

Background: Flavonoids are naturally occurring compounds that can regulate certain transcription factors.
Results: We identified the flavonoid luteolin as a repressor of HNF4α and revealed that dietary luteolin improves high-fat diet-induced metabolic disorder in mice.
Conclusion: HNF4α is a target of luteolin.
Significance: Modulation of HNF4α activity through luteolin may be of therapeutic value in atherosclerotic disease.
Keywords: apolipoprotein, flavonoid, hepatocyte nuclear factor 4 (HNF4), metabolic syndrome, nuclear receptor, luteolin, microsomal triglyceride transfer protein (MTP)
Abstract
Hepatocyte nuclear factor 4α (HNF4α) is a nuclear receptor that regulates the expression of genes involved in the secretion of apolipoprotein B (apoB)-containing lipoproteins and in glucose metabolism. In the present study, we identified a naturally occurring flavonoid, luteolin, as a repressor of HNF4α by screening for effectors of the human microsomal triglyceride transfer protein (MTP) promoter. Luciferase reporter gene assays revealed that the activity of the MTP gene promoter was suppressed by luteolin and that the mutation of HNF4α-binding element abolished luteolin responsiveness. Luteolin treatment caused a significant decrease in the mRNA levels of HNF4α target genes in HepG2 cells and inhibited apoB-containing lipoprotein secretion in HepG2 and differentiated Caco2 cells. The interaction between luteolin and HNF4α was demonstrated using absorption spectrum analysis and luteolin-immobilized beads. Luteolin did not affect the DNA binding of HNF4α to the promoter region of its target genes but suppressed the acetylation level of histone H3 in the promoter region of certain HNF4α target genes. Short term treatment of mice with luteolin significantly suppressed the expression of HNF4α target genes in the liver. In addition, long term treatment of mice with luteolin significantly suppressed their diet-induced obesity and improved their serum glucose and lipid parameters. Importantly, long term luteolin treatment lowered serum VLDL and LDL cholesterol and serum apoB protein levels, which was not accompanied by fat accumulation in the liver. These results suggest that the flavonoid luteolin ameliorates an atherogenic lipid profile in vivo that is likely to be mediated through the inactivation of HNF4α.
Introduction
Hepatocyte nuclear factor 4α (HNF4α),4 a member of the nuclear receptor family, forms a homodimer to function as a transcriptional regulator (1). HNF4α is expressed at high levels in the liver and expressed at a lesser degree in the intestine, kidney, and pancreas (2). The HNF4α homodimer binds to the HNF4α-responsive element in DNA, consisting of a direct repeat of the core sequence AGGTCA, separated by either one or two nucleotides, referred to as direct repeat 1 (DR-1) or direct repeat 2 (DR-2), respectively. HNF4α regulates the expression of genes involved in several metabolic pathways, including drug metabolism, lipid transport, and gluconeogenesis (3).
It is well established that the overproduction of hepatic apolipoprotein B (apoB)-containing lipoproteins confers an increased atherosclerotic risk (4). Microsomal triglyceride transfer protein (MTP) is critical for the formation and secretion of apoB-containing lipoproteins from cells of the liver and intestine (5). Because the inhibition of MTP through pharmacotherapy results in the reduction of apoB-containing lipoproteins in circulation, it is considered to be a promising drug target for atherosclerotic disease (6). HNF4α regulates the expression of genes involved in apoB-containing lipoprotein secretion, such as MTP and apoB; liver-specific HNF4α-deficient mice exhibit greatly reduced serum cholesterol and triglyceride levels, possibly because of decreased expression of genes encoding apoB and MTP (7).
Although HNF4α contains a putative ligand-binding domain (LBD), the endogenous ligand of HNF4α was unclear for a long time. Recently, linoleic acid (LA) was identified as an endogenous ligand for HNF4α; however, the binding of LA to HNF4α does not affect its transcriptional activity (8). More recently, small synthetic molecules, such as BIM5078 and BI6015, were identified as antagonists for HNF4α. The binding of their antagonists to HNF4α resulted in the suppression of HNF4α activity (9), suggesting that exogenous small molecules could control HNF4α activity. This finding led us to consider additional investigations to find HNF4α antagonists.
Luteolin is one of the most common flavonoids in plants and is classified as a flavone. Luteolin-containing plants are used as a food and traditional medicine to treat various pathologies (10). Luteolin exhibits several pharmacological activities, such as anti-cancer, anti-inflammatory, anti-microbial, and anti-diabetic activities (10, 11). Although the molecular mechanism by which luteolin exhibits anti-cancer activities has been extensively investigated, the mechanism underlying the anti-diabetic effect of luteolin is largely unknown.
In the present study, we identified the flavonoid luteolin as a repressor of HNF4α. Luteolin bound LBD of HNF4α and suppressed its activity. Luteolin potently suppressed apoB-containing lipoprotein secretion in cultured cells. Dietary luteolin suppressed obesity and decreased lipid levels in the serum and liver as well as improved glucose tolerance in mice fed a high-fat diet (HFD).
Experimental Procedures
Reagents
Luteolin used for cell treatment and the animal diet was purchased from TCI and Ark Pharm Inc., respectively. DMEM was from Wako. Isopropyl β-d-thiogalactopyranoside was obtained from Nacalai Tesque. LB broth, luteolin 7-glucoside, and isoorientin were purchased from Sigma. The information on other companies from which we obtained other compounds used for screening is available upon request. HEK293, HepG2, and Caco2 cells were obtained from ATCC.
Cell Culture
HEK293 and HepG2 cells were maintained in medium A (DMEM supplemented with 10% fetal bovine serum (FBS), containing 100 units/ml penicillin and 100 μg/ml streptomycin). Caco2 cells were maintained in medium B (DMEM supplemented with 10% FBS and non-essential amino acids, containing 100 units/ml penicillin and 100 μg/ml streptomycin). Cells were incubated at 37 °C under 5% CO2 atmosphere. Caco2 cells that had been cultured for 14 days after reaching confluence were considered to be differentiated.
Plasmid Constructs
The reporter plasmids containing the human MTP promoter (−204 to +33), pMTP204-Luc, and HNF4α-responsive element-mutated MTP promoter (HNF4α B site: AGTTTGGAGTCTG → AGTGCGGCCGCTG), pMTP204-HNF4α-mut-Luc, and expression plasmids for the GAL4 DNA-binding domain (DBD)-HNF4α LBD fusion protein (pGAL4 DBD-HNF4α LBD) and pFLAG-HNF4α were described previously (12, 13). A reporter plasmid, pInsig1-Luc, was constructed by inserting a 2.8-kb NheI-HindIII PCR fragment coding the 5′-promoter region (−2782/+84) of mouse insulin-induced gene 1 (Insig1) into the same restriction sites of the pGL4.10 vector. pET-28-hHNF4α-LBD (amino acid residues 148–377) was constructed by inserting a PCR fragment that encodes human HNF4α into pET-28a (Novagen).
Food Factor Screening Using Luciferase Assays
HepG2 cells were plated in 12-well plates at a density of 2 × 105 cells/well, cultured with medium A for 24 h, and transfected with 200 ng of pMTP204-Luc (a reporter plasmid) and 200 ng of pCMV-β-Gal (an expression plasmid for β-galactosidase) using the calcium phosphate method. Twenty-four hours later, the test compounds (100 μm) or control vehicle were added to the medium. All of the test compounds were dissolved in DMSO. The final DMSO concentration of the cultured medium was 0.1%. After incubation for another 12 h, luciferase and β-galactosidase activities were determined as described previously (14). Normalized luciferase values were determined by dividing the luciferase activity by the β-galactosidase activity.
Luciferase Assays
HEK293 and HepG2 cells were plated in 12-well plates at a density of 1 × 105 and 2 × 105 cells/well, respectively, cultured with medium A for 24 h, and transfected with plasmids, as indicated in the figure legends. Twenty-four hours later, the test compound or control vehicle was added to the medium. The final DMSO concentration of the cultured medium was 0.1%. After incubation for another 12 h, the luciferase and β-galactosidase activities were determined as described above.
Small Interfering RNA (siRNA) Experiments
siRNAs (150 pmol/6-well plate) for human HNF4α (Santa Cruz Biotechnology) and control (pGL2 luciferase; Bonac) were transfected using Lipofectamine RNAiMAX (Invitrogen) into HepG2 cells according to the manufacturer's instructions.
Real-time PCR
Total RNA was extracted using Isogen (Nippon Gene) according to the manufacturer's instructions. cDNA was synthesized and amplified from total RNA using a high capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed using the Applied Biosystems 7000 sequence detection system. Relative mRNA levels were determined by normalizing to the 36B4 transcript. The TaqMan ID number and sequences of the primer sets are available upon request.
Chromatin Immunoprecipitation (ChIP) Assay
HepG2 cells were grown in 10-cm dishes with medium A to confluence, followed by treatment with vehicle or 50 μm luteolin for 3 h. The cells were then processed for the ChIP assay using a reagent kit (Upstate Biotechnology) according to the manufacturer's instructions. Immunoprecipitation (IP) was performed with normal rabbit IgG, acetyl-histone H3, or anti-HNF4α antibody. Real-time PCR used the following primers: human MTP DR-1, 5′-GTGAGAGACTGAAAACTGCAGC-3′ and 5′-CATCCAGTGCCCAGCTAGGAG-3′; human HNF1α DR-1, 5′-CATGATGCCCCTACAAGGTT-3′ and 5′-AGCTGGGGAAATTCTCCAAG-3′; human G6Pase DR-1, 5′-AACCTACTGGTGATGCACCT-3′ and 5′-TGCTCTGCTATGAGTCTGTG-3′; and human HNF1α distal region, 5′-CATATTGGTCAGGGTGGTCT-3′ and 5′-TGTGGCTACCAAGCACTTGA-3′.
Immunoblotting and Antibodies
HepG2 or differentiated Caco2 cells were treated as described in the figure legends. After treatment, cells and media were harvested, and immunoblotting was performed as described previously (15). Polyclonal anti-HNF4α (H-171) and anti-PGC1 (H-300) were obtained from Santa Cruz Biotechnology. Monoclonal anti-β-actin (AC-15) was obtained from Sigma. Polyclonal anti-apoB (ab7616) was obtained from Abcam. Normal rabbit IgG and acetyl-histone H3 (Lys-9) antibody used for ChIP assays were obtained from Millipore.
Binding Assay Using Luteolin-conjugated Beads
Luteolin-conjugated agarose beads and control beads were kindly provided by RIKEN NPDepo (16). HepG2 cells were treated with 50 μm luteolin or vehicle for 3 h, harvested and washed twice with PBS, and then resuspended in binding buffer (10 mm Tris-HCl (pH 7.6), 50 mm KCl, 5 mm MgCl2, 1 mm EDTA, and a protease inhibitor mixture). After cell lysis by homogenization with a syringe, the supernatant was collected as a cell lysate. After the cell lysate (1 mg of protein) was preincubated with control beads (10 μl) for 1 h at 4 °C, the cleared cell lysate was incubated with luteolin beads or control beads (15 μl) for 12 h at 4 °C. The reacted beads were washed with binding buffer, and the coprecipitated proteins were detected by immunoblotting.
Spectroscopic Assay for the Detection of HNF4α-Luteolin Interactions
Plasmid pET-28-hHNF4α-LBD was transformed in Escherichia coli strain BL21 (DE3). Cells harboring the expression plasmid for pET-28-hHNF4α-LBD were grown in LB-kanamycin (50 μg/ml) medium until A600 reached 0.6–0.8 at 37 °C. Following this, 0.25 mm isopropyl β-d-thiogalactopyranoside was added, and the cells were grown overnight at 30 °C. The cells were then collected and suspended in equilibration buffer (50 mm Tris-HCl, pH 7.5), disrupted by sonication (duty cycle 50%, output 5.0), and centrifuged at 11,000 rpm for 30 min at 4 °C. After centrifugation, the supernatant was equilibrated with HIS-Select nickel affinity gel. The column was washed with Tris buffer (50 mm Tris-HCl, pH 7.5), and the proteins were eluted with elution buffer (50 mm Tris-HCl, 500 mm imidazole, pH 7.5). The eluted sample was checked by Coomassie Brilliant Blue staining. Purified HNF4α protein solution was incubated with or without 1 mm luteolin overnight at 4 °C and centrifuged at 11,000 rpm for 10 min at 4 °C. The supernatant was washed twice by ultrafiltration with 5 mm Tris-HCl, pH 7.5, and washed by gel filtration (column: Hiload 16/60 Superdex 200 prep grade) with wash buffer (20 mm Tris-HCl, pH 7.5, 250 mm NaCl). After gel filtration, the absorption spectra of HNF4α (0.8 mg/ml) solutions with or without luteolin treatment were recorded using a JASCO V-560 spectrophotometer.
Mice and Diet
All experiments were performed according to the guidelines of the Animal Usage Committee of the University of Tokyo. Male 5-week-old C57BL/6J mice obtained from Clea Japan were housed in the animal care facility under controlled temperature and humidity conditions with a 12-h light/dark cycle. The mice were given free access to water and were initially fed a standard pelleted diet (Labo MR Stock, Nosan Corp. Bio Department) during a 7-day acclimation period. A pelleted HFD with 60% energy supplied by fat (D12492) was purchased from Research Diet.
Short Term Administration (for 3 Days) of a Luteolin-supplemented HFD
Mice (n = 10) were fed an HFD with mealtime restricted to 1000–1200 h for 7 days to acclimate them to time-limited feeding and were then divided into two groups (n = 5/group). For 3 days, the mice were fed an HFD or an HFD with 0.6% (w/w) luteolin. Food intake was measured each day. Body weight was measured at the start and end points. The mice were sacrificed at 1400 h (after 2 h of fasting) under anesthesia. Liver samples were rapidly excised, frozen in liquid nitrogen, and stored at −80 °C until further processing. Blood samples were also taken, and the serum was separated and stored at −80 °C until further processing.
Long Term Administration (for 57 Days) of a Luteolin-supplemented HFD
The mice (n = 24) were fed ad libitum a pelleted HFD for 11 weeks and then divided into three groups with similar average body weight and blood glucose levels. For 57 days, mice (n = 8/group) were fed ad libitum HFD, HFD with 0.6% (w/w) luteolin, or HFD with 1.5% (w/w) luteolin. Food intake and body weight were measured every other day. At 35, 44, and 49 days after the initiation of luteolin supplementation, a blood glucose assay, computed tomography (CT), and an oral glucose tolerance test (OGTT) were performed, respectively. The mice were fasted for 4 h and sacrificed under anesthesia. The livers and intestines were rapidly excised, frozen in liquid nitrogen, and stored at −80 °C until further processing. Blood samples were obtained, and the serum was separated and stored at −80 °C until further processing.
Blood Glucose Assay
At day 35 after luteolin supplementation, blood glucose levels were measured after a 4-h period of fasting. Using a handheld glucometer (Ascensia Breeze 2, Bayer Diagnostics), the blood glucose concentration was determined using a drop of blood from the tail.
Abdominal Fat Composition Analysis
At day 44 after luteolin supplementation, abdominal fat weight was analyzed by CT. Mice were anesthetized by inhalation of isoflurane and then scanned using the LaTheta (LCT-100) experimental animal CT system (Aloka). Abdominal scans refer to the area between the proximal end of the first vertebra and the distal end of the tibia. For abdominal fat composition analysis, CT scanning was performed at 2-mm intervals from the diaphragm to the bottom of the abdominal cavity. Visceral and subcutaneous fats were distinguished and quantitatively evaluated using LaTheta software (17).
OGTT
OGTT was performed at 49 days after luteolin supplementation. After a 16-h period of food deprivation, glucose (2 g/kg body weight) in water was orally administered by gavage. Using a handheld glucometer (Ascensia Breeze 2, Bayer Diagnostics), plasma glucose levels were measured from the tail blood before and 15, 30, 45, 60, 90, 120, and 150 min after glucose administration.
Serum Biochemistry
Serum ApoB was analyzed by immunoblotting. The serum was diluted to 1:20 and then detected by immunoblotting. The signals on the membrane were quantified by ImageQuant LAS 400 mini (GE Healthcare). Serum triglycerides, total cholesterol, non-esterified fatty acids (NEFAs), total bile acid, alanine aminotransferase, and aspartate aminotransferase levels were determined using kits purchased from Wako Chemicals. The serum insulin concentration was determined using the Lbis Insulin-mouse-T ELISA kit purchased from Shibayagi. Serum lipoproteins were analyzed using a high-performance liquid chromatography system from Skylight Biotech (LipoSEARCH).
Hepatic Lipid Extraction
An ∼150-mg piece of frozen liver tissue was immersed in 4 ml of chloroform/methanol (2:1, v/v) homogenized for 20 s. Tissue homogenates were vortexed and stored at room temperature for 30 min. In total, 1 ml of 50 mm NaCl was added to all samples. All samples were vortexed for 10 s and centrifuged at 1500 × g at 4 °C for 30 min. The upper aqueous phase was removed and discarded. The lower lipid-containing phase of each sample was mixed with 1 ml of 0.36 mol/liter CaCl2/methanol (1:1, v/v), vortexed, and centrifuged at 1500 × g at 4 °C for 10 min. The upper aqueous phase was removed. The lower lipid-containing phase was washed once more with 1 ml of 0.36 mol/liter CaCl2/methanol (1:1), lipid extracts were transferred to a 5-ml mess flask, and chloroform was added to the mark to prepare a sample.
Triglyceride and Cholesterol Assays
In total, 10 μl of 50% Triton X-100 was added to 200 μl of lipids extracted with 40 μl of double-distilled water and 40 μl of standard in a glass tube, vortexed, and air-dried in a fume hood overnight. In total, 500 μl of color reagent for triglyceride/cholesterol was added, vortexed, incubated at 37 °C for 5 min, and centrifuged at 13,000 rpm at room temperature for 3 min. Supernatants were transferred to 96-well plates, and the level of triglyceride/cholesterol was calculated from the results of sample absorbance spectrophotometrically measured at 595 nm. This level was normalized to the weight of the mouse liver from which the sample was derived.
Statistical Analysis
All data are represented as mean ± S.E. Statistical analysis was performed using Ekuseru-Toukei version 2.0 (Social Survey Research Information). Comparisons between treatments were made by Student's t test for two groups. One-way analysis of variance followed by the Bonferroni procedure was used to compare more than two groups. Differences were considered significant at p < 0.05.
Docking Simulation
GOLD version 5.2.2 software (18) was used to search probable complex structures of human HNF4α LBD and luteolin. The x-ray crystal structure by Rha et al. (Protein Data Bank code 3fs1) (19) was used as human HNF4α LBD. In this crystal structure, myristic acid and PGC1α are bound to the protein. For docking, PGC1α was removed, whereas myristic acid was not removed. The structure of luteolin was built by Torch software, and its force field parameters were determined by AmberTools version 1.2. One hundred complex structures were generated by genetic algorithms with default parameters of slow mode. The GOLD score was used for evaluation of the structures.
Results
Identification of Luteolin as a Food Compound That Suppresses HNF4α Activity
We previously reported that the MTP promoter is under the control of HNF4α and contains an HNF4α-binding element (12). In the present study, we conducted luciferase reporter assays using the MTP promoter region between −204 and +33 to identify small molecules that suppress HNF4α activity. Approximately 140 food components were used as test compounds. Human hepatoma HepG2 cells were transfected with the reporter plasmid and then treated with the test compounds (100 μm) for 12 h. As shown in Table 1 and supplemental Table S1, several food components suppressed the MTP promoter activity. The flavones (luteolin, apigenin, chrysin, acacetin, and nobiletin) and flavonols (fisetin, kaempferol, and galangin) were ranked as top suppressors of the MTP promoter activity (Table 1 and Fig. 1). We focused on the effect of luteolin in additional experiments because luteolin is widely contained in various fruits, vegetables, and seeds. Although it is well known that luteolin exhibits various pharmacological properties, the effect of luteolin on lipid metabolism is largely unknown. To further validate the suppressive effect of luteolin on the MTP promoter activity, we performed luciferase assays with lower concentrations of luteolin (30 and 50 μm). These concentrations of luteolin significantly suppressed the MTP promoter activity, whereas 50 μm daidzein and genistein (isoflavones) did not (Fig. 2A, left). Luteolin did not suppress the activity of the Insig1 promoter, which does not contain the HNF4α-binding element, indicating that luteolin does not affect the general transcription (Fig. 2A, right). The mutation of the HNF4α-binding element of the MTP promoter resulted in the inhibition of luteolin responsiveness accompanied by a decrease in the basal MTP promoter activity (Fig. 2B). The suppressive effect of luteolin was not observed in HEK293 cells, which do not express HNF4α. The stimulated MTP promoter activity, due to the expression of exogenous HNF4α, was significantly suppressed by treatment with luteolin (Fig. 2C). Consistent with this observation, luteolin suppressed the Gal4 DBD-fused HNF4α LBD (Fig. 2D). These results indicate that luteolin suppressed HNF4α activity by affecting HNF4α LBD.
TABLE 1.
Effect of the test compounds (100 μm) on MTP promoter activity
Results are mean ± S.E. (n = 3).
| Compounds | Percentage of relative luciferase activitya |
|---|---|
| % | |
| DMSO (control) | 100 ± 0.3 |
| Luteolin | 17.5 ± 2.3 |
| Apigenin | 18.0 ± 1.0 |
| Chrysin | 20.0 ± 0.0 |
| Acacetin | 20.0 ± 0.9 |
| Fisetin | 39.3 ± 9.7 |
| Kaempferol | 42.0 ± 6.0 |
| Nobiletin | 46.3 ± 0.8 |
| Galangin | 47.4 ± 1.6 |
| Genistein | 104.0 ± 35.9 |
| Daidzein | 145.7 ± 16.8 |
a Luciferase assays were performed using pMTP204-Luc as described under “Experimental Procedures”. Table 1 shows part of the data. Complete data are shown in supplemental Table S1.
FIGURE 1.

Structures of flavonoids and their suppressive effects on MTP promoter activity. The percentages represent the effect of the respective compounds on MTP promoter activity. These values are obtained from Table 1.
FIGURE 2.
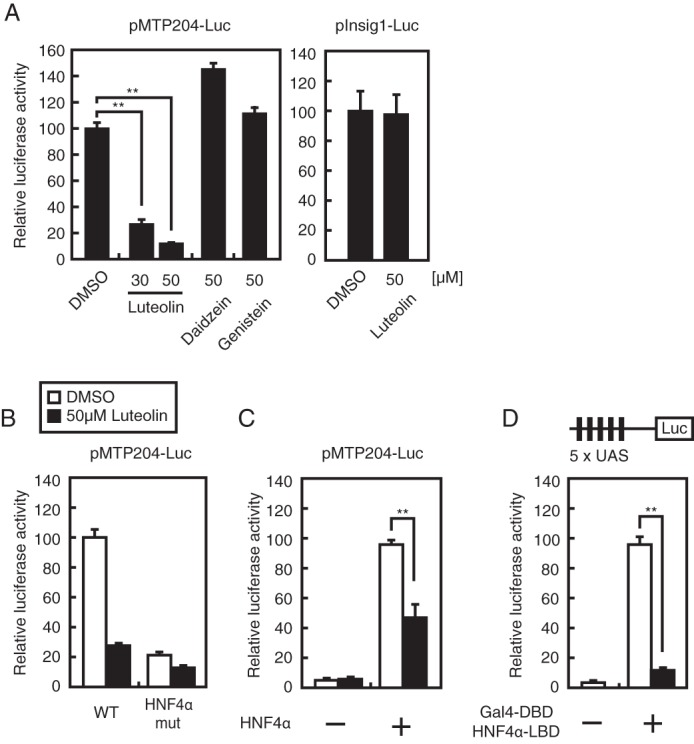
Luteolin suppresses HNF4α activity. HepG2 cells (A, B, and D) and HEK293 cells (C) were transfected with 200 ng of the indicated reporter constructs and 200 ng of pCMV-β-Gal (A–D) and 200 ng of pFLAG-HNF4α (C) or 200 ng of pGal4 DBD-HNF4α LBD (D). The cells were cultured with medium A for 24 h and then treated with the indicated compound for 12 h. Luciferase assays were performed as described under “Experimental Procedures.” All data are expressed as means ± S.E. (error bars) (n = 3). **, p < 0.01.
Luteolin Suppresses HNF4α Target Gene Expression
HepG2 cells were treated with luteolin for 12 h. Quantitative real-time PCR analyses revealed that 10 and 50 μm luteolin treatment caused a significant decrease in the mRNA levels of HNF4α targets, including MTP, apoB, PEPCK, G6Pase, HNF1α, and COUP-TFII, but not in the levels of IRE1, which is independent of HNF4α; in contrast, 1 μm luteolin treatment did not alter the expression of these genes (Fig. 3). It has been reported that HNF4α expression is self-regulated both directly and indirectly. HNF4α directly binds to the enhancer element of the HNF4α gene and stimulates its own expression (20). In addition, the transcription factors HNF1α and COUP-TFII are target genes of HNF4α; HNF1α and COUP-TFII stimulate HNF4α gene expression (20, 21). These observations led us to investigate whether the suppression of HNF4α activity by treatment with luteolin caused a decrease in the mRNA level of HNF4α itself in HepG2 cells. In HepG2 cells, the mRNA level of the HNF4α gene was significantly decreased by luteolin treatment for 3 h, whereas the expression level of the HNF4α protein did not decrease at that time but decreased 6 and 12 h later (Fig. 4, A and B).
FIGURE 3.
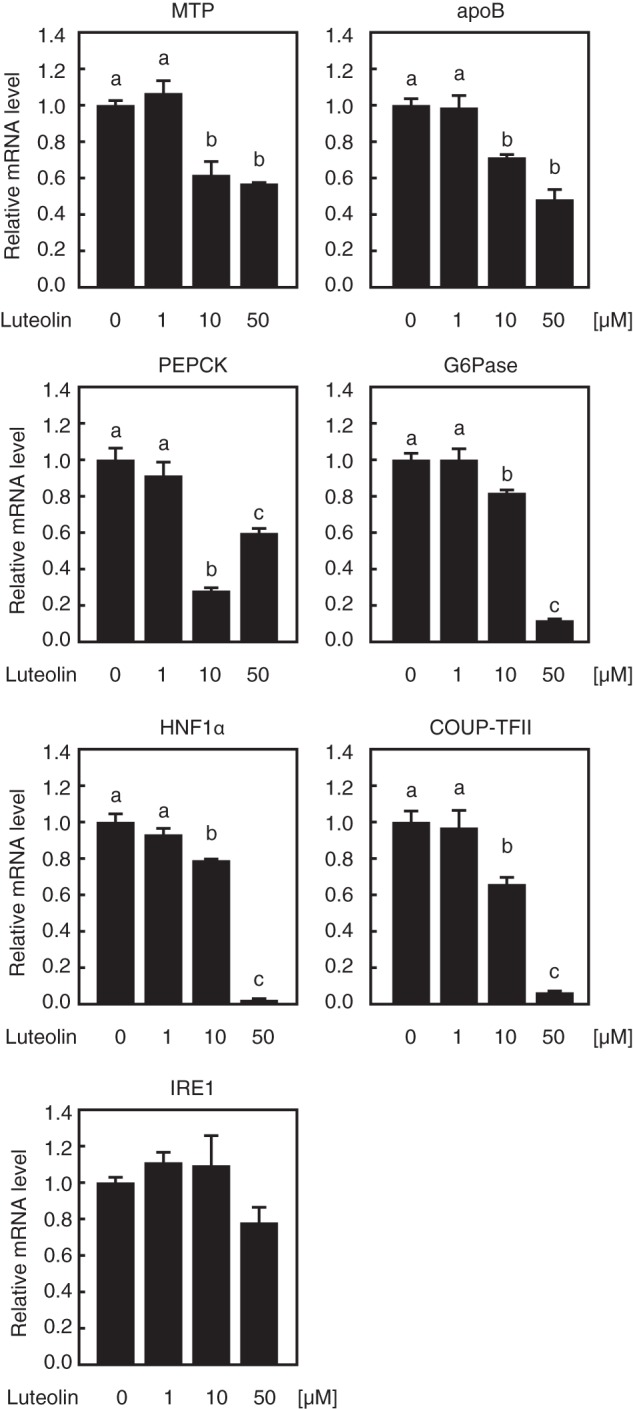
Luteolin suppresses the gene expression of endogenous HNF4α targets in HepG2 cells. HepG2 cells were cultured with the indicated concentration of luteolin for 12 h, and total RNA was isolated. Real-time PCR analysis was performed, and relative mRNA levels were obtained after normalization of 36B4 mRNA. The mRNA levels without luteolin addition are represented as 1. All data are expressed as means ± S.E. (error bars) (n = 3). Means without a common letter differ. p < 0.05.
FIGURE 4.
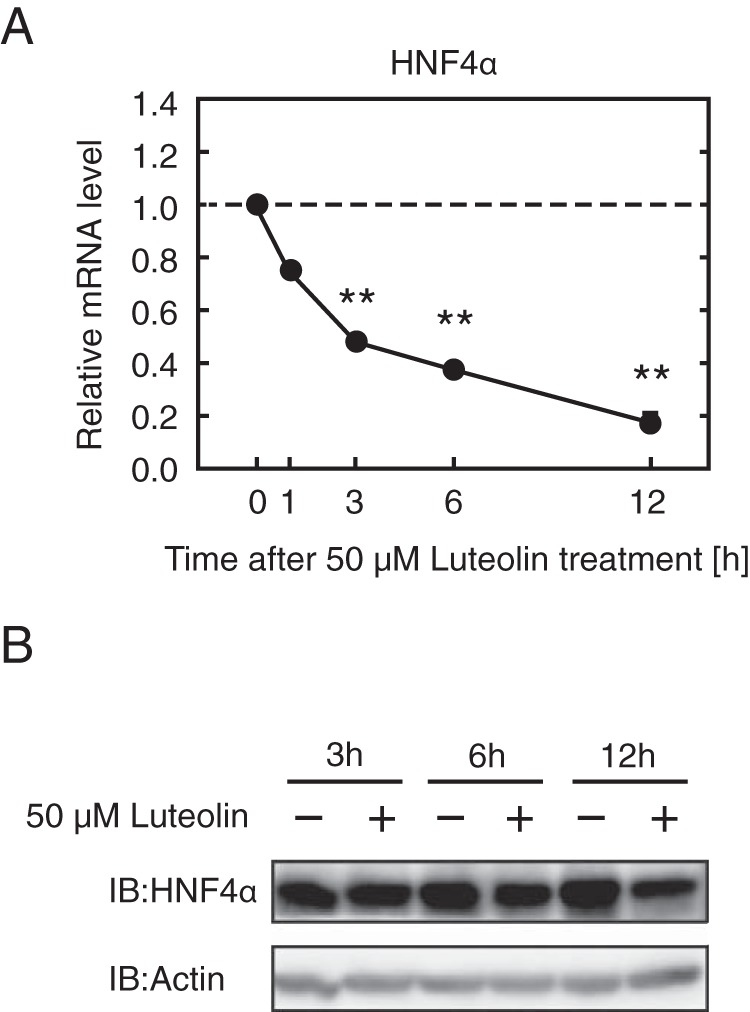
Luteolin suppresses the expression of HNF4α at the mRNA and protein levels in HepG2 cells. A and B, HepG2 cells were cultured with luteolin (50 μm) for the indicated periods of time, and total RNA and whole cell extracts were isolated. A, real-time PCR analysis was performed, and relative mRNA levels were obtained after normalization of 36B4 mRNA. The mRNA levels without luteolin addition at each time point are represented as 1. All data are expressed as means ± S.E. (n = 3). **, different from control (without luteolin addition at each time point); p < 0.01. B, the whole cell extracts were subjected to SDS-PAGE and immunoblotting (IB) with anti-HNF4α or anti-β-actin antibodies. The same results were obtained in three separate experiments.
Luteolin Suppresses ApoB Secretion
Following this, we sought to confirm whether luteolin caused a reduction of apoB secretion. HepG2 cells were incubated with 50 μm luteolin, and the cultured medium was evaluated for the apoB protein level by immunoblotting. As shown in Fig. 5A, the secreted apoB protein levels were suppressed by treatment with luteolin for 12 h, accompanied by the reduction of intracellular apoB protein levels, indicating that luteolin suppresses apoB secretion from HepG2 cells. Treatment with luteolin for 3 h also suppressed secreted apoB protein levels but did not suppress MTP and apoB gene expression (Fig. 5, B and C). These results imply that the presence of multiple mechanisms suppresses apoB secretion by luteolin rather than the transcriptional regulation of apoB and MTP genes. When endogenous HNF4α expression in HepG2 cells was reduced with gene-specific siRNA, apoB secretion was suppressed (Fig. 5D, lanes 1 and 2), indicating that the suppression of HNF4α activity results in the inhibition of apoB secretion. Importantly, treatment with luteolin for 9 h further suppressed apoB secretion from siHNF4α-treated HepG2 cells (Fig. 5D, lanes 2 and 4). These results support the idea that luteolin regulates multiple stages to suppress apoB secretion. Following this, we examined whether luteolin suppresses apoB secretion from intestinal cells. As shown in Fig. 5, E and F, luteolin treatment for 12 h suppressed the mRNA level of the MTP gene, intracellular apoB protein, and secreted apoB protein from differentiated Caco-2 cells.
FIGURE 5.

Luteolin suppresses apoB secretion from HepG2 cells and differentiated Caco2 cells. HepG2 cells (A–C) or Caco2 cells (E and F) were cultured with the indicated concentration of luteolin for 12 h (A, E, and F) or 3 h (B and C), and total RNA, whole cell extracts, and media were isolated. A, B, and F, the samples were subjected to SDS-PAGE and immunoblotting (IB) with anti-apoB or anti-β-actin antibodies. The same results were obtained in three separate experiments. C and E, real-time PCR analysis was performed, and relative mRNA levels were obtained after normalization of 36B4 mRNA. The mRNA levels without luteolin addition are represented as 1. All data are expressed as means ± S.E. (error bars) (n = 3). *, p < 0.05. D, HepG2 cells were transfected with either control (siCon) or HNF4α siRNA oligonucleotides (siHNF4α), cultured with medium A for 48 h, and re-fed the medium containing luteolin (50 μm) for 9 h before harvest. The whole cell extracts and media were isolated and subjected to SDS-PAGE and immunoblotting with anti-apoB, anti-HNF4α, or anti-β-actin antibodies. The same results were obtained in three separate experiments.
Luteolin Glucosides Do Not Suppress MTP Gene Expression and ApoB Secretion from HepG2 Cells
We examined the effect of luteolin glucosides, luteolin 7-glucoside, and isoorientin (Fig. 6A) on MTP gene expression and apoB secretion. Treatment with luteolin glucosides did not affect the MTP promoter activity (Fig. 6B), MTP gene expression (Fig. 6C), and apoB secretion (Fig. 6D), implying that the luteolin glucosides have no effect on HNF4α activity in HepG2 cells. Because cells have been shown to be impermeable to luteolin glucosides, possibly because of the hydrophilicity of glucoside, these results imply that luteolin functions within cells.
FIGURE 6.
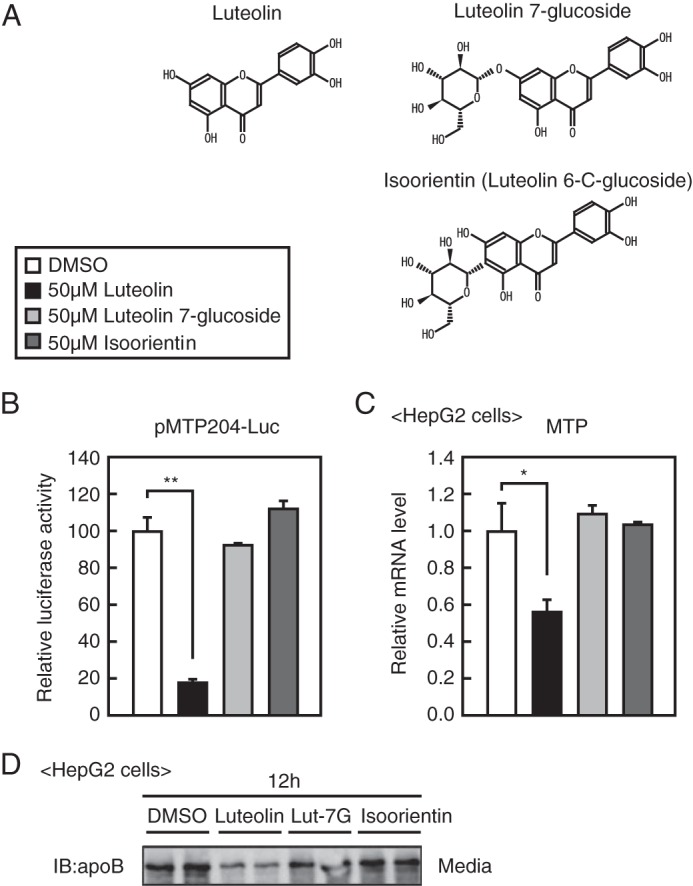
Luteolin glucosides do not suppress apoB secretion from HepG2 cells. A, structure of luteolin, luteolin 7-glucoside, and isoorientin. B, HepG2 cells were transfected with plasmids as described in Fig. 1A and then treated with the indicated compounds for 12 h. Luciferase assays were performed as described under “Experimental Procedures.” C, HepG2 cells were cultured with the indicated compounds for 12 h, and total RNA was isolated. Real-time PCR analysis was performed, and relative mRNA levels were obtained after normalization of 36B4 mRNA. The mRNA levels without luteolin addition are represented as 1. All data are expressed as means ± S.E. (error bars) (n = 3). *, p < 0.05; **, p < 0.01. D, HepG2 cells were cultured with the indicated compounds for 12 h, and media were isolated and subjected to SDS-PAGE and immunoblotting (IB) with anti-apoB antibodies. The same results were obtained in three separate experiments.
Luteolin Directly Binds to LBD of HNF4α
There are a couple of possible explanations for the luteolin-mediated suppression of HNF4α activity. The most likely ones are the direct binding of luteolin to LBD of HNF4α and the alteration of signaling pathways, such as phosphorylation, by luteolin. We therefore attempted to determine whether luteolin binds to HNF4α LBD. As a first step, we examined whether luteolin binds to purified recombinant HNF4α LBD protein that is expressed in E. coli. The purified HNF4α protein was incubated with or without luteolin, subjected to gel filtration for removing the unbound luteolin, and analyzed using the UV-visible spectra. Absorption spectrum analysis showed that the main absorption band was around 280 nm, which indicates protein absorption, and it was comparable despite luteolin incubation (Fig. 7A). The intensity of absorbance between 300 and 450 nm was increased by treatment with luteolin (Fig. 7A, inset). Fig. 7B shows the absorption spectrum of HNF4α subtracted from that of HNF4α treated with luteolin. The spectrum was caused by luteolin binding to HNF4α and represents a bathochromic shift in comparison with free luteolin (Fig. 7B). It has been known that human serum albumin is the most abundant carrier protein in the blood that can bind to luteolin and that the binding causes the bathochromic shift of the absorption spectrum of luteolin (22). These results imply that there may be an interaction between luteolin and HNF4α LBD.
FIGURE 7.

Luteolin can bind to LBD of HNF4α. A, UV-visible absorption spectra of LBD of HNF4α or luteolin-treated LBD of HNF4α. LBD of HNF4α was incubated with luteolin and then subjected to gel filtration for removing the unbound luteolin. The absorption spectra were recorded in 20 mm Tris-HCl buffer (pH 7.5). B, UV-visible absorption spectra of free luteolin and luteolin that bound to LBD of HNF4α. The bound luteolin absorption spectra were calculated by the subtraction of the absorbance of LBD of HNF4α form that of LBD of HNF4α treated with luteolin. The absorption spectra of free luteolin and bound luteolin had λmax ∼355 nm and λmax ∼392 nm, respectively. C, structure of the photoaffinity linker. D, HepG2 cells were treated with luteolin (50 μm) for 3 h, and whole cell extracts were isolated. The extracts were incubated with luteolin beads or control beads, and coprecipitated proteins were detected by immunoblotting (IB) with anti-HNF4α and anti-PGC1α antibodies. The same results were obtained in three separate experiments. E, results of the docking simulation of luteolin to LBD of HNF4α. The protein is displayed in the ribbon model, and luteolin with the highest GOLD score is shown as magenta sticks. Black sticks indicate the position of myristic acid.
As a next step, to examine whether luteolin binds to HNF4α, we used a luteolin affinity matrix using a recently developed photocross-linking method. Luteolin was immobilized on agarose beads through a photoaffinity linker (Fig. 7C). The cell lysates of HepG2 cells were preincubated with control beads, which do not immobilize luteolin, and then unbound proteins were further incubated with luteolin beads. The reacted beads were washed, and coprecipitated proteins were analyzed by immunoblotting using antibodies against HNF4α. Whereas proteins coprecipitated with control beads did not include HNF4α proteins, those coprecipitated with luteolin beads included a large amount of HNF4α proteins (Fig. 7D, lanes 3 and 5). In addition, the amount of HNF4α proteins coprecipitated with luteolin beads was decreased by the pretreatment of HepG2 cells with luteolin for 3 h (Fig. 7D, lanes 5 and 6), suggesting that free luteolin competes with luteolin beads for HNF4α binding. The proteins coprecipitated with control and luteolin beads did not include PGC1α proteins (Fig. 7D). These results suggest that luteolin can form a complex with HNF4α, and HNF4α LBD contributes to the binding.
To further test the binding mode of luteolin to LBD of HNF4α, computational analysis was conducted with the GOLD docking algorithm. We used the crystal structure of HNF4α LBD, which contains a free fatty acid, myristic acid, at the ligand-binding pocket (Protein Data Bank code 3fs1). Computational analysis predicts the possible binding mode of luteolin to LBD of HNF4α in atomic resolution. As shown in Fig. 7E, luteolin can bind to a pocket created by helix 1, 3, and 5, which is far from the hydrophobic pocket occupied by myristic acid. All of the generated complex structures had high GOLD scores (larger than 56) and bound to almost similar positions.
Luteolin Does Not Suppress the DNA Binding of HNF4α
To investigate whether luteolin treatment influences the DNA binding of HNF4α, we employed the ChIP assay. We used HepG2 cells treated with luteolin for 3 h because long term treatment caused a decrease in the expression of HNF4α (Fig. 4B). In HepG2 cells, HNF1α and G6Pase gene expression was decreased, and MTP gene expression was unaltered by treatment with luteolin for 3 h (Figs. 5C and 8A); in contrast, MTP gene expression was decreased by treatment with luteolin for 12 h (Fig. 3), suggesting that treatment with luteolin for 3 h was sufficient to affect the expression of certain genes. As expected, the ChIP assay revealed that HNF4α could bind to the promoter region of its target genes, such as MTP, HNF1α, and G6Pase (Fig. 8B). Luteolin treatment did not alter the recruitment of HNF4α to the promoter region of these genes, whereas it reduced the recruitment of acetyl-histone H3 to the promoter region of HNF1α and G6Pase genes but not the MTP gene (Fig. 8B). It should be noted that HNF4α and acetyl-histone H3 did not bind to the distal region of the HNF1α promoter, and luteolin did not influence these bindings (Fig. 8B). These results suggest that the luteolin-mediated suppression of HNF4α activity is not caused by the alteration of DNA binding of HNF4α to the promoter region of its target gene.
FIGURE 8.
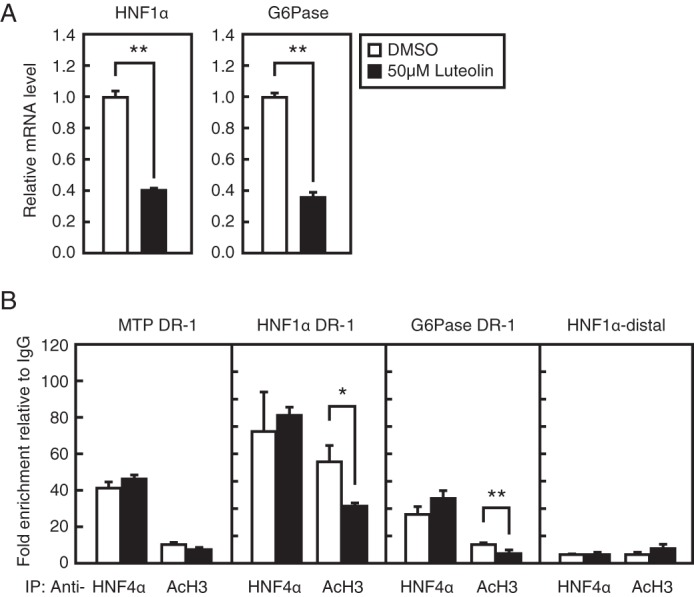
Luteolin does not suppress the DNA binding of HNF4α in HepG2 cells. A, HepG2 cells were cultured with luteolin (50 μm) for 3 h, and total RNA was isolated. Real-time PCR analysis was performed, and relative mRNA levels were obtained after normalization of 36B4 mRNA. The mRNA levels without luteolin addition are represented as 1. B, HepG2 cells were cultured with luteolin (50 μm) for 3 h and processed for ChIP analyses as described under “Experimental Procedures.” After immunoprecipitation (IP) with anti-HNF4α, anti-acetyl-histone H3, or control IgG, real-time PCR analysis was performed with a primer set covering the HNF4α-binding region (DR-1) in the human MTP, HNF1α, and G6Pase promoter or distal region in the human HNF1α promoter. The results are represented by -fold enrichment relative to control IgG. All data are expressed as means ± S.E. (error bars) (n = 3). *, p < 0.05; **, p < 0.01.
Luteolin Reduces the Expression of HNF4α Target Genes in the Mouse Liver
Next, we investigated the effect of luteolin in mice. The mice were fed an HFD or an HFD with 0.6% luteolin in a set 2-h-long mealtime for 3 days. Food intake and body weight as well as several serum parameters were not affected by short term dietary luteolin supplementation (Table 2). On the other hand, the expression of HNF4α target genes, such as HNF4α, MTP, ApoB, HNF1α, PEPCK, and G6Pase, significantly decreased in the livers of mice fed an HFD supplemented with luteolin when compared with the HFD-only control group (Table 3). These results indicate that luteolin suppresses HNF4α activity in the mouse liver, but this effect is not reflected in the serum glucose, total cholesterol, or total triglyceride levels in short term administration of luteolin.
TABLE 2.
Food intake, body weight, and serum biochemistry in C57BL/6J mice fed HFD or HFD + luteolin diets for 3 days
Results are mean ± S.E. (n = 5).
| HFD | HFD + 0.6% luteolin | |
|---|---|---|
| Food intake (g/mouse/day) | 1.53 ± 0.10 | 1.52 ± 0.12 |
| Body weight (g) (day 0) | 21.6 ± 0.9 | 22.1 ± 0.6 |
| Body weight (g) (day 3) | 21.4 ± 1.0 | 21.5 ± 0.9 |
| Serum | ||
| Aspartate aminotransferase (IU/liter) | 64.7 ± 2.2 | 69.7 ± 6.4 |
| Alanine aminotransferase (IU/liter) | 44.7 ± 3.7 | 45.0 ± 5.8 |
| Glucose (mg/dl) | 164 ± 3.7 | 162 ± 4.8 |
| NEFA (milliequivalents/liter) | 1.02 ± 0.06 | 1.22 ± 0.07 |
| Total cholesterol (mg/dl) | 110 ± 0.6 | 111 ± 1.4 |
| Total triglyceride (mg/dl) | 111 ± 15.6 | 100 ± 9.5 |
TABLE 3.
Expression of HNF4α target genes in the liver of C57BL/6J mice fed HFD or HFD + luteolin diets for 3 days
Results are mean ± S.E. (n = 5). *, p < 0.05; **, p < 0.01.
| Gene | Relative mRNA expression (% of HF)a |
|
|---|---|---|
| HFD | HFD + 0.6% luteolin | |
| % | % | |
| HNF4α | 100 ± 11.0 | 64.9 ± 5.1* |
| MTP | 100 ± 3.2 | 80.0 ± 5.7* |
| ApoB | 100 ± 3.5 | 66.0 ± 6.9** |
| HNF1α | 100 ± 8.0 | 70.8 ± 8.2* |
| PEPCK | 100 ± 6.3 | 40.4 ± 17.0* |
| G6Pase | 100 ± 10.8 | 49.4 ± 10.7* |
a The relative amount of each transcript was normalized to the amount of 36B4 transcript.
Dietary Luteolin for 57 Days Suppresses Diet-induced Obesity
To verify the effect of luteolin on diet-induced obese mice, the mice were fed HFD for 11 weeks and subsequently fed HFD or HFD supplemented with luteolin (HFD + 0.6% luteolin or HFD + 1.5% luteolin) for 57 days. The food intake and the parameters representing hepatic toxicity, such as aspartate aminotransferase and alanine aminotransferase, were not affected by dietary luteolin supplementation (Table 4), indicating that the 1.5 and 0.6% luteolin-containing diet had no toxic effect. Compared with the HFD group, the HFD + 0.6% luteolin group had a lower body weight at day 36 after luteolin supplementation, although it did not reach statistical significance. On the other hand, the HFD + 1.5% luteolin group had a significantly lower body weight at day 30 after luteolin supplementation; this effect was observed until sacrifice (Fig. 9A). This decreased body weight was accounted for by a decrease in fat mass, as measured by CT (Fig. 9B) at day 44 after luteolin supplementation (represented by the arrow in Fig. 9A). These results indicate that long term treatment (LTT) with luteolin (for 57 days) suppressed diet-induced obesity, and the decreased fat weight was, at least in part, responsible for the suppressive effect.
TABLE 4.
Food intake, serum biochemistry, liver weight, and liver lipid content in C57BL/6J mice fed HFD or HFD + luteolin diets for 57 days
Results are mean ± S.E. (n = 8). Values within a row without a common letter differ significantly (p < 0.05).
| HFD | HFD + 0.6% luteolin | HFD + 1.5% luteolin | |
|---|---|---|---|
| Food intake (g/mouse/day) | 2.85 ± 0.03 | 2.79 ± 0.07 | 2.84 ± 0.11 |
| Serum | |||
| Aspartate aminotransferase (IU/liter) | 76.9 ± 6.6 | 77.6 ± 9.0 | 53.4 ± 6.8 |
| Alanine aminotransferase (IU/liter) | 40.0 ± 3.5 | 41.9 ± 6.3 | 25.0 ± 5.1 |
| Glucose (mg/dl) (day 35) | 281 ± 11.3A | 246 ± 13.6AB | 215 ± 14.2B |
| Insulin (ng/dl) | 9.47 ± 1.37A | 4.90 ± 0.68B | 4.52 ± 0.90B |
| NEFA (milliequivalents/liter) | 1.00 ± 0.06 | 0.90 ± 0.06 | 0.87 ± 0.05 |
| Total bile acid (μmol/liter) | 50.1 ± 4.2 | 58.0 ± 6.5 | 40.3 ± 4.7 |
| Cholesterol (mg/dl) | |||
| Total | 205 ± 8.8A | 201 ± 4.5A | 171 ± 18B |
| CM | 0.42 ± 0.13 | 0.36 ± 0.04 | 0.34 ± 0.12 |
| VLDL | 2.38 ± 0.37A | 1.97 ± 0.42AB | 1.53 ± 0.41B |
| LDL | 56.0 ± 1.9A | 54.4 ± 5.3A | 37.8 ± 10.4B |
| HDL | 146 ± 6.6A | 145 ± 1.8AB | 132 ± 9.4B |
| Triglyceride (mg/dl) | |||
| Total | 24.7 ± 4.4A | 20.3 ± 2.5AB | 16.5 ± 4.6B |
| CM | 3.02 ± 0.81 | 2.58 ± 0.43 | 2.77 ± 1.00 |
| VLDL | 10.3 ± 1.82 | 9.03 ± 1.66 | 8.28 ± 1.97 |
| LDL | 9.32 ± 1.83A | 7.09 ± 1.21AB | 4.48 ± 1.33B |
| HDL | 2.13 ± 0.58A | 1.57 ± 0.52AB | 0.95 ± 0.47B |
| Liver | |||
| Weight (g) | 2.18 ± 0.10A | 2.20 ± 0.19A | 1.59 ± 0.12B |
| Triglyceride (mg/g) | 916 ± 119A | 910 ± 70A | 409 ± 109B |
| Cholesterol (mg/g) | 7.38 ± 0.24 | 6.43 ± 0.45 | 6.20 ± 0.36 |
FIGURE 9.

Dietary luteolin suppresses diet-induced obesity and improves glucose tolerance. A, change in the body weight of male mice fed HFD or HFD with 0.6 and 1.5% luteolin for 57 days (n = 8). The arrows represent the points when blood glucose measurement, CT, and OGTT were performed. B, at day 44 after luteolin supplementation, CT was performed as described under “Experimental Procedures.” C, at day 57 after luteolin supplementation, the mice were fasted for 4 h and sacrificed. Proteins in serum were analyzed by immunoblotting (IB) with anti-apoB antibodies (n = 8). The typical samples (n = 3) were represented. D, the signals (n = 8) detected on the membrane in C were quantified. The signals of the HFD group are represented as 1. E, at day 49 after luteolin supplementation, OGTT was performed as described under “Experimental Procedures.” F, the average of area under the curve (AUC). All data are expressed as means ± S.E. (error bars) (n = 8). Means without a common letter differ (p < 0.05).
LTT with Luteolin Improves the Glucose and Lipid Parameters in Diet-induced Obese Mice
The blood glucose level showed a tendency to decrease in the HFD + 0.6% luteolin group and significantly decreased in the HFD + 1.5% luteolin group (Table 4). Insulin levels were significantly reduced in the HFD + 0.6% luteolin and 1.5% luteolin groups, whereas no change in serum NEFA and total bile acid levels was found (Table 4). Serum total cholesterol and triglyceride levels were significantly reduced in the HFD + 1.5% luteolin group (Table 4). VLDL and LDL cholesterol levels were reduced, whereas the chylomicron (CM) cholesterol level was not affected by 1.5% luteolin supplementation (Table 4). Similar results were observed in serum triglyceride levels (Table 4). Serum apoB100 protein levels were significantly lowered by 1.5 and 0.6% luteolin supplementation, whereas serum apoB48 protein level was significantly lowered by only 1.5% luteolin supplementation (Fig. 9, C and D). These results imply that LTT with luteolin suppresses VLDL and CM secretion from the liver and small intestine, respectively.
The Effect of LTT with Luteolin on the Expression of HNF4α Target Genes
LTT with luteolin did not reduce the expression of HNF4α and PEPCK genes in the mouse liver, whereas the expression of HNF4α target genes, such as MTP, apoB, HNF1α, and G6Pase, was decreased in the HFD + 1.5% luteolin group (Table 5), implying that LTT with luteolin suppresses HNF4α activity in the mouse liver. Dietary luteolin reduced the expression of HNF1α and G6Pase genes in the mouse duodenum (Table 6). The expression of HNF4α target genes in the jejunum was not reduced by dietary luteolin, with the exception of the G6Pase gene, whereas the expression of apoB, PEPCK, and G6Pase genes in the ileum was decreased in the HFD + 1.5% luteolin group (Tables 7 and 8). Although LTT with luteolin tended to decrease the expression of certain HNF4α target genes in the mouse liver and intestine, these effects did not reach statistical significance. The expression of genes involved in fatty acid synthesis, such as fatty acid synthase (FAS) and acetyl-CoA carboxylase 1 (ACC1), was significantly decreased in the HFD + 1.5% luteolin group in the liver, whereas the expression of genes involved in fatty acid oxidation, such as carnitine palmitoyltransferase 1A (CPT1A) and peroxisome proliferator-activated receptor α (PPARα), was unaltered (Table 5).
TABLE 5.
Expression of HNF4α target genes in the liver of C57BL/6J mice fed HFD or HFD + luteolin diets for 57 days
Results are mean ± S.E. (n = 8). Values within a row without a common letter differ significantly (p < 0.05).
| Gene | Relative mRNA expression (% of HFD)a |
||
|---|---|---|---|
| HFD | HFD + 0.6% luteolin | HFD + 1.5% luteolin | |
| % | % | % | |
| HNF4α | 100 ± 57.4 | 117 ± 8.7 | 128 ± 15 |
| MTP | 100 ± 6.6 | 105 ± 11 | 79.9 ± 6.3 |
| ApoB | 100 ± 8.2 | 106 ± 12 | 85 ± 5.2 |
| HNF1α | 100 ± 15 | 120 ± 14 | 88.7 ± 9.2 |
| PEPCK | 100 ± 7.9 | 123 ± 16 | 143 ± 17 |
| G6Pase | 100 ± 12 | 113 ± 17 | 66.3 ± 10 |
| FAS | 100 ± 12A | 111 ± 7.4A | 59.4 ± 4.2B |
| ACC1 | 100 ± 6.1A | 117 ± 11A | 67.9 ± 6.2B |
| CPT1A | 100 ± 5.6 | 123 ± 15 | 88.2 ± 6.7 |
| PPARα | 100 ± 6.8 | 129 ± 15 | 93.6 ± 9.7 |
a The relative amount of each transcript was normalized to the amount of 36B4 transcript.
TABLE 6.
Expression of HNF4α target genes in the duodenum of C57BL/6J mice fed HFD or HFD + luteolin diets for 57 days
Results are mean ± S.E. (n = 8).
| Gene | Relative mRNA expression (% of HFD)a |
||
|---|---|---|---|
| HFD | HFD + 0.6% luteolin | HFD + 1.5% luteolin | |
| % | % | % | |
| HNF4α | 100 ± 17 | 117 ± 29 | 94.3 ± 24 |
| MTP | 100 ± 17 | 110 ± 26 | 97 ± 22 |
| ApoB | 100 ± 17 | 164 ± 41 | 100 ± 18 |
| HNF1α | 100 ± 20 | 127 ± 35 | 84.4 ± 36 |
| PEPCK | 100 ± 23 | 138 ± 35 | 132 ± 32 |
| G6Pase | 100 ± 15 | 103 ± 18 | 69 ± 13 |
a The relative amount of each transcript was normalized to the amount of 36B4 transcript.
TABLE 7.
Expression of HNF4α target genes in the jejunum of C57BL/6J mice fed HFD or HFD + luteolin diets for 57 days
Results are mean ± S.E. (n = 8).
| Gene | Relative mRNA expression (% of HFD)a |
||
|---|---|---|---|
| HFD | HFD + 0.6% luteolin | HFD + 1.5% luteolin | |
| % | % | % | |
| HNF4α | 100 ± 7.8 | 132 ± 19 | 137 ± 21 |
| MTP | 100 ± 8.4 | 187 ± 35 | 154 ± 29 |
| ApoB | 100 ± 9.4 | 148 ± 25 | 113 ± 24 |
| HNF1α | 100 ± 8.8 | 189 ± 32 | 190 ± 40 |
| PEPCK | 100 ± 24 | 155 ± 29 | 129 ± 25 |
| G6Pase | 100 ± 13 | 155 ± 39 | 69 ± 11 |
a The relative amount of each transcript was normalized to the amount of 36B4 transcript.
TABLE 8.
Expression of HNF4α target genes in the ileum of C57BL/6J mice fed HFD or HFD + luteolin diets for 57 days
Results are mean ± S.E. (n = 8). Values within a row without a common letter differ significantly (p < 0.05).
| Gene | Relative mRNA expression (% of HFD)a |
||
|---|---|---|---|
| HFD | HFD + 0.6% luteolin | HFD + 1.5% luteolin | |
| % | % | % | |
| HNF4α | 100 ± 12 | 150 ± 24 | 116 ± 11 |
| MTP | 100 ± 15 | 122 ± 21 | 98 ± 13 |
| ApoB | 100 ± 15 | 113 ± 23 | 63 ± 7.7 |
| HNF1α | 100 ± 11 | 125 ± 20 | 118 ± 28 |
| PEPCK | 100 ± 38 | 74.4 ± 14 | 72.3 ± 15 |
| G6Pase | 100 ± 11A | 120 ± 26A | 54.4 ± 4.2B |
a The relative amount of each transcript was normalized to the amount of 36B4 transcript.
LTT with Luteolin Attenuates Hepatic Lipid Accumulation and Improves Glucose Tolerance
The liver weight was significantly reduced in the HFD + 1.5% luteolin group (Table 4). Quantitative analysis of the liver lipid contents revealed that liver cholesterol tended to decrease, whereas liver triglyceride was significantly decreased in the HFD + 1.5% luteolin group (Table 4). OGTT (2 g of glucose/kg) performed on day 49 after luteolin supplementation revealed that the HFD + 1.5% luteolin group had enhanced glucose tolerance (Fig. 9, E and F).
Discussion
Our results demonstrate that the flavonoid luteolin suppressed HNF4α activity and the secretion of apoB-containing lipoproteins in cultured cells. Luteolin was able to form a complex with HNF4α, thereby leading to the suppression of HNF4α activity. Dietary luteolin in mice suppressed diet-induced obesity and lowered serum cholesterol and triglyceride levels, accompanied by a reduction in serum apoB protein levels. Although most of our conclusions are based on cellular studies and luteolin supplementation to HFD in mice, the novel modulation of HNF4α activity by food components reveals a possible effective method of prevention and improvement of atherosclerotic diseases.
In the present study, we identified food components that suppress the MTP gene promoter activity from our library containing ∼140 food components in addition to several flavones and flavonols that suppress the MTP promoter activity. Flavones showed a stronger effect than flavonols (Table 1 and Fig. 1). The difference between flavones and flavonols is a hydroxyl group at the C3 position (Fig. 1). The difference in the suppressive effects seems to be determined by the position of the hydroxyl group rather than by the number of these groups. Both luteolin (flavone) and kaempferol (flavonol) contain four hydroxyl groups. Three hydroxyl groups are located at the same positions; however, one group is located at the B5′ and C3 positions in luteolin and kaempferol, respectively (Fig. 1). Therefore, the presence of the hydroxyl group at the C3 position may have influenced the suppressive effect of flavonoid on the MTP promoter activity. Apigenin (flavone), the structure of which is identical to that of kaempferol, except for the detachment of a hydroxyl group at the C3 position, also effectively suppressed the MTP gene promoter activity. Isoflavones, which contain the B-ring at the C3 position, had no effect on the MTP promoter activity (Table 1 and Fig. 1). This evidence implies that the position of the B-ring also influences the suppressive effect. Clearly, further studies are required to determine the structure-activity relationship of flavonoids in the suppression of MTP promoter activity.
There is evidence that genistein (isoflavone) and naringenin (flavanone) reduce the gene expression and activity of MTP, thereby inhibiting apoB secretion from HepG2 cells (23, 24). In the present study, these flavonoids did not suppress the MTP promoter activity. However, this is not surprising because we used the MTP promoter region between −204 and +33 to examine HNF4α activity. Therefore, these flavonoids may affect the transcription factor that binds to an MTP promoter region other than the region used in the present study. Our results indicate that luteolin treatment for 12 h decreased HNF4α target genes and inhibited apoB secretion in HepG2 cells (Figs. 3 and 5A). In addition, we demonstrated that luteolin suppressed apoB secretion from HNF4α-knocked down HepG2 cells (Fig. 5D). These results suggest that luteolin suppresses apoB secretion through HNF4α-dependent and -independent mechanisms. At present, the HNF4α-independent mechanism by which luteolin suppresses apoB secretion is unclear; however, the inhibition of MTP activity by luteolin may contribute to this, as in the case of genistein and naringenin. It has been reported that MTP inhibitor treatment is associated with increased degradation of the apoB protein due to its incomplete lipidation (25). Consistent with this, treatment with genistein and naringenin decreased intracellular apoB protein levels and apoB secretion (23, 24). In the present study, the 3-h luteolin treatment caused a reduction of intracellular apoB protein levels in HepG2 cells, which was not accompanied by a reduction of apoB mRNA expression, although apoB secretion was reduced (Fig. 5, B and C); this finding suggests that luteolin suppresses MTP activity and stimulates intracellular apoB protein degradation. Alternatively, luteolin may stimulate apoB degradation independent of its lipidation levels. It has been known that the degradation of apoB protein is mediated by multiple pathways, including proteasomal degradation and autophagy (26, 27). Several reports indicate that luteolin could induce autophagy in several cell types (28–30). Thus, it is plausible that luteolin-mediated autophagy contributes to the apoB protein degradation. In this regard, luteolin functions at multiple levels to suppress apoB secretion, including the reduction of gene expression through the HNF4α antagonism as well as non-transcriptional regulation, such as MTP inhibition or induction of autophagy-mediated apoB protein degradation. Further studies are required to determine the identity of non-transcriptional regulation of luteolin on the suppression of apoB secretion.
Several reports have revealed flavonoid-mediated regulation of nuclear receptor activity, such as the estrogen receptor and PPARγ (31–34). Structural studies have revealed that luteolin along with myristic acid occupies a ligand-binding pocket of PPARγ (35). The binding pocket occupied by luteolin is identical to that of the synthetic ligands thiazolidinediones, although each binding mode of PPARγ differs (35). The x-ray crystal structure studies using purified recombinant HNF4α protein revealed that the ligand-binding pocket of HNF4α contains a mixture of fatty acids, including lauric, myristic, and palmitic acids (36). More recently, it has been shown that exogenously expressed HNF4α in COS-7 cells binds to LA, and this binding is exchangeable. However, LA does not exhibit a significant effect on the HNF4α transcriptional activity (8). In the present study, we showed that luteolin is capable of binding to HNF4α and that the binding pocket of HNF4α occupied by luteolin is far from that occupied by the fatty acid. Luteolin did not affect the DNA binding of HNF4α but suppressed the histone H3 acetylation on its target gene promoter. These results imply that the binding of luteolin alters the cofactor recruitment to HNF4α. Although we have examined the effect of luteolin on the binding between HNF4α and its cofactor PGC1α, the binding was unaltered by luteolin treatment.5 The precise mechanism by which luteolin suppresses HNF4α activity remains unknown. Recently, Kiselyuk et al. (9) reported the synthetic HNF4α antagonist BI6015. They showed that BI6015 binds to the ligand-binding pocket of HNF4α in a position similar to that of the pocket occupied by fatty acid with the GOLD docking algorithm. Importantly, the binding pockets of HNF4α with luteolin and BI6015 are different. This result suggests the possibility that luteolin and BI6015 can bind to HNF4α concurrently and that the binding results in augmentation of the suppressive effect. Further studies are required to determine the molecular mechanism by which luteolin suppresses the activity of HNF4α and whether the suppressive effect of luteolin is augmented by BI6015.
Shimoi et al. (37) reported that luteolin 7-glucoside, unlike luteolin, cannot be detected in the rat plasma after the administration of luteolin 7-glucoside by gastric intubation. They also demonstrated that luteolin 7-glucoside is hardly absorbed using the rat everted small intestine model (37). In the present study, we demonstrated that luteolin glucosides do not suppress MTP gene expression and apoB secretion in HepG2 cells and concluded that luteolin functions within cells after absorption. However, we cannot exclude the possibility that the receptor localized to the plasma membrane senses luteolin and that the addition of glucoside makes it inaccessible to the receptor. It has been reported that luteolin enhances the phosphorylation of AMP-activated protein kinase in HepG2 cells (38), and AMP-activated protein kinase directly phosphorylates HNF4α and represses its transcriptional activity (39). Thus, it is conceivable that luteolin indirectly suppressed HNF4α activity through the stimulation of AMP-activated protein kinase signaling in addition to direct binding to its LBD as an antagonist.
In the present study, LTT with luteolin lowered the serum total cholesterol and triglyceride levels in HFD-induced obese mice. CM triglyceride showed a tendency toward reduction, whereas VLDL and LDL cholesterol levels were significantly reduced by dietary luteolin. These results imply that the suppressive effect of luteolin on apoB secretion was more effective in the liver than in the intestine. Indeed, LTT with luteolin tended to suppress the gene expression of MTP and apoB in the liver but not in the intestine (Tables 6–8). However, these alterations did not reach statistical significance despite the significant suppressive effect of short term treatment of luteolin on HNF4α target gene expression in the liver of mice (Table 3). At present, the reason for this difference is unclear; however, LTT with luteolin exhibited various beneficial effects, such as anti-obesity and anti-hyperglycemia, and these phenomena may weaken the luteolin effect. LTT with luteolin tended to increase the mRNA level of HNF4α (Table 5), which may obscure the suppressive effect of luteolin on HNF4α target gene expression. In addition, there is a possibility that the plasma concentrations of luteolin cannot reach sufficient levels to affect certain gene expression. It has been reported that after administration of 14.3 mg/kg luteolin or 92.3 mg/kg peanut hull extract (equivalent to 14.3 mg/kg luteolin) by oral gavage in rats, the maximum plasma concentration of luteolin reached 1.97 μg/ml (6.88 μm) or 8.34 μg/ml (29.1 μm) (40). In the present study, we used different methods to administer luteolin to the mice. Dietary supplementation of 1.5 and 0.6% luteolin are estimated to be ∼1050 mg/kg/day and 420 mg/kg/day of luteolin. Although different administration methods (oral gavage and supplementation to the diet) make it difficult to convert plasma concentrations of luteolin, oral intake of luteolin can result in micromolar orders of plasma luteolin concentration. Given that 10 μm, but not 1 μm, luteolin treatment caused a significant decrease in the mRNA levels of HNF4α targets in HepG2 cells (Fig. 3), it is probable that dietary luteolin can influence the activity of HNF4α in vivo.
MTP inhibitors that suppress the secretion of apoB-containing lipoproteins are promising drugs for atherosclerosis; however, the critical side effects, such as fatty liver, prohibit their clinical use (6). LTT with luteolin lowered triglyceride levels in the liver despite decreased serum triglyceride levels. Luteolin suppressed the hepatic expression of lipogenic genes, including FAS and ACC1, but showed no effect on the expression of genes related to fatty acid β-oxidation, including CTP1A and PPARα. Therefore, it is probable that the suppression of fatty acid synthesis was responsible for the reduction of triglyceride in the liver.
We demonstrated that LTT with luteolin suppressed the body weight in HFD-induced obese mice. Luteolin supplementation reduced the weight of the liver and adipose tissue, and this could be responsible for the loss of body weight. At present, the reason why dietary luteolin suppresses diet-induced obesity is unclear. JTT-130, an intestine-specific inhibitor of MTP, has been shown to suppress HFD-induced obesity in rats with suppressed food intake and fat absorption (41). Thus, dietary luteolin appears to suppress apoB secretion from the intestine, thereby suppressing fat absorption, and this could contribute to the anti-obesity effect of luteolin. However, although the CM triglyceride level tended to be lower in the HFD + 0.6% luteolin group than in the HFD + 1.5% group when compared with the HFD group, weight gain was significantly suppressed only in the HFD + 1.5% group. Therefore, it is likely that the suppression of fat absorption has limited contribution to the anti-obesity effect of luteolin. More recently, Kwon et al. (42) reported that dietary luteolin significantly decreased body weight in HFD-induced obese mice. They reported that dietary luteolin up-regulated the expression of genes controlling lipolysis and the tricarboxylic acid cycle prior to lipid droplet formation in the adipose tissue, thereby reducing adiposity. Thus, it is likely that luteolin modulates multiple pathways in both the small intestine and adipose tissue to ameliorate diet-induced obesity. It has been reported that liver-specific HNF4α-deficient mice exhibit lower body weight than wild-type mice, suggesting that the suppression of HNF4α activity in the liver can cause the body weight loss (7). Therefore, dietary luteolin may reduce the body weight, at least in part, through the inactivation of HNF4α in the liver.
We also demonstrated that LTT with luteolin improves fasting hyperinsulinemia and glucose intolerance in HFD-induced obese mice. An improved fasting glucose level despite the low level of insulin in mice fed dietary luteolin suggests that luteolin improves insulin sensitivity, although HNF4α has been known as a causative gene for maturity onset diabetes of the young 1, characterized by defective insulin secretion of the pancreatic β cells (43). Maturity onset diabetes of the young is an autosomal dominant inherited disease in humans; thus, it is considered that HNF4α activity remains relatively low throughout a patient's lifetime. On the other hand, inactivation of HNF4α by its repressor, such as luteolin and BI6015, is transient. Therefore, it is doubtful whether the transient suppression of HNF4α causes diabetes. Indeed, genetic deletion of HNF4α in pancreatic β cells did not show abnormalities in islet architecture and β cell mass (44), but administration of BI6015 to normal mice for 2 weeks resulted in β cell proliferation (45). These data imply that the difference in the modes of suppression of HNF4α activity (constitutive and transient) could result in different phenotypes. However, it is not clear whether suppression of HNF4α activity by luteolin contributes to the improvement of glucose tolerance in diet-induced obese mice. Obesity accelerates adipose tissue dysfunction and causes insulin resistance (46). Therefore, it is likely that dietary luteolin improves insulin sensitivity by the amelioration of obesity. It is also probable that the decreased expression of the G6Pase gene in the liver and the small intestine suppresses gluconeogenesis, and this contributes to the amelioration of the fasting blood glucose level. Kwon et al. (42) also reported that dietary luteolin improves hepatic insulin sensitivity by suppressing the expression of sterol regulatory element-binding protein 1, which modulates Irs2 expression through negative feedback and gluconeogenesis. In addition, dietary luteolin increases PPARγ protein expression, which may be linked to the improvement in circulating fatty acid levels by enhancing fatty acid uptake genes in adipose tissue (42). These results suggest that dietary luteolin functions at multiple levels to improve insulin sensitivity. Clearly, further studies are required to determine whether suppression of HNF4α activity by luteolin can contribute to the amelioration of glucose metabolism.
Collectively, we demonstrated that luteolin suppresses HNF4α activity and apoB secretion in HepG2 cells. In addition, luteolin ameliorates an atherogenic lipid profile in vivo that is likely to be mediated through the inactivation of HNF4α. Although luteolin is one of the common flavonoids in plants, supplemental intake of luteolin is suitable for its metabolic improvement because of the low content of luteolin in plants. The amelioration of the atherogenic lipid profile by dietary luteolin accompanied by the improvement of fatty liver indicates that luteolin and related compounds may be a novel treatment for atherosclerotic diseases and fatty liver.
Author Contributions
J. I. and R. S. conceived the project. J. L. and J. I. designed the study. J. L., J. M. C., Z. Y., H. K., and T. H. performed the experiments. J. L., J. I., S. N., S. F., H. K., M. S., and R. S. analyzed the data. J. I. wrote the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We thank Kaori Honda and Drs. Yasumitsu Kondoh, Tamio Saito, and Hiroyuki Osada for providing luteolin-conjugated agarose beads. We thank Enago for the English language review.
This work was supported by research grants from the Japan Science and Technology Agency (Research for Promoting Technological Seeds) (to J. I.); the Tojuro Iijima Foundation for Food Science and Technology (to J. I.); the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to S. N.); the Japanese Council for Science, Technology and Innovation; and the Cross-ministerial Strategic Innovation Promotion Program (SIP Project 14533567). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
J. Li, J. Inoue, and R. Sato, unpublished data.
- HNF4α and HNF1α
- hepatocyte nuclear factor 4α and 1α, respectively
- apo
- apolipoprotein
- CM
- chylomicron
- CT
- computed tomography
- DR
- direct repeat
- DBD
- DNA-binding domain
- HFD
- high-fat diet
- LBD
- ligand-binding domain
- LA
- linoleic acid
- LTT
- long term treatment
- NEFA
- non-esterified fatty acid
- OGTT
- oral glucose tolerance test.
References
- 1. Sladek F. M., Zhong W. M., Lai E., Darnell J. E. Jr. (1990) Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 4, 2353–2365 [DOI] [PubMed] [Google Scholar]
- 2. Drewes T., Senkel S., Holewa B., Ryffel G. U. (1996) Human hepatocyte nuclear factor 4 isoforms are encoded by distinct and differentially expressed genes. Mol. Cell. Biol. 16, 925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gonzalez F. J. (2008) Regulation of hepatocyte nuclear factor 4α-mediated transcription. Drug Metab. Pharmacokinet. 23, 2–7 [DOI] [PubMed] [Google Scholar]
- 4. Abbasi F., Brown B. W. Jr., Lamendola C., McLaughlin T., Reaven G. M. (2002) Relationship between obesity, insulin resistance, and coronary heart disease risk. J. Am. Coll. Cardiol. 40, 937–943 [DOI] [PubMed] [Google Scholar]
- 5. Hussain M. M., Shi J., Dreizen P. (2003) Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 44, 22–32 [DOI] [PubMed] [Google Scholar]
- 6. Burnett J. R., Watts G. F. (2007) MTP inhibition as a treatment for dyslipidaemias: time to deliver or empty promises? Expert Opin. Ther. Targets 11, 181–189 [DOI] [PubMed] [Google Scholar]
- 7. Hayhurst G. P., Lee Y. H., Lambert G., Ward J. M., Gonzalez F. J. (2001) Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21, 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuan X., Ta T. C., Lin M., Evans J. R., Dong Y., Bolotin E., Sherman M. A., Forman B. M., Sladek F. M. (2009) Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One 4, e5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiselyuk A., Lee S. H., Farber-Katz S., Zhang M., Athavankar S., Cohen T., Pinkerton A. B., Ye M., Bushway P., Richardson A. D., Hostetler H. A., Rodriguez-Lee M., Huang L., Spangler B., Smith L., Higginbotham J., Cashman J., Freeze H., Itkin-Ansari P., Dawson M. I., Schroeder F., Cang Y., Mercola M., Levine F. (2012) HNF4α antagonists discovered by a high-throughput screen for modulators of the human insulin promoter. Chem. Biol. 19, 806–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. López-Lázaro M. (2009) Distribution and biological activities of the flavonoid luteolin. Mini Rev. Med. Chem. 9, 31–59 [DOI] [PubMed] [Google Scholar]
- 11. Kritas S. K., Saggini A., Varvara G., Murmura G., Caraffa A., Antinolfi P., Toniato E., Pantalone A., Neri G., Frydas S., Rosati M., Tei M., Speziali A., Saggini R., Pandolfi F., Cerulli G., Theoharides T. C., Conti P. (2013) Luteolin inhibits mast cell-mediated allergic inflammation. J. Biol. Regul. Homeost. Agents 27, 955–959 [PubMed] [Google Scholar]
- 12. Hirokane H., Nakahara M., Tachibana S., Shimizu M., Sato R. (2004) Bile acid reduces the secretion of very low density lipoprotein by repressing microsomal triglyceride transfer protein gene expression mediated by hepatocyte nuclear factor-4. J. Biol. Chem. 279, 45685–45692 [DOI] [PubMed] [Google Scholar]
- 13. Misawa K., Horiba T., Arimura N., Hirano Y., Inoue J., Emoto N., Shimano H., Shimizu M., Sato R. (2003) Sterol regulatory element-binding protein-2 interacts with hepatocyte nuclear factor-4 to enhance sterol isomerase gene expression in hepatocytes. J. Biol. Chem. 278, 36176–36182 [DOI] [PubMed] [Google Scholar]
- 14. Sato R., Miyamoto W., Inoue J., Terada T., Imanaka T., Maeda M. (1999) Sterol regulatory element-binding protein negatively regulates microsomal triglyceride transfer protein gene transcription. J. Biol. Chem. 274, 24714–24720 [DOI] [PubMed] [Google Scholar]
- 15. Inoue J., Ito Y., Shimada S., Satoh S. I., Sasaki T., Hashidume T., Kamoshida Y., Shimizu M., Sato R. (2011) Glutamine stimulates the gene expression and processing of sterol regulatory element binding proteins, thereby increasing the expression of their target genes. FEBS J. 278, 2739–2750 [DOI] [PubMed] [Google Scholar]
- 16. Kawatani M., Okumura H., Honda K., Kanoh N., Muroi M., Dohmae N., Takami M., Kitagawa M., Futamura Y., Imoto M., Osada H. (2008) The identification of an osteoclastogenesis inhibitor through the inhibition of glyoxalase I. Proc. Natl. Acad. Sci. U.S.A. 105, 11691–11696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lubura M., Hesse D., Neumann N., Scherneck S., Wiedmer P., Schürmann A. (2012) Non-invasive quantification of white and brown adipose tissues and liver fat content by computed tomography in mice. PLoS One 7, e37026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones G., Willett P., Glen R. C. (1995) Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245, 43–53 [DOI] [PubMed] [Google Scholar]
- 19. Rha G. B., Wu G., Shoelson S. E., Chi Y. I. (2009) Multiple binding modes between HNF4α and the LXXLL motifs of PGC-1α lead to full activation. J. Biol. Chem. 284, 35165–35176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bailly A., Torres-Padilla M. E., Tinel A. P., Weiss M. C. (2001) An enhancer element 6 kb upstream of the mouse HNF4α1 promoter is activated by glucocorticoids and liver-enriched transcription factors. Nucleic Acids Res. 29, 3495–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perilhou A., Tourrel-Cuzin C., Zhang P., Kharroubi I., Wang H., Fauveau V., Scott D. K., Wollheim C. B., Vasseur-Cognet M. (2008) The MODY1 gene for hepatocyte nuclear factor 4α and a feedback loop control COUP-TFII expression in pancreatic β cells. Mol. Cell. Biol. 28, 4588–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jurasekova Z., Marconi G., Sanchez-Cortes S., Torreggiani A. (2009) Spectroscopic and molecular modeling studies on the binding of the flavonoid luteolin and human serum albumin. Biopolymers 91, 917–927 [DOI] [PubMed] [Google Scholar]
- 23. Borradaile N. M., de Dreu L. E., Wilcox L. J., Edwards J. Y., Huff M. W. (2002) Soya phytoestrogens, genistein and daidzein, decrease apolipoprotein B secretion from HepG2 cells through multiple mechanisms. Biochem. J. 366, 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilcox L. J., Borradaile N. M., de Dreu L. E., Huff M. W. (2001) Secretion of hepatocyte apoB is inhibited by the flavonoids, naringenin and hesperetin, via reduced activity and expression of ACAT2 and MTP. J. Lipid Res. 42, 725–734 [PubMed] [Google Scholar]
- 25. Benoist F., Grand-Perret T. (1997) Co-translational degradation of apolipoprotein B100 by the proteasome is prevented by microsomal triglyceride transfer protein: synchronized translation studies on HepG2 cells treated with an inhibitor of microsomal triglyceride transfer protein. J. Biol. Chem. 272, 20435–20442 [DOI] [PubMed] [Google Scholar]
- 26. Fisher E., Lake E., McLeod R. S. (2014) Apolipoprotein B100 quality control and the regulation of hepatic very low density lipoprotein secretion. J. Biomed. Res. 28, 178–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher E. A. (2012) The degradation of apolipoprotein B100: multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta 1821, 778–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu J., Wang H., Lu X., Ding K., Zhang L., He J., Wei W., Wu Y. (2014) Posttraumatic administration of luteolin protects mice from traumatic brain injury: implication of autophagy and inflammation. Brain Res. 1582, 237–246 [DOI] [PubMed] [Google Scholar]
- 29. Huang H. C., Syu K. Y., Lin J. K. (2010) Chemical composition of Solanum nigrum linn extract and induction of autophagy by leaf water extract and its major flavonoids in AU565 breast cancer cells. J. Agric. Food Chem. 58, 8699–8708 [DOI] [PubMed] [Google Scholar]
- 30. Park S. H., Park H. S., Lee J. H., Chi G. Y., Kim G. Y., Moon S. K., Chang Y. C., Hyun J. W., Kim W. J., Choi Y. H. (2013) Induction of endoplasmic reticulum stress-mediated apoptosis and non-canonical autophagy by luteolin in NCI-H460 lung carcinoma cells. Food Chem. Toxicol. 56, 100–109 [DOI] [PubMed] [Google Scholar]
- 31. Dang Z., Löwik C. W. (2004) The balance between concurrent activation of ERs and PPARs determines daidzein-induced osteogenesis and adipogenesis. J. Bone Miner. Res. 19, 853–861 [DOI] [PubMed] [Google Scholar]
- 32. Dang Z. C., Audinot V., Papapoulos S. E., Boutin J. A., Löwik C. W. (2003) Peroxisome proliferator-activated receptor γ (PPARγ) as a molecular target for the soy phytoestrogen genistein. J. Biol. Chem. 278, 962–967 [DOI] [PubMed] [Google Scholar]
- 33. Liang Y. C., Tsai S. H., Tsai D. C., Lin-Shiau S. Y., Lin J. K. (2001) Suppression of inducible cyclooxygenase and nitric oxide synthase through activation of peroxisome proliferator-activated receptor-γ by flavonoids in mouse macrophages. FEBS Lett. 496, 12–18 [DOI] [PubMed] [Google Scholar]
- 34. Ding L., Jin D., Chen X. (2010) Luteolin enhances insulin sensitivity via activation of PPARγ transcriptional activity in adipocytes. J. Nutr. Biochem. 21, 941–947 [DOI] [PubMed] [Google Scholar]
- 35. Puhl A. C., Bernardes A., Silveira R. L., Yuan J., Campos J. L., Saidemberg D. M., Palma M. S., Cvoro A., Ayers S. D., Webb P., Reinach P. S., Skaf M. S., Polikarpov I. (2012) Mode of peroxisome proliferator-activated receptor γ activation by luteolin. Mol. Pharmacol. 81, 788–799 [DOI] [PubMed] [Google Scholar]
- 36. Dhe-Paganon S., Duda K., Iwamoto M., Chi Y. I., Shoelson S. E. (2002) Crystal structure of the HNF4 α ligand binding domain in complex with endogenous fatty acid ligand. J. Biol. Chem. 277, 37973–37976 [DOI] [PubMed] [Google Scholar]
- 37. Shimoi K., Okada H., Furugori M., Goda T., Takase S., Suzuki M., Hara Y., Yamamoto H., Kinae N. (1998) Intestinal absorption of luteolin and luteolin 7-O-β-glucoside in rats and humans. FEBS Lett. 438, 220–224 [DOI] [PubMed] [Google Scholar]
- 38. Liu J. F., Ma Y., Wang Y., Du Z. Y., Shen J. K., Peng H. L. (2011) Reduction of lipid accumulation in HepG2 cells by luteolin is associated with activation of AMPK and mitigation of oxidative stress. Phytother. Res. 25, 588–596 [DOI] [PubMed] [Google Scholar]
- 39. Hong Y. H., Varanasi U. S., Yang W., Leff T. (2003) AMP-activated protein kinase regulates HNF4α transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem. 278, 27495–27501 [DOI] [PubMed] [Google Scholar]
- 40. Zhou P., Li L. P., Luo S. Q., Jiang H. D., Zeng S. (2008) Intestinal absorption of luteolin from peanut hull extract is more efficient than that from individual pure luteolin. J. Agric. Food Chem. 56, 296–300 [DOI] [PubMed] [Google Scholar]
- 41. Hata T., Mera Y., Tadaki H., Kuroki Y., Kawai T., Ohta T., Kakutani M. (2011) JTT-130, a novel intestine-specific inhibitor of microsomal triglyceride transfer protein, suppresses high fat diet-induced obesity and glucose intolerance in Sprague-Dawley rats. Diabetes Obes. Metab. 13, 446–454 [DOI] [PubMed] [Google Scholar]
- 42. Kwon E. Y., Jung U. J., Park T., Yun J. W., Choi M. S. (2015) Luteolin attenuates hepatic steatosis and insulin resistance through the interplay between the liver and adipose tissue in mice with diet-induced obesity. Diabetes 64, 1658–1669 [DOI] [PubMed] [Google Scholar]
- 43. Ryffel G. U. (2001) Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J. Mol. Endocrinol. 27, 11–29 [DOI] [PubMed] [Google Scholar]
- 44. Gupta R. K., Vatamaniuk M. Z., Lee C. S., Flaschen R. C., Fulmer J. T., Matschinsky F. M., Duncan S. A., Kaestner K. H. (2005) The MODY1 gene HNF-4α regulates selected genes involved in insulin secretion. J. Clin. Invest. 115, 1006–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee S. H., Piran R., Keinan E., Pinkerton A., Levine F. (2013) Induction of β-cell replication by a synthetic HNF4α antagonist. Stem Cells 31, 2396–2407 [DOI] [PubMed] [Google Scholar]
- 46. Guilherme A., Virbasius J. V., Puri V., Czech M. P. (2008) Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]