules demonstrate conformational heterogeneity in solution associated with cis-trans-isomerization of the Arg-32–Pro-33 peptide bond. Therefore, we focused our structural studies on a P33A mutant that has one structural form in solution and demonstrates functional properties similar to rWTX (Fig. 2, B–D). Computer modeling revealed the substantially different modes of the rWTX(P33A) binding to M1- and M3-mAChRs. In the case of M3-mAChR (Fig. 8A), the toxin interacts with all three ECLs of the receptor by residues from loops II and III (Table 2) without penetrating into the allosteric ligand-binding site. In the rWTX(P33A)·M1-mAChR complex (Fig. 8C), the toxin interacts with the receptor by residues from the loops I and II and the C terminus (Table 2) and occupies the outer part of the receptor's allosteric site. Less tight organization of the toxin·M3-mAChR complex and the inability to insert the toxin's loops into the receptor's allosteric site could be explained by the structural properties of this site. Indeed, the analysis of ECL sequences of mAChRs (Fig. 8E) revealed that the extracellular part of the M1 receptor is negatively charged (total charge of three ECLs is −3), although the corresponding region of M3 receptor is uncharged. This can explain (at least partly) the preferential binding of the cationic toxin molecule (overall charge +6) to the M1 receptor as compared with M3. The positions of Glu-170 and Glu-397 in the ECL2- and ECL3-TM7 of M1-mAChR, with which the toxin forms ionic bonds by the positively charged residues from loop II and the C terminus, corresponds to Lys-213 and Lys-523 in ECL2- and ECL3-TM7 of M3-mAChR (Fig. 8E). Thus, the favorable toxin/receptor interactions that stabilize the complex with M1-mAChR appear to be destroyed in the case of M3-mAChR. The negative influence of the Lys-523 residue on the lower affinity of cationic allosteric ligands toward the M3 receptors was discussed recently (43). The electrostatic properties of other mAChR subtypes (Fig. 8E) correlate with the observed activity of rWTX. The M2 receptor, similarly to M1, has the anionic vestibule (overall charge of ECLs is −2), and negative mode of rWTX interaction with NMS binding was observed at both M1 and M2 receptors. At the same time, the M4 and M5 receptors, where no interference with NMS binding was observed, have almost neutral allosteric sites (overall charges are 0 and −1, respectively).
The differences between the modeled complexes provide the necessary structural framework for rationalization of target-specific positive/negative allosteric regulation. We could speculate that inhibition of the NMS binding to M1-mAChR and M2-mAChR observed in the presence of rWTX is governed by the direct blockage of the entrance into the orthosteric receptor-binding site by the toxin molecule. In contrast, the rWTX molecule does not block the ligand-binding vestibule of the M3 receptor but could fix the ECLs in the conformation that facilitates a ligand access into the orthosteric site. This hypothesis provides an explanation to the observed positive cooperativity between rWTX and NMS binding at M3-mAChR.
Position of the toxin in other predicted models of its complex with the M3 receptor (cluster 3, Fig. 8B) partially coincides with one of the dimerization interfaces of the receptor (TM4/TM5) (44) pointing to a possible participation of the homodimeric M3-mAChR in the interaction with rWTX. Nowadays, the role of the homodimerization in functioning of mAChRs and other GPCRs is a matter of broad discussion (45); nevertheless, we cannot exclude it in the case of WTX. For example, the probable role of mAChRs homodimerization was discussed recently in the mutagenesis/modeling study of the interaction between muscarinic toxin MT7 and M1-mAChR (13). Notably, the rWTX(P33A) molecule in the models from cluster 3 (similarly to the situation with the cluster 2) does not immerse into the allosteric binding site but could “fasten” together the receptor's ECLs, thus being in agreement with the proposed “fixing” model of the positive allosteric regulation.
According to recent MD and mutagenesis data, a “cage” of aromatic residues in M2-mAChR forms the main site of binding for di-cationic allosteric modulators (43). The homologous cluster of aromatic residues (Trp-91 at ECL1, Trp-164 at TM4, Tyr-179 at ECL2, and Trp-400 and Tyr-404 at TM7) is situated just “below” the rWTX(P33A) position in the complex with M1-mAChR. Although the π-cation interactions between the arginine residues located in the toxin's loop II and the aromatic moieties of the receptor were not observed in the modeled complex, they still may occur upon binding. Moreover, the mode observed for rWTX(P33A) binding to M1-mAChR resembles the interaction between the low molecular weight allosteric modulator LY2119620 and M2-mAChR (46). Similarly to rWTX and P33A mutant, this compound demonstrates mild negative cooperativity with the NMS at the M2 receptor. Like the toxin's loop II, the aromatic moiety of compound LY2119620 occupies the cavity inside the receptor's TM α-helical bundle and contacts Trp-422 and Tyr-426 residues from the TM7 (Trp-400 and Tyr-404 in the case of M1-mAChR). Moreover, similarly to the toxin's C-terminal residues, the LY2119620 piperidine moiety participates in the electrostatic interaction with Glu-172 from the ECL2 (Glu-170 for M1-mAChR). Thus, rWTX(P33A)-binding site on the M1 receptor coincides with the binding site of low molecular weight allosteric ligands.
The similar activity of WTX and rWTX observed at M3-mAChRs (Fig. 2A) suggests that the native and recombinant toxins have the same mode of interaction with this type of receptor (Fig. 8, A and B). At the same time, the mode of wild-type WTX interaction with M1 receptor could be substantially different from the model obtained here for rWTX(P33A)·M1-mAChR (Fig. 8C). Positive cooperativity between native WTX and NMS binding at M1- and M3-mAChRs suggests that the mode of toxin/receptor interaction should be very similar in both cases. Insertion of N-terminal Met into the toxin molecule provokes the conformational changes in the head and C terminus of rWTX revealed by NMR analysis (Fig. 5D). These changes might promote additional contacts of the toxin's loop I and C terminus with the ECL2 of the M1 receptor (Table 2 and Fig. 8C). This in turn could result in changing of the mutual orientation of the toxin and ECLs and stabilize the toxin in the position immersed into the ligand-binding vestibule switching the pharmacological properties of rWTX.
In summary, we studied the activity of a series of mutant variants of rWTX on M1-, M2-, and M3-mAChRs. To the best of our knowledge, this is the first work on the site-directed mutagenesis of nonconventional toxins. We determined the spatial structure for one of the rWTX mutants having the same activity as rWTX and revealed a high mobility of the toxin loop II. Based on the structure and mutagenesis data, models of the rWTX complexes with M1- and M3-mAChRs were built. These models confirmed the key role of the toxin's loop II in the binding to mAChRs and provided a structural framework for rationalization of target-specific positive/negative allosteric regulation.
Author Contributions
E. N. L. initiated and supervised the project and wrote the article. Z. O. S. analyzed structural and biochemical data and wrote the article. M. A. S. made the gene constructs and performed recombinant protein production. A. S. P. performed NMR experiments and the calculation of spatial structure. A. O. C. performed the MD simulations and docking. R. G. E. managed MD simulations and docking and wrote the paper. H. J. and E. D. performed functional tests. V. D. analyzed and managed functional tests. A. S. A. supervised the NMR study. Y. N. U. purified wild-type WTX from snake venom. V. I. T. contributed valuable discussions during this study and wrote the article. D. A. D. supervised protein expression. M. P. K. supervised the project. All co-authors contributed to revising the article.
Acknowledgments
We thank Russian Scientific Foundation (Project 14-14-00255 issued to E. N. L., Z. O. S., M. A. S., A. S. P., A. O. C., and D. A. D.) for support of the work on the production of recombinant WTX, its functional tests, and modeling of rWTX·mAChR complexes.
This work was supported by Russian Academy of Sciences (Program Molecular and Cellular Biology), President of the Russian Federation Grants SS-1766.2014.4 and YC-6310.2015.4, Russian Foundation for Basic Research Grants 13-04-00825 and 14-04-31634, Grant Agency of the Czech Republic Grant 14-05696S (to V. D.), Institute of Physiology of the Academy of Sciences of the Czech Republic Project RVO:67985823 (to H. J., E. D., and V. D.), and Russian Scientific Foundation Project 14-14-00255 (to E. N. L., Z. O. S., M. A. S., A. S. P., A. O. C., and D. A. D.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
The atomic coordinates and structure factors (code 2MJ0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- nAChR
- nicotinic acetylcholine receptor
- α-NT
- α-neurotoxins
- GPCR
- G protein coupled receptors
- mAChR
- muscarinic acetylcholine receptor
- MD
- molecular dynamics
- MT
- muscarinic toxin
- NMS
- N-methylscopolamine
- r-WTX
- recombinant analogue of weak toxin from Naja kaouthia venom
- TM
- transmembrane
- ECL
- extracellular loop.
References
- 1. Tsetlin V. I., Hucho F. (2004) Snake and snail toxins acting on nicotinic acetylcholine receptors: fundamental aspects and medical applications. FEBS Lett. 557, 9–13 [DOI] [PubMed] [Google Scholar]
- 2. Kini R. M., Doley R. (2010) Structure, function and evolution of three-finger toxins: mini proteins with multiple targets. Toxicon 56, 855–867 [DOI] [PubMed] [Google Scholar]
- 3. Papke R. L. (2014) Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol. 89, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kruse A. C., Kobilka B. K., Gautam D., Sexton P. M., Christopoulos A., Wess J. (2014) Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat. Rev. Drug Discov. 13, 549–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wess J., Eglen R. M., Gautam D. (2007) Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 6, 721–733 [DOI] [PubMed] [Google Scholar]
- 6. Jakubik J., El-Fakahany E. E. (2010) Allosteric modulation of muscarinic acetylcholine receptors. Pharmaceuticals 3, 2838–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pillet L., Trémeau O., Ducancel F., Drevet P., Zinn-Justin S., Pinkasfeld S., Boulain J. C., Ménez A. (1993) Genetic engineering of snake toxins. Role of invariant residues in the structural and functional properties of a curare mimetic toxin, as probed by site-directed mutagenesis. J. Biol. Chem. 268, 909–916 [PubMed] [Google Scholar]
- 8. Bourne Y., Talley T. T., Hansen S. B., Taylor P., Marchot P. (2005) Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 24, 1512–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lyukmanova E. N., Shenkarev Z. O., Schulga A. A., Ermolyuk Y. S., Mordvintsev D. Y., Utkin Y. N., Shoulepko M. A., Hogg R. C., Bertrand D., Dolgikh D. A., Tsetlin V. I., Kirpichnikov M. P. (2007) Bacterial expression, NMR, and electrophysiology analysis of chimeric short/long-chain α-neurotoxins acting on neuronal nicotinic receptors. J. Biol. Chem. 282, 24784–24791 [DOI] [PubMed] [Google Scholar]
- 10. Nirthanan S., Gopalakrishnakone P., Gwee M. C., Khoo H. E., Kini R. M. (2003) Non-conventional toxins from Elapid venoms. Toxicon 41, 397–407 [DOI] [PubMed] [Google Scholar]
- 11. Fry B. G., Wüster W., Kini R. M., Brusic V., Khan A., Venkataraman D., Rooney A. P. (2003) Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 57, 110–129 [DOI] [PubMed] [Google Scholar]
- 12. Servent D., Blanchet G., Mourier G., Marquer C., Marcon E., Fruchart-Gaillard C. (2011) Muscarinic toxins. Toxicon 58, 455–463 [DOI] [PubMed] [Google Scholar]
- 13. Marquer C., Fruchart-Gaillard C., Letellier G., Marcon E., Mourier G., Zinn-Justin S., Ménez A., Servent D., Gilquin B. (2011) Structural model of ligand-G protein-coupled receptor (GPCR) complex based on experimental double mutant cycle data: MT7 snake toxin bound to dimeric hM1 muscarinic receptor. J. Biol. Chem. 286, 31661–31675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Utkin Y. N., Kukhtina V. V., Kryukova E. V., Chiodini F., Bertrand D., Methfessel C., Tsetlin V. I. (2001) “Weak toxin” from Naja kaouthia is a nontoxic antagonist of α7 and muscle-type nicotinic acetylcholine receptors. J. Biol. Chem. 276, 15810–15815 [DOI] [PubMed] [Google Scholar]
- 15. Mordvintsev D. Y., Polyak Y. L., Rodionov D. I., Jakubik J., Dolezal V., Karlsson E., Tsetlin V. I., Utkin Y. N. (2009) Weak toxin WTX from Naja kaouthia cobra venom interacts with both nicotinic and muscarinic acetylcholine receptors. FEBS J. 276, 5065–5075 [DOI] [PubMed] [Google Scholar]
- 16. Mordvitsev D. Y., Polyak Y. L., Kuzmin D. A., Levtsova O. V., Tourleigh Y. V., Utkin Y. N., Shaitan K. V., Tsetlin V. I. (2007) Computer modeling of binding of diverse weak toxins to nicotinic acetylcholine receptors. Comput. Biol. Chem. 31, 72–81 [DOI] [PubMed] [Google Scholar]
- 17. Lyukmanova E. N., Shulepko M. A., Tikhonov R. V., Shenkarev Z. O., Paramonov A. S., Wulfson A. N., Kasheverov I. E., Ustich T. L., Utkin Y. N., Arseniev A. S., Tsetlin V. I., Dolgikh D. A., Kirpichnikov M. P. (2009) Bacterial production and refolding from inclusion bodies of a “weak” toxin, a disulfide-rich protein. Biochemistry 74, 1142–1149 [DOI] [PubMed] [Google Scholar]
- 18. Jakubík J., El-Fakahany E. E., Dolezal V. (2006) Differences in kinetics of xanomeline binding and selectivity of activation of G proteins at M(1) and M(2) muscarinic acetylcholine receptors. Mol. Pharmacol. 70, 656–666 [DOI] [PubMed] [Google Scholar]
- 19. Cavanagh J., Fairbrother W. J., Palmer A. G., Skelton N. J., Rance M. (2006) Protein NMR Spectroscopy Principles and Practice, 2nd Ed., pp. 781–817, Academic Press, New York [Google Scholar]
- 20. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 21. Dosset P., Hus J. C., Blackledge M., Marion D. (2000) Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR 16, 23–28 [DOI] [PubMed] [Google Scholar]
- 22. García de la Torre J., Huertas M. L., Carrasco B. (2000) HYDRONMR: Prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn Reson. 147, 138–146 [DOI] [PubMed] [Google Scholar]
- 23. Martí-Renom M. A., Stuart A. C., Fiser A., Sánchez R., Melo F., Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 24. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and stable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 25. Ryckaert J.-P., Bellemans A. (1975) Molecular dynamics of a liquid n-butane near its boiling point. Chem. Phys. Lett. 30, 123–125 [Google Scholar]
- 26. Berger O., Edholm O., Jähnig F. (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72, 2002–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen R., Li L., Weng Z. (2003) ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 28. Chaudhury S., Lyskov S., Gray J. J. (2010) PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26, 689–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pyrkov T. V., Chugunov A. O., Krylov N. A., Nolde D. E., Efremov R. G. (2009) PLATINUM: a web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics 25, 1201–1202 [DOI] [PubMed] [Google Scholar]
- 30. Eletsky A. V., Maslennikov I. V., Kukhtina V. V., Utkin Yu. N., Tsetlin V. I., Arseniev A. S. (2001) Structure and conformational heterogeneity of the weak toxin from the cobra Naja kaouthia venom. Russian J. Bioorg. Chem. 27, 72–83 [PubMed] [Google Scholar]
- 31. MacArthur M. W., Thornton J. M. (1991) Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412 [DOI] [PubMed] [Google Scholar]
- 32. Utkin Y. N., Kukhtina V. V., Maslennikov I. V., Eletsky A. V., Starkov V. G., Weise C., Franke P., Hucho F., Tsetlin V. I. (2001) First tryptophan-containing weak neurotoxin from cobra venom. Toxicon 39, 921–927 [DOI] [PubMed] [Google Scholar]
- 33. Shulepko M. A., Lyukmanova E. N., Kasheverov I. E., Dolgikh D. A., Tsetlin V. I., Kirpichnikov M. P. (2011) Bacterial expression of the water-soluble domain of Lynx1, an endogenous neuromodulator of human nicotinic receptors. Russian J. Bioorg. Chem. 37, 543–549 [DOI] [PubMed] [Google Scholar]
- 34. Shulepko M. A., Lyukmanova E. N., Paramonov A. S., Lobas A. A., Shenkarev Z. O., Kasheverov I. E., Dolgikh D. A., Tsetlin V. I., Arseniev A. S., Kirpichnikov M. P. (2013) Human neuromodulator SLURP-1: bacterial expression, binding to muscle-type nicotinic acetylcholine receptor, secondary structure, and conformational heterogeneity in solution. Biochemistry (Mosc) 78, 204–211 [DOI] [PubMed] [Google Scholar]
- 35. Lyukmanova E. N., Shulepko M. A., Bychkov M. L., Shenkarev Z. O., Paramonov A. S., Chugunov A. O., Arseniev A. S., Dolgikh D. A., Kirpichnikov M. P. (2014) Human SLURP-1 and SLURP-2 proteins acting on nicotinic acetylcholine receptors reduce proliferation of human colorectal adenocarcinoma HT-29 cells. Acta Naturae 6, 60–66 [PMC free article] [PubMed] [Google Scholar]
- 36. Kruse A. C., Hu J., Pan A. C., Arlow D. H., Rosenbaum D. M., Rosemond E., Green H. F., Liu T., Chae P. S., Dror R. O., Shaw D. E., Weis W. I., Wess J., Kobilka B. K. (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Efremov R. G., Chugunov A. O., Pyrkov T. V., Priestle J. P., Arseniev A. S., Jacoby E. (2007) Molecular lipophilicity in protein modeling and drug design. Curr. Med. Chem. 14, 393–415 [DOI] [PubMed] [Google Scholar]
- 38. Pyrkov T. V., Kosinsky Y. A., Arseniev A. S., Priestle J. P., Jacoby E., Efremov R. G. (2007) Complementarity of hydrophobic properties in ATP-protein binding: a new criterion to rank docking solutions. Proteins 66, 388–398 [DOI] [PubMed] [Google Scholar]
- 39. Rosano G. L., Ceccarelli E. A. (2014) Recombinant protein expression in microbial systems. Front. Microbiol. 5, 1–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He Y., Wang K., Yan N. (2014) The recombinant expression systems for structure determination of eukaryotic membrane proteins. Protein Cell 5, 658–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lazareno S., Gharagozloo P., Kuonen D., Popham A., Birdsall N. J. (1998) Subtype-selective positive cooperative interactions between brucine analogues and acetylcholine at muscarinic receptors, radioligand binding studies. Mol. Pharmacol. 53, 573–589 [DOI] [PubMed] [Google Scholar]
- 42. Lyukmanova E. N., Shenkarev Z. O., Shulepko M. A., Mineev K. S., D'Hoedt D., Kasheverov I. E., Filkin S. Y., Krivolapova A. P., Janickova H., Dolezal V., Dolgikh D. A., Arseniev A. S., Bertrand D., Tsetlin V. I., Kirpichnikov M. P. (2011) NMR structure and action on nicotinic acetylcholine receptors of water-soluble domain of human LYNX1. J. Biol. Chem. 286, 10618–10627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dror R. O., Green H. F., Valant C., Borhani D. W., Valcourt J. R., Pan A. C., Arlow D. H., Canals M., Lane J. R., Rahmani R., Baell J. B., Sexton P. M., Christopoulos A., Shaw D. E. (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 [DOI] [PubMed] [Google Scholar]
- 44. Hu J., Hu K., Liu T., Stern M. K., Mistry R., Challiss R. A., Costanzi S., Wess J. (2013) Novel structural and functional insights into M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 288, 34777–34790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Redka D. S., Morizumi T., Elmslie G., Paranthaman P., Shivnaraine R. V., Ellis J., Ernst O. P., Wells J. W. (2014) Coupling of G proteins to reconstituted monomers and tetramers of the M2 muscarinic receptor. J. Biol. Chem. 289, 24347–24365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P. M., Christopoulos A., Felder C. C., Gmeiner P., Steyaert J., Weis W. I., et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kudryavtsev D. S., Shelukhina I. V., Son L. V., Ojomoko L. O., Kryukova E. V., Lyukmanova E. N., Zhmak M. N., Dolgikh D. A., Ivanov I. A., Kasheverov I. E., Starkov V. G., Ramerstorfer J., Sieghart W., Tsetlin V. I., Utkin Y. N. (2015) Neurotoxins from snake venoms and α-conotoxin ImI inhibit functionally active ionotropic GABA receptors. J. Biol. Chem. 10.1074/jbc.M115.648824 [DOI] [PMC free article] [PubMed] [Google Scholar]