

Background: Myosin II heavy chain kinase A (MHCK-A) is a member of the atypical α-kinase family.
Results: The catalytic and nucleotide binding properties of the MHCK-A kinase domain are characterized.
Conclusion: α-Kinases convert ATP to adenosine via formation of an aspartyl phosphate intermediate.
Significance: The α-kinase active site has unique functions that may aid in the design of specific inhibitors.
Keywords: adenosine, catalysis, enzyme kinetics, protein kinase, x-ray crystallography, α-kinase, aspartyl phosphate, atypical protein kinase, myosin-II heavy chain kinase
Abstract
The α-kinases are a widely expressed family of serine/threonine protein kinases that exhibit no sequence identity with conventional eukaryotic protein kinases. In this report, we provide new information on the catalytic properties of the α-kinase domain of Dictyostelium myosin-II heavy chain kinase-A (termed A-CAT). Crystallization of A-CAT in the presence of MgATP yielded structures with AMP or adenosine in the catalytic cleft together with a phosphorylated Asp-766 residue. The results show that the β- and α-phosphoryl groups are transferred either directly or indirectly to the catalytically essential Asp-766. Biochemical assays confirmed that A-CAT hydrolyzed ATP, ADP, and AMP with kcat values of 1.9, 0.6, and 0.32 min−1, respectively, and showed that A-CAT can use ADP to phosphorylate peptides and proteins. Binding assays using fluorescent 2′/3′-O-(N-methylanthraniloyl) analogs of ATP and ADP yielded Kd values for ATP, ADP, AMP, and adenosine of 20 ± 3, 60 ± 20, 160 ± 60, and 45 ± 15 μm, respectively. Site-directed mutagenesis showed that Glu-713, Leu-716, and Lys-645, all of which interact with the adenine base, were critical for nucleotide binding. Mutation of the highly conserved Gln-758, which chelates a nucleotide-associated Mg2+ ion, eliminated catalytic activity, whereas loss of the highly conserved Lys-722 and Arg-592 decreased kcat values for kinase and ATPase activities by 3–6-fold. Mutation of Asp-663 impaired kinase activity to a much greater extent than ATPase, indicating a specific role in peptide substrate binding, whereas mutation of Gln-768 doubled ATPase activity, suggesting that it may act to exclude water from the active site.
Introduction
Protein kinases that phosphorylate serine, threonine, or tyrosine residues regulate a myriad of cellular signaling events in eukaryotic cells and, as such, represent important therapeutic targets for a variety of diseases and disorders. Most protein kinases belong to the eukaryotic protein kinase (ePK)4 superfamily and contain a highly conserved and extensively characterized catalytic domain (1–4). A smaller number of protein kinases, often termed “atypical” protein kinases, have catalytic domains that exhibit no detectable sequence similarity to the ePKs. One widespread family of atypical protein kinases, present in organisms ranging from single-celled protozoa to mammals, is named the α-kinases (5, 6). The α-kinases phosphorylate serine and threonine residues, with some members displaying a high degree of specificity for threonine (7, 8). Well studied α-kinases include Dictyostelium myosin-II heavy chain kinase-A (MHCK-A), which phosphorylates the tail of myosin-II to control the timing and spatial location of myosin-II bipolar filament assembly (9–12), and the eukaryotic elongation factor 2 kinase, which phosphorylates eEF2 to inhibit ribosomal protein translation elongation and control cell growth (13–15). The transient receptor potential melastatin-like 7 (TRPM7) ion channel contains a C-terminal α-kinase domain that, like MHCK-A, regulates myosin-II filament assembly (16, 17). Proteolytic cleavage of the α-kinase domain from TRPM7 allows the kinase domain to translocate to the nucleus, phosphorylate histones, and regulates gene expression (18).
X-ray crystal structures for the α-kinase domains of TRPM7 and MHCK-A have been solved (19, 20). The α-kinase domain, like the ePK domain, is bi-lobed with the active site located in a wide cleft at the interface between the two lobes. The α-kinase N-lobe and the ePK N-lobe have a similar architecture composed primarily of β strands, whereas the two kinase families have distinct C-lobes. One unusual feature of the α-kinase C-lobe is that it contains a tightly bound zinc atom that is essential for stability. Despite these differences, the α-kinase and ePK active sites share many features in common and contain a fairly well conserved set of catalytic residues. Thus, the α-kinases can be considered divergent members of the protein kinase-like (PKL) superfamily (1, 3).
The roof of the α-kinase catalytic cleft is formed by the β5, β6, and β7 strands in the N-lobe. The β5 and β6 strands are connected by the P-loop, which helps to bind and orient the nucleotide β- and/or γ-phosphoryl groups. In contrast to the P-loop in ePKs, the α-kinase P-loop is not glycine-rich and contains an invariant basic residue (Arg-592; all numbering is for MHCK-A) that makes electrostatic contacts with the nucleotide phosphoryl groups. The β7 strand contains a second invariant basic residue (Lys-645) that forms a salt bridge with an invariant glutamic acid residue (Glu-713) in the hinge loop connecting the N- and C-lobes. This interaction positions the Lys-645 side chain between the adenine base and the α-phosphoryl group. Lys-645 is the counterpart of the essential lysine residue in the β3 strand of ePKs, which is Lys-72 in the cAMP-dependent protein kinase (PKA) (4, 21). In active ePKs, the side chain of Lys-72 forms a salt bridge with an invariant glutamic acid residue (Glu-91) in the αC-helix and is positioned between the α- and β-phosphoryl groups. The αC-helix represents another major point of difference between the α-kinases and ePKs. In the α-kinases the αC-helix runs obliquely from the top of the N-lobe to the bottom of the C-lobe, and thus it functions as a supporting scaffold that limits the ability of the two lobes to move relative to one another.
The bottom of the α-kinase catalytic cleft is formed by residues in the C-lobe. An extended sequence that runs along the outer edge of the catalytic cleft contains a highly conserved lysine residue (Lys-722), and inside of this is a catalytic loop containing three invariant residues (Asp-756, Gln-758, and Asp-766). The functions of Asp-756, Lys-722, and Gln-758 have not been examined in detail, but they take up positions similar to the conserved Asp-166, Lys-168, and Asn-171 residues in the PKA catalytic loop. Asp-766 is essential for catalysis and, unexpectedly, is phosphorylated in some crystal structures of the α-kinase domain of MHCK-A (termed A-CAT) (19). A structure of A-CAT crystallized in the presence of MgATP contains ADP and a phosphorylated Asp-766 (Asp(P)-766), showing that the γ-phosphate can be transferred to Asp-766. The-Asp(P)-766 phosphoryl group is directed away from the nucleotide, which binds into the left-hand side of the catalytic cleft, toward the right-hand side of the cleft where it is exposed to solvent. The right-hand side of the catalytic cleft is adjacent to a structure unique to the α-kinases, termed the N/D-loop, that contains an invariant asparagine/aspartate residue of unknown function. The N/D-loop is involved in binding the protein substrate and is proposed to undergo a conformational change that regulates access of the protein substrate to the active site (19, 22). Thus, Asp-766 may function as an intermediate to transfer phosphoryl groups from the nucleotide to the protein substrate.
The counterpart to Asp-766 in PKA is Asp-184. Asp-184 is part of the highly conserved DFG motif and functions to chelate an essential Mg2+ ion that bridges the β- and γ-phosphoryl groups of ATP (23). It is well established that PKA catalyzes a direct in-line transfer of the γ-phosphate to the protein substrate hydroxyl group without forming a phosphoenzyme intermediate (23, 24). Interestingly, however, the residue equivalent to Asp-184/Asp-766 is phosphorylated in crystal structures of two eukaryotic RIO kinases (25, 26). The RIO kinases are an ancient atypical protein kinase family present in prokaryotes and eukaryotes that play a role in small ribosomal subunit biogenesis. It is thought that the primary purpose of aspartyl phosphate formation in the RIO kinases is to drive a conformational change in the kinase domain, but a role in protein substrate phosphorylation cannot be excluded (27). These results highlight the possibility that despite sharing similar active sites, the functions of conserved catalytic residues in some atypical protein kinases may diverge from those of their well studied counterparts in the ePKs.
In this study, we present structural and biochemical data that show that A-CAT can hydrolyze ATP to adenosine and that Asp-766 sequentially accepts the γ-, β-, and α-phosphoryl groups from the nucleotide. Evidence is also presented that A-CAT can use ADP to phosphorylate protein and peptide substrates. The nucleotide binding properties of A-CAT have been characterized using fluorescent nucleotide analogs, and site-directed mutagenesis is used to define the roles played by several conserved and nonconserved active site residues in nucleotide binding, ATPase activity, and kinase activity.
Experimental Procedures
Chemicals and Reagents
ATP, ADP, AMP, GTP, UTP, ITP, 2′-deoxy-ATP, and 3′-deoxy-ATP were obtained from Sigma. 2′,3′-O-(N-Methylanthraniloyl)-ADP (mant-ADP), mant-ATP, and ATPγS were obtained from Jena Bioscience. Radioactively labeled [γ-32P]ATP and [35S]ATPγS were obtained from PerkinElmer Life Sciences.
Plasmid Constructs, Protein Expression, and Purification
A-CAT, comprising residues 552–841 of MHCK-A, was cloned into the pET-28a vector (Novagen) as reported previously(28). Site-directed mutagenesis was performed using the QuikChange II XL kit (Stratagene). Wild-type and mutant forms of A-CAT, containing an N-terminal His tag and tobacco etch virus protease site, were expressed in Escherichia coli BL21(DE3) cells and purified at 4 °C using His-Bind resin (Novagen) and DE53 and SP-Sepharose Fast Flow columns (GE Biosciences) (19). Yields from a 4-liter bacterial culture ranged from 0.4 mg for the A-CAT-E713A mutant to 5 mg for the A-CAT-D663A mutant. For crystallization, the His tag was cleaved off by overnight incubation at 4 °C with His-tagged AcTEV protease (Life Technologies, Inc.). The protease was removed by chromatography over a His-Bind column, and A-CAT was concentrated to 0.25 mm using a 10,000 molecular weight cutoff Ultrafree-4 centrifugal filtration unit (Millipore).
Crystallization Procedures and Structural Determination
A-CAT was crystallized at 4 °C using the hanging-drop vapor diffusion method. Diffraction-quality crystals of A-CAT in a complex with AMP (A-CAT·AMP·P) were obtained by mixing 0.25 mm A-CAT with 2 mm MgCl2, 2 mm ATP, and 1.25 mm of the 9-mer peptide substrate AAYKTKKKK (8). Crystallization drops were then set up by mixing 1 μl of A-CAT with 1 μl of mother liquor solution (200 mm NaH2PO4, 20% (w/v) polyethylene glycol 8000, and 100 mm Tris-HCl, pH 7.5). Diffraction quality crystals of A-CAT in a complex with adenosine (A-CAT·ADN·P) were obtained by mixing 0.25 mm A-CAT with 2 mm MgCl2, 2 mm ATP, and 2 mm of the 16-mer peptide substrate MH-1 (RKKFGESEKTKTKEFL) (28). Crystallization drops were then set up by mixing 1 μl of A-CAT with 1 μl of mother liquor solution (200 mm NaH2PO4, 18–20% (w/v) polyethylene glycol 8000, and 100 mm sodium cacodylate, pH 7.4). Crystals were frozen in liquid nitrogen after being dipped into buffer solution containing 25% (v/v) ethylene glycol as cryoprotectant. Diffraction data were collected on the F1 beamline at the Cornell High Energy Synchrotron Source (Cornell, New York). Data were integrated and merged using the HKL2000 suite program (29). The structures were solved by the molecular replacement method using the known structure of A-CAT (Protein Data Bank code 3LKM) devoid of nucleotide, Zn2+, Mg2+, and H2PO4− ions and the AutoMR program from the PHASER crystallography software suite (30). Manual model rebuilding was performed using Coot (31), and refinement was carried out using CNS, PHENIX, and REFMAC5 (32–34). After refinement, the quality of the model was checked using PROCHECK (35). The electron density for A-CAT·ADN·P chains A and B was clear and continuous except for the absence of Ala-651 and residues C-terminal to Gly-810 (chain A) or Gly-822 (chain B). For A-CAT·AMP·P electron density was lacking for residues 652–654 and 653 in chains A and B, respectively, for residues 702–703 (chain B) and for residues C-terminal to Lys-808. No electron density was visible for the substrate included in the crystallization buffer. Graphics were produced using The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC.
Nucleotide Binding Assays
Assays were carried out in 50 mm NaCl, 1 mm dithiothreitol, 20 mm Tris, pH 7.0, with or without 2 mm MgCl2. Each binding experiment involved addition of 24 different concentrations of mant-ADP or mant-ATP to sample wells on a 96-well plate in the presence or absence of 5 μm A-CAT. The final assay volume was 100 μl. Fluorescence spectra were recorded using a SpectraMax Gemini XS fluorescence plate reader (Molecular Devices) with excitation and emission slits set to 2 and 5 nm, respectively. Mant-nucleotide concentration was determined using a 340-nm excitation wavelength and measuring emission at 431 nm with a 530-nm cutoff filter. Fluorescence resonance energy transfer (FRET) from A-CAT to the mant-nucleotide was measured using a 280-nm excitation wavelength and a 431-nm emission wavelength. Corrections were made to compensate for the inner filter effect due to mant-nucleotide absorbance. The increase in fluorescence (ΔF) in the presence of A-CAT was used to calculate the dissociation constant (Kd) for the mant-nucleotide using Equation 1,
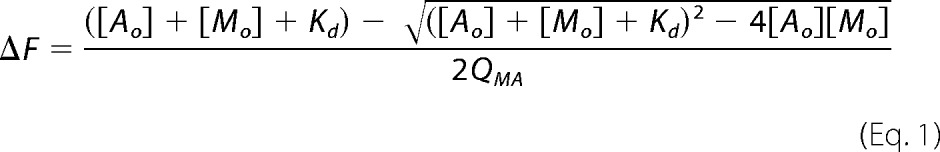 |
where [Ao] and [Mo] are the initial concentrations of A-CAT and mant-nucleotide, respectively, and QMA is a multiplicative constant that reflects both the photophysical properties of the probe and the sensitivity of the fluorescence reader. Values for Kd and QMA were obtained by fitting the ΔF versus [Mo] curve using Excel software (Microsoft). To limit inaccuracies caused by corrections for the inner filter effect, the maximum mant-ATP concentration that was used in the assays was 150 μm. Therefore, Kd values could not be accurately estimated for A-CAT mutants that were less than 60% saturated at 150 μm mant-ATP. Competition experiments were performed by titrating mant-ADP into solutions containing a fixed concentration of nucleotide. Kd values were calculated using Equation 2,
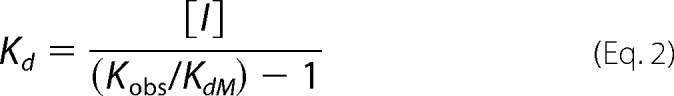 |
where [I] is the concentration of the competing nucleotide, and Kd, M and Kobs are the dissociation constants for mant-ADP observed in the absence and presence of the nucleotide. Each titration was repeated three times using three different concentrations of the competing nucleotide.
Kinase and Hydrolytic Assays
Kinase and hydrolytic assays were carried out at 22 °C in a buffer containing 2 mm MgCl2, 1 mm dithiothreitol, and 20 mm TES, pH 7.0. Kinase assays contained 20 μm myelin basic protein (MBP), 0.5 μm A-CAT, and except where indicated, 200 μm [γ-32P]ATP (specific activity of 100 cpm/pmol). Incorporation of 32P into MBP was quantified by spotting 20-μl aliquots of the reaction mixture onto squares of Whatman P81 phosphocellulose paper. The paper squares were washed in 1% phosphoric acid, immersed in Scintiverse Universal LS Mixture (Fisher Scientific) and counted using a Beckman LS 9000 scintillation counter. Assays were also carried out in which ADP was used as the substrate. The assays contained 500 μm ADP, 1 μm A-CAT or kinase-dead A-CAT-D766S, and either 250 μm YAYDTRYRR peptide or 12 μm MBP that had been dephosphorylated prior to use by treatment with alkaline phosphatase-agarose (P0762, Sigma Aldrich). Assays containing the YAYDTRYRR peptide were allowed to proceed for 12 h and were analyzed by electrospray ionization (ESI)-MS/MS using a Thermo Scientific Orbitrap Velos Pro mass spectrometer. All identified peaks had a mass error less than 5 ppm (where ppm = (Mcal − Mexp)/(Mcal) × 10−6). Assays containing MBP were stopped by addition of SDS sample loading buffer and boiled for 5 min. Samples were analyzed by electrophoresis on 10% SDS-polyacrylamide gels polymerized in the presence or absence of 50 μm Phos-tag acrylamide (Phos-tag Consortium, Hiroshima, Japan) and in the presence of 0.1 mm MnCl2. Nucleotide hydrolysis assays contained 10–1200 μm nucleotide, with routine measurements of ATPase activity carried out using 250 μm ATP. Release of inorganic phosphate (Pi) was measured using the PiColorLock Gold phosphate detection system (Innova Biosciences). Data from the initial linear portion of the time courses were used to calculate kinase and hydrolytic activity. Results represent the mean and standard deviation of at least three separate experiments.
Results
Crystal Structures of A-CAT Bound to AMP and Adenosine
A-CAT consists of the core α-kinase domain of MHCK-A plus a short C-terminal extension that harbors an autophosphorylation site (Thr-825) required for activity (28, 36). Crystallization of A-CAT in the presence of MgATP has yielded structures with ADP (A-CAT·ADP·P) and AMP (A-CAT·AMP) bound to the inter-lobe cleft (Table 1) (19). Asp-766 is phosphorylated in the A-CAT·ADP·P structure but not the A-CAT·AMP structure. Here, we report crystal structures for A-CAT bound to AMP (A-CAT·AMP·P) and to adenosine (A-CAT·ADN·P), both obtained by crystallization from a buffer containing MgATP (Fig. 1A). The A-CAT·AMP·P and A-CAT·ADN·P structures were solved to a resolution of 2.4 and 2.0 Å, respectively. The diffraction data statistics for the two structures are summarized in Table 2. The asymmetric unit contained two molecules of A-CAT·AMP·P or A-CAT·ADN·P, arranged in a back-to-back manner as described for the A-CAT·ADP·P structure (19). In both the A-CAT·AMP·P and A-CAT·ADN·P structures, there was extra electron density associated with the side chain of Asp-766 (Fig. 1, B and C). The density allowed a phosphoryl group to be unambiguously placed in a covalent bond with Asp-766. Taken together, the results demonstrate that A-CAT has the ability to sequentially remove the three phosphoryl groups from ATP to generate adenosine. Moreover, it would appear that each phosphoryl group is transferred from the nucleotide to generate a phosphorylated Asp-766 (Asp(P)-766) residue.
TABLE 1.
Summary of A-CAT crystal structure features
| A-CATa | PDB code | Name | Pep.b | Catalytic cleftc |
Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Nucleotide | P-D766 | Pi1 | Mg2+ | |||||
| WT | 3LMH | A-CAT·ADP·P | P1 | ADP | + | − | − | 19 |
| WT | 3LKM | A-CAT·AMP | AMP | − | + | Mg1,2 | 19 | |
| WT | 3LLA | A-CAT-AMPPCP | AMPPCP | − | − | − | 19 | |
| D766A | 3LMI | A-CAT-D766A | ATP | − | − | Mg1 | 19 | |
| Δ809 | 3PDT | A-CAT-Δ809 | ADP | − | − | Mg2 | 36 | |
| WT | 4ZME | A-CAT·ADN·P | MH1 | Adenosine | + | + | − | TWd |
| WT | 4ZMF | A-CAT·AMP·P | P1 | AMP | + | − | − | TW |
a Construct was crystallized; Δ809 indicates truncated at residue 809.
b Pep. indicates peptide included in crystallization buffer as follows: P1, AAYKTKKKK; MH1, RKKFGESEKTKTKEFL.
c Nucl. indicates identity of bound nucleotide; P-D766 indicates presence (+) or absence (−) of phosphorylated Asp-766; Pi1 indicates presence (+) or absence (−) of Pi bound to P-loop.
d TW indicates this work.
FIGURE 1.

Structure of A-CAT bound to adenosine or AMP. A, schematic representation of A-CAT bound to adenosine (A-CAT·ADN·P) with the N-terminal lobe (residues 552–655) colored salmon, the central core (residues 656–712) colored cyan, and the C-terminal lobe and tail (residues 713–842) colored blue. The N and C termini, β-sheets, and α-helices are labeled. Phosphate (Pi1), Asp(P)-766 (P-D766), and adenosine (ADN) are shown as sticks with the adenosine carbon atoms colored yellow. The zinc atom is shown as a magenta sphere. B and C, close-up views showing the catalytic cleft of A-CAT·ADN·P and A-CAT·AMP·P. Adenosine, AMP, Pi1, and Asp(P)-766 are shown as sticks. The mesh shows the 2Fo − Fc electron density map contoured at the 2σ level. D, alignment of the A-CAT·ADN·P (gray), A-CAT·AMP·P (salmon), and A-CAT·ADP·P (blue) structures shows that there is little or no change in the positions of the P-loop, N/D-loop, or key catalytic residues. E, view of the A-CAT·ADN·P active site showing the interactions (dashed lines) made by Asp(P)-766 and Pi1.
TABLE 2.
Data collection and refinement statistics for the A-CAT structures
| Data set | A-CAT·AMP·P | A-CAT·ADN·P |
|---|---|---|
| Data collection | ||
| Space group | P21212 | P21212 |
| Molecules per asymmetric unit | 2 | 2 |
| Unit cell parameters (Å) | a = 83.1 | a = 83.19 |
| b = 109.96 | b = 111.29 | |
| c = 79.49 | c = 79.56 | |
| Resolution limits (Å) | 30.0–2.40 (2.49–2.0) | 30.0–2.0 (2.07–2.0) |
| Unique reflections | 29,130 | 51,052 |
| Multiplicity | 9.5 (8.1) | (6.9 (4.7) |
| Mean 〈I/σ〉 (I) > | 27.04 (4.66) | 6.93 (6.45) |
| Completeness (%) | 99.8 (100) | 99.5 (99.9) |
| Rmerge (%)a | 0.10 (0.45) | 0.09 (0.44) |
| Refinement | ||
| Resolution range (Å) | 79.06–2.39 | 28.75–1.98 |
| No. of reflections in the working set | 27,601 | 48,960 |
| Rwork/Rfreeb | 0.20/0.22 | 0.21/0.23 |
| R.M.S.D. bonds (Å) | 0.008 | 0.008 |
| Root mean square deviation angle (°) | 1.51 | 1.27 |
| Average B factor (Å2) | 42.51 | 37.37 |
| Nucleotide | AMP | Adenosine |
| Auxiliary molecules | 2 aspartyl phosphate | 2 aspartyl phosphate |
| No. of refined atoms | ||
| Protein | 4034 | 4193 |
| Solvent | 194 | 463 |
| Ions | 2 H2PO4−, 2 Zn2+ | 4 H2PO4−, 2 Zn2+ |
| Ramachandran plot | ||
| Allowed (%) | 100.0 | 100.0 |
| Outlier (%) | 0.0 | 0.0 |
a Rmerge = |Iobs − 〈I〉|/Iobs, where Iobs is the intensity measurement and 〈I〉 is the mean intensity for multiply recorded reflections.
b Rwork and Rfree = |Fobs − Fcalc|/|Fobs| for reflections in the working and test sets, respectively. Rfree is the cross-validation R factor calculated for the test set (5%) of reflections omitted in model refinement.
Alignments of the A-CAT·ADN·P, A-CAT·AMP·P, and A-CAT·ADP·P structures showed that they are nearly identical, with root mean square deviations ranging from 0.24 to 0.34 Å for the Cα atoms. Notably, the positions of the P-loop and key active site residues, including Asp(P)-766, are essentially the same in all three structures (Fig. 1D). Thus, the nucleotide α- and β-phosphoryl groups do not play an important role in organizing the active site. The Asp(P)-766 side chain is directed away from the nucleotide toward the N/D-loop. The side chain adopts alternate configurations in the two A-CAT·ADN·P and A-CAT·AMP·P chains; in one chain, the unphosphorylated Oδ2 atom points in toward the αC helix, and in the other chain, it points outs toward the solvent where it forms a hydrogen bond with the Gln-758 side chain (Fig. 1, D and E). In both configurations, the Asp(P)-766 phosphoryl group interacts with the side chains of Arg-592, Asp-663, Asp-756, and Gln-768. Between 1 and 6 water molecules are within 4 Å of the phosphoryl group in the A-CAT·ADN·P and A-CAT·AMP·P structures.
A free Pi molecule (Pi1) is bound to the P-loop in the A-CAT·ADN·P structure but not in the A-CAT·AMP·P structure (Fig. 1, B and C). Pi1 forms hydrogen bonds with the main chain amides of Ala-590, Leu-591, and Arg-592 and makes an electrostatic interaction with the side chain of Arg-592 (Fig. 1E). A free Pi molecule bound to the P-loop was previously detected in the A-CAT·AMP structure (Table 1). When the catalytic cleft contains ADP, the P-loop binds the β-phosphoryl group (Fig. 1D) (19, 20).
Catalytic Activity of A-CAT
A-CAT has been shown to convert ATP to AMP, but its ability to generate adenosine has not been described previously (19). The rates at which A-CAT catalyzes hydrolysis of ATP, ADP, and AMP were measured using an assay that detects formation of Pi. A-CAT hydrolyzed ATP with a kcat of 1.92 min−1, which is similar to the value previously determined by following release of 32Pi from [γ32P]ATP (Fig. 2 and Table 3) (36). ADP and AMP were hydrolyzed at rates 3- and 6-fold slower than ATP, respectively (Fig. 2 and Table 3). The assays yielded Km values for ATP, ADP, and AMP of 35 ± 1, 71 ± 23, and 270 ± 90 μm, respectively.
FIGURE 2.

A-CAT can hydrolyze ATP, ADP, and AMP and is able to use ADP to phosphorylate peptide and protein substrates. A, hydrolysis of ATP (▴), ADP (○), and AMP (●) by A-CAT. The initial rate of Pi generation at each nucleotide concentration was measured as described under “Experimental Procedures.” Kinetic constants, obtained by fitting the data to a hyperbolic curve, are listed in Table 3. B, Phos-tag gel analysis of MBP phosphorylation. Kinase assays were carried out using A-CAT and MBP in the absence of nucleotide or with 0.5 mm ATP or 0.5 mm ADP as described under “Experimental Procedures.” Samples were taken at the indicated times (ON = 12 h) and electrophoresed on SDS gels polymerized in the presence (+) or absence (−) of the Phos-tag molecule. The gels were stained with Coomassie Blue to visualize proteins. Phosphorylated forms of MBP (P-MBP) electrophorese with a reduced mobility in the presence of the Phos-tag molecule. C and D, mass spectrometry analysis of the YAYDTRYRR peptide incubated with ADP and kinase-dead A-CAT-D766S (C) or A-CAT (D). The full MS spectra of [M + 2H]2+ in the positive mode is shown. A peak corresponding to the unphosphorylated peptide (m/z = 652.83) was detected in the sample incubated with A-CAT-D766S. An abundant peak corresponding to the phosphorylated peptide (m/z = 692.81) was detected in the sample incubated with A-CAT. Kinase assays were performed as described under “Experimental Procedures.”
TABLE 3.
Hydrolytic and kinase activities for wild-type and mutant A-CAT
| A-CAT | Nucleotide | Hydrolysis activity |
Kinase activity |
||
|---|---|---|---|---|---|
| kcat | Km, ATP | kcat | Km, ATP | ||
| min−1 | μm | min−1 | μm | ||
| WT | ATP | 1.92 ± 0.09 | 35 ± 4 | 5.8 ± 0.4 | 23 ± 5 |
| WT | ADP | 0.61 ± 0.05 | 71 ± 23 | ||
| WT | AMP | 0.32 ± 0.04 | 270 ± 90 | ||
| R592L | ATP | 0.54 ± 0.03 | 48 ± 7 | 1.8 ± 0.2 | 58 ± 18 |
| K722N | ATP | 0.32 ± 0.04 | 70 ± 10 | 1.0 ± 0.2 | 63 ± 27 |
The ability of A-CAT to use ADP to phosphorylate protein and peptide substrates was examined. In one experiment, A-CAT was incubated with MBP in the absence of nucleotide or in the presence of 0.5 mm ATP or 0.5 mm ADP. The samples were then electrophoresed on an SDS gel containing a polyacrylamide-bound Mn2+- Phos-tag molecule that causes phosphorylated proteins to migrate with a reduced mobility (37). Lower mobility, phosphorylated forms of MBP were detected in the samples incubated in the presence of ATP or ADP but not in samples without nucleotide (Fig. 2A). MBP phosphorylation did, however, appear to be slower and less extensive when ADP was used as the substrate. In a second experiment, the YAYDTRYRR peptide was incubated with A-CAT or a kinase-dead A-CAT-D766S mutant in the presence of ADP. Mass spectrometry was used to detect the incorporation of a phosphoryl group into the peptide. The control sample incubated with A-CAT-D766S contained only a peak corresponding to the unphosphorylated peptide (Fig. 2C). In contrast, an abundant peak representing the phosphorylated peptide was detected in the sample incubated with ADP and A-CAT (Fig. 2D). The results show that A-CAT can utilize ADP to phosphorylate both peptides and proteins.
Nucleotide Binding Properties of A-CAT
The nucleotide binding properties of A-CAT were investigated using derivatives of ATP and ADP that have a fluorescent methylanthraniloyl group coupled to the ribose 2′- or 3′-hydroxyl group (mant-ATP and mant-ADP) (38). Binding of mant-ATP and mant-ADP to A-CAT increased the fluorescence emission at 430 nm by 4-fold when the excitation wavelength was 280 nm (Fig. 3A). This increase can be attributed to fluorescence resonance energy transfer (FRET) from aromatic residues in A-CAT to the mant group. Titration curves based on the increase in fluorescence intensity at 430 nm yielded Kd values for mant-ATP of 4.8 and 14 μm and for mant-ADP of 15 and 70 μm in the presence and absence of 2 mm MgCl2, respectively (Fig. 3, B and C and Table 4).
FIGURE 3.
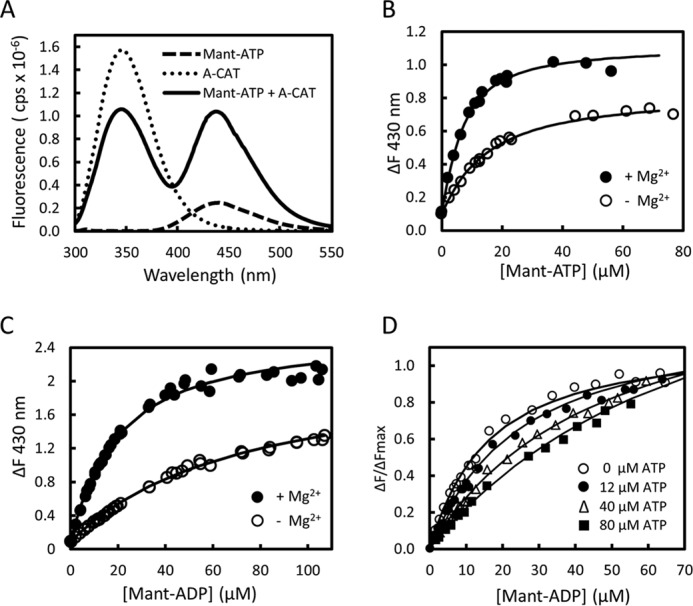
Binding of mant-ADP and mant-ATP to A-CAT. A, fluorescence emission spectra obtained upon excitation at 280 nm were recorded for 28 μm mant-ATP (dashed line), 2 μm A-CAT (dotted line), and a mixture of 28 μm mant-ATP and 2 μm A-CAT (black line). Assays contained 2 mm MgCl2 and were carried out as described under “Experimental Procedures.” In the presence of A-CAT, there is a large increase in the fluorescence emission of mant-ATP in the 400–500 nm region. B and C, change in fluorescence emission at 430 nm (ΔF) was measured as mant-ATP (B) or mant-ADP (C) and titrated into a solution containing 2 μm A-CAT in the presence (●) or absence (○) of 2 mm MgCl2. The lines show the best fit of the data to a hyperbolic binding curve. D, change in fluorescence emission at 430 nm (ΔF) was measured as mant-ADP was titrated into solutions containing 2 μm A-CAT and 2 mm MgCl2 in the presence of 9, 12, 40, and 80 μm ATP. Decreased ΔF values reflect competition between ATP and mant-ADP for binding to A-CAT. The binding constants determined by fitting the data in B–D are listed in Table 4.
TABLE 4.
Binding affinity of A-CAT for nucleotides and nucleosides
| Nucleotide | Kd + Mg2+ | Kd − Mg2+ |
|---|---|---|
| μm | μm | |
| Mant-ATP | 4.8 ± 0.6 | 14 ± 4 |
| Mant-ADP | 15 ± 2 | 70 ± 10 |
| ATP | 20 ± 3 | 160 ± 30 |
| ADP | 60 ± 20 | 150 ± 20 |
| AMP | 160 ± 60 | 110 ± 30 |
| Adenosine | 45 ± 15 | 50 ± 15 |
| cAMP | 50 ± 20 | |
| AMPPNP | 21 ± 8 | |
| ATPγS | 41 ± 8 | |
| ITP | 420 ± 170 | |
| GTP | 170 ± 50 | |
| d3′ATP | 42 ± 12 |
Competition binding assays with mant-ADP were used to measure the binding affinity of A-CAT for other nucleotides. The assays yielded a Kd value for ATP of 20 ± 3 μm in the presence of 2 mm MgCl2 (Fig. 3D and Table 4). ADP and AMP bound 3- and 8-fold less tightly than ATP, respectively (Table 4). The Kd values measured for the nucleotides are somewhat lower than the Km values determined in the hydrolysis assays but follow a very similar trend.
A-CAT bound adenosine with a Kd value of 45 ± 15 μm (Table 4). The relatively tight binding of adenosine shows that the phosphoryl groups play a relatively minor role in the binding interaction. Indeed, the α-phosphoryl group of AMP represents a strong negative binding determinant. A-CAT bound cAMP with a Kd value similar to adenosine, showing that cyclization eliminates the negative interactions of the α-phosphate (Table 4). AMPPNP, which has an imido group in place of the β-γ bridging oxygen, bound with an affinity comparable with ATP. ATPγS, which has a sulfur atom in place of one of the γ-phosphate oxygen atoms, bound about 2-fold less tightly than ATP. Nevertheless, kinase assays carried out using ATPγ35S demonstrated that A-CAT cannot use ATPγS as a substrate to phosphorylate proteins.
Residues Involved in Nucleotide Binding and Catalytic Activity
Several residues in the active site of A-CAT were mutated to investigate their roles in nucleotide binding, ATPase, and kinase activities. Mant-ATP binding was most strongly impaired by mutation of two residues (Glu-713 and Lys-645) that directly interact with the adenine base (Fig. 4 and Table 5). Circular dichroism spectrum showed that the E713A and K645R mutants were properly folded but had a 5–7% lower α-helical content, suggesting that these mutations may have both direct and indirect effects on nucleotide binding. The binding of mant-ATP to these mutants was too weak to be accurately measured but was estimated to be greater than 300 μm. The results indicate that the interaction between the adenine N6 amino group and Glu-713 plays a key role in nucleotide recognition by A-CAT. This interaction was further examined by using ITP and GTP in competition binding assays. ITP and GTP bound 10–20-fold less tightly than ATP, demonstrating that the N6 amino group is critical for binding to A-CAT (Table 4). As expected, the Glu-713 and Lys-645 mutants exhibited very low kinase and ATPase activities (Fig. 5A).
FIGURE 4.
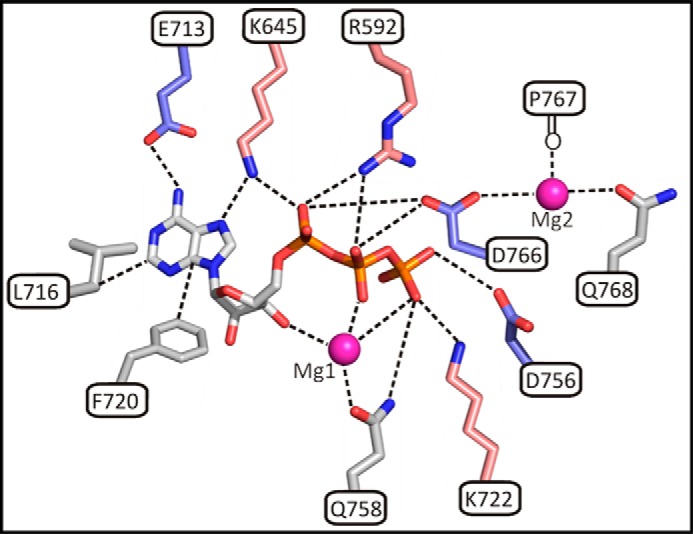
Schematic diagram showing A-CAT interactions with ATP and Mg2+ ions. Residues in A-CAT that interact with ATP, Mg1, and Mg2 (magenta spheres) and that were examined in this study by site-directed mutagenesis are shown as sticks. Acidic residues are colored blue, and basic residues salmon and other residues are gray.
TABLE 5.
Effect of mutations on the binding affinity of A-CAT for mant-ATP
| Mutation | Kd |
|---|---|
| μm | |
| WT | 4.8 ± 0.6 |
| R592L | 15 ± 2 |
| K645R | >300 |
| D663A | 3.2 ± 0.9 |
| E713A | >300 |
| E713D | >300 |
| L716S | 60 ± 10 |
| F720S | 6.0 ± 0.2 |
| K722N | 9 ± 1 |
| D756A | 2.2 ± 0.9 |
| Q758A | 6.6 ± 0.6 |
| D766S | 3.9 ± 0.8 |
| Q768A | 5.0 ± 0.6 |
FIGURE 5.

Kinase and ATPase activities of wild-type and mutant A-CAT. A, kinase and ATPase activity of wild-type (WT) A-CAT and the indicated mutants were assayed as described under “Experimental Procedures.” B, ATPase and kinase activities for the WT A-CAT and the R592L and K722N mutants were assayed at varying ATP concentrations. The kinetic constants obtained by fitting the data to a hyperbolic curve are listed in Table 3.
Leu-716 and Phe-720 make hydrophobic interactions with the adenine base (Fig. 4). Mutation of Leu-716 to serine decreased mant-ATP binding affinity by 12-fold and strongly inhibited kinase and ATPase activities, indicating that Leu-716 plays an important role in forming the hydrophobic pocket that binds the adenine base (Table 5 and Fig. 5A). In contrast, the F720S mutation had little effect on mant-ATP binding. Nevertheless, the F720S mutation abolished kinase activity and strongly inhibited ATPase activity, showing that Phe-720 plays a critical role in catalysis that is independent of nucleotide binding.
A-CAT contains two highly conserved basic residues that interact with the nucleotide phosphoryl groups: Arg-592 in the P-loop and Lys-722 in the C-lobe. Mutation of Arg-592 or Lys-722 only modestly reduced the binding affinity of mant-ATP (Table 5). Consistent with these results, steady-state kinetic analysis of the R592L and K722N mutants showed a small 2–3-fold increase in the Km for ATP (Fig. 5B and Table 3). The turnover numbers for the R592L and K722N mutants in the ATPase and kinase assays were 3- and 6-fold lower than that of the wild-type enzyme, respectively. Thus, the Arg-592 and Lys-722 residues enhance the efficiency of catalysis but do not play crucial roles in the phosphotransferase reaction.
The phosphoryl group of Asp(P)-766 forms hydrogen bonds with the invariant Asp-756 residue and the poorly conserved Asp-663 and Gln-758 residues (Fig. 1E). The D663A, D756A, and D766S mutants bound mant-ATP somewhat more tightly than the wild-type enzyme, suggesting that this cluster of negatively charged residues may be involved in unfavorable charge interactions with the γ-phosphoryl group (Table 5). The D756A mutation abolished kinase activity and reduced ATPase activity by 10-fold (Fig. 5A) (19). The D766S mutation completely eliminated kinase and ATPase activity (Fig. 5A). Moreover, the D766S mutant was unable to hydrolyze ADP and AMP, showing that Asp-766 plays an essential role in the removal of all three phosphoryl groups.
Interestingly, the D663A mutation inhibited kinase activity to a much greater extent than ATPase activity (Fig. 5A). These results suggest that Asp-663 plays a specific role in the recognition and/or the phosphorylation of the protein substrate.
Role of Mg2+ Ion-chelating Groups
A-CAT requires Mn2+ or Mg2+ for activity (28). In the absence of divalent cations, mant-ATP, mant-ADP, ATP, and ADP bound 3–8-fold less tightly to A-CAT (Table 4). The absence of divalent cations did not, however, impair the binding of AMP or adenosine. The effects of divalent cations therefore depend on interactions with the nucleotide β- and γ-phosphoryl groups.
In some, but not all, A-CAT structures, a Mg2+ ion (Mg1) is coordinated by the ribose 3′-hydroxyl, the nucleotide phosphoryl groups, and Gln-758, and a second Mg2+ (Mg2) is ligated by Asp-766, Gln-768, and the backbone carbonyl of Pro-767 (Fig. 4) (19). To test whether Mg1 is required for catalysis, we carried out assays with 3′-deoxy-ATP (d3′ATP) and mutated Gln-758 to alanine. The absence of the ribose 3′-hydroxyl group and the Q758A mutation had only a modest effect (1.5–2-fold) on nucleotide binding (Tables 4 and 5). A-CAT hydrolyzed d3′ATP with a kcat of 0.66 ± 0.03 min−1, which is only about 3-fold lower than the rate of ATPase hydrolysis. Conversely, the Q758A mutation abolished ATPase activity and reduced kinase activity by 20-fold (Fig. 5A). It can be concluded that chelation of Mg1 by the invariant Gln-758 residue, but not by the ribose 3′-hydroxyl group, plays an essential role in catalysis.
The Mg2-binding site was probed by mutating Gln-768 to alanine. The Q768A mutation had no effect on the binding of mant-ATP (Table 5) but, surprisingly, increased ATPase activity by 2-fold while at the same time reducing kinase activity by 30% (Fig. 5A). The results indicate that Gln-768 may play an important role in excluding water from the active site and thus enhancing the stability of Asp(P)-766.
Discussion
A-CAT is an excellent model in which to investigate the basic properties of the α-kinases, because it consists of little more than a core α-kinase domain, is catalytically active, and is structurally well characterized. The α-kinase active site is highly conserved so that the catalytic properties of A-CAT are likely to be typical of those of other family members. We show here that A-CAT has the unusual ability to catalyze the hydrolysis of ATP to adenosine and we provide evidence that each of the three phosphoryl groups is transferred sequentially to Asp-766. We also describe the nucleotide binding properties of A-CAT and have defined the roles played by key active site residues in nucleotide binding, ATPase activity, and kinase activity.
A-CAT Catalytic Mechanism
A-CAT is a very active protein kinase with a turnover rate as high as 10 s−1 and is a much less active ATPase with a turnover rate of about 1.9 min−1 (8, 19, 28). A-CAT is also able to hydrolyze ADP to AMP, and we show here that it can hydrolyze AMP to adenosine. The turnover rates for these two activities were determined to be ∼0.6 and 0.3 min−1, respectively. Consequently, A-CAT crystallized from solutions containing MgATP may have ADP, AMP, or adenosine bound to the catalytic cleft (Table 1). The identity of the bound nucleotide presumably depends on factors such as the time required for crystal growth and the composition of the crystallization solution. We also demonstrate that A-CAT can use ADP to phosphorylate proteins and peptides. These catalytic properties distinguish A-CAT from members of the ePK superfamily, such as PKA, which employ only ATP as a substrate.
In previous work, crystallization of A-CAT from a buffer containing MgATP resulted in a structure with ADP bound to the catalytic cleft and a phosphorylated Asp-766 (Asp(P)-766) residue (19). This result demonstrated that the ATP γ-phosphoryl group could be transferred, either directly or indirectly, to Asp-766. The structures described here now show that Asp-766 is phosphorylated when AMP or adenosine are bound to the inter-lobe cleft. The most straightforward interpretation of these results is that the hydrolysis of ADP to AMP and AMP to adenosine also results in the transfer of the phosphoryl group to Asp-766. Thus, as ATP is hydrolyzed to adenosine, the three phosphoryl groups are transferred in a stepwise manner to Asp-766. This result is consistent with the finding that mutation of Asp-766 to serine eliminated the ATPase, ADPase, and AMPase activities of A-CAT and that ADP can function as a substrate for protein phosphorylation. The A-CAT·AMP structure indicates that a direct transfer of the AMP α-phosphate to Asp-766 is feasible. The Asp-766 side chain carboxylate and the α-phosphorus atom are 3.4 Å apart. A small clockwise rotation of the Asp-766 side chain would reduce this distance to 3.0 Å and position the carboxylate for a direct in-line attack on the α-phosphorus atom. We have not, however, been able to demonstrate that A-CAT can phosphorylate proteins using AMP as a substrate. It is less clear how the β- and γ-phosphoryl groups might be transferred to Asp-766. The only structure of A-CAT bound to ADP has a phosphorylated Asp-766, so that the position of the Asp-766 side chain in relation to the β-phosphoryl group prior to the phosphorylation reaction is not known (19). A structure with AMPPCP in the active site shows that the γ-phosphate is directed away from the Asp-766 side chain (19). It is noteworthy that all A-CAT structures that contain Asp(P)-766 have been crystallized in the presence of a peptide substrate. Although the peptide substrate does not appear in the final crystal structure, it is possible that the transient binding of the peptide is required for the nucleotide phosphoryl groups to take up positions suitable for transfer to Asp-766.
Conformational Changes in A-CAT
The crystal structures of the atypical eukaryotic Rio1 and Rio2 kinases show that the active site contains an aspartyl phosphate residue (25, 26). The residue phosphorylated is the equivalent to Asp-766 in A-CAT. The RIO kinases hydrolyze ATP at a rate similar to A-CAT (kcat of 0.9 min−1) but have very low kinase activity, estimated to be 50–100-fold lower than their ATPase activity (27). It has been proposed that the RIO kinases function primarily as ATPases, with phosphorylation of aspartic acid being used to drive a conformational change in the RIO kinase domain that regulates its binding interactions. The A-CAT·AMP·Pi structure reported here, together with the previously described A-CAT·AMP structure, allows a direct comparison of two A-CAT structures bound to AMP that differ in the phosphorylation state of Asp-766. An alignment of the two structures shows that the phosphorylation of Asp-766 causes the P-loop to shift upwards and the N/D-loop to move outwards by 2–3 Å. These conformational changes open up the right-hand side of the inter-lobe cleft, which could potentially alter protein substrate binding. In addition, the presence of Asp(P)-766 changes the electrostatic properties of the catalytic cleft, which could further influence the selection of protein substrates. Nevertheless, the conformational changes are small and are localized to the putative protein substrate-binding site. It seems unlikely that the primary function of Asp-766 phosphorylation in A-CAT is to drive a conformational change.
A crystal structure of an archaeal Rio1 kinase obtained in the absence of added nucleotides has adenosine in the active site (39). This result implies that the RIO kinases, like A-CAT, are able to hydrolyze ATP to adenosine. Rio1 bound to adenosine has a significantly different conformation than Rio1 bound to ADP, indicating that removal of the α- and β-phosphoryl groups could be of functional relevance (39). In contrast, the structure of A-CAT remains essentially the same whether bound to ADP, AMP, or adenosine.
Asp-663 and Gln-768 Modulate Catalytic Activity
The phosphoryl group of Asp(P)-766 is directed away from the nucleotide to interact with Asp-756, Asp-663, and Gln-768 on the right-hand side of the catalytic cleft (Fig. 1E). This region of the catalytic cleft is thought to form the docking site for protein substrates with the invariant Asp-756 residue fulfilling the function of the catalytic base (19). Asp-663, which is conserved in about 50% of α-kinases, has been proposed to play a role in the recognition of basic residues in the peptide substrate (19). This proposal is supported by the finding that mutation of Asp-663 to alanine inhibited kinase activity to a much greater extent than ATPase activity. In eukaryotic elongation factor 2 kinase, a double mutation of Glu-183 and Asp-184 (the residue equivalent to Asp-663) to alanine inhibits kinase activity, again consistent with a role in protein substrate recognition (22).
The position of Gln-768 in the A-CAT active site is similar to that of Phe-187, the DFG+1 residue, in PKA (Fig. 6) (40). Phe-187 helps to exclude water from the active site when PKA is in the active, closed conformation (41). Mutation of Gln-768 to alanine doubled the ATPase activity of A-CAT but reduced kinase activity by 30%, suggesting that Gln-768 functions to protect Asp(P)-766 from hydrolysis. In ePKs, the DFG+1 residue is a major determinant governing serine or threonine phosphorylation site specificity (40). We therefore examined whether the Q768A mutation altered the high degree of threonine specificity exhibited by A-CAT. Kinase assays with pairs of peptides containing either serine or threonine at the phosphoacceptor site showed that the A-CAT-Q768A mutant strongly preferred to phosphorylate the threonine-containing peptides (data not shown). Thus, the molecular basis for the threonine specificity exhibited by A-CAT remains unexplained.
FIGURE 6.
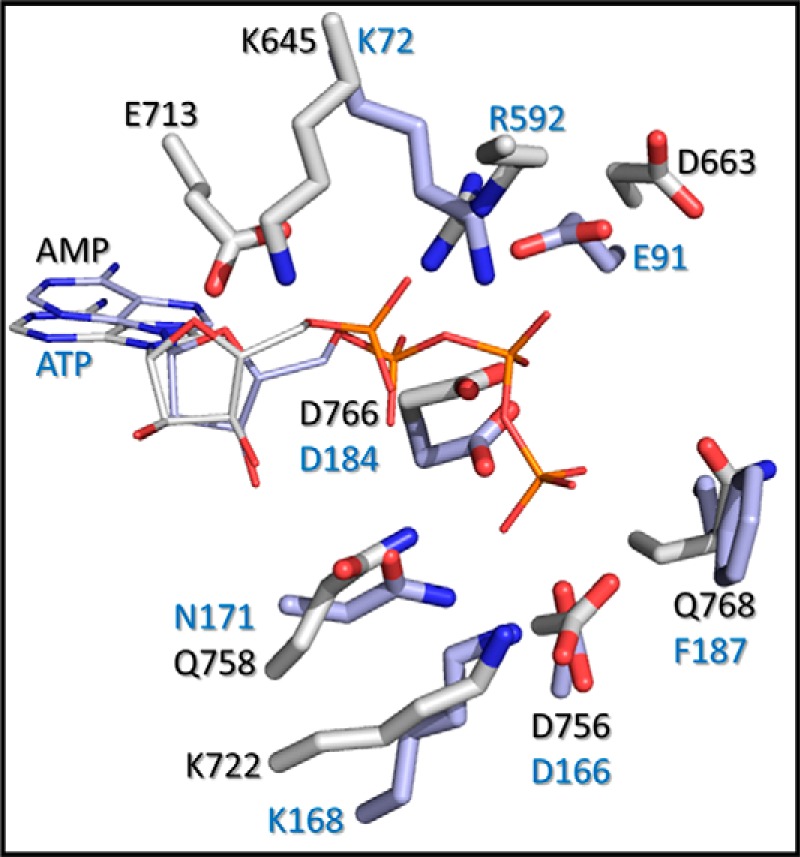
Comparison of the A-CAT and PKA active sites. Active site residues in A-CAT (Protein Data Bank code 3LKM) (gray) and PKA (Protein Data Bank code 1ATP) (blue) were aligned using PyMOL. The A-CAT structure contains AMP, and the PKA structure contains ATP.
Lys-645 and Glu-713 Play Critical Roles in Nucleotide Binding
The nucleotide binding properties of A-CAT differ somewhat from those described for PKA. In the presence of Mg2+, A-CAT bound ATP 2–3-fold more tightly than ADP and exhibited a higher affinity for mant-ATP than mant-ADP. In contrast, PKA binds ATP and ADP with the same affinity (Kd = 10 μm) and prefers to bind mant-ADP (Kd = 9 μm) rather than mant-ATP (Kd = 36 μm) (38, 42). A-CAT also exhibited a significantly higher affinity for AMP (Kd = 160 μm) than PKA (Kd =640 μm) (42). The altered distribution of positively charged residues in the two active sites is likely to account for these differences (Fig. 6). In particular, the presence of the highly conserved Arg-592 residue in the P-loop provides A-CAT with an extra basic residue with which to neutralize the negative charges on the nucleotide phosphoryl groups.
A-CAT bound adenosine almost as tightly as ATP (Kd values of 45 and 20 μm, respectively). The primary energy for nucleotide binding is therefore derived from interactions with the adenine ring. In agreement with this, the binding of mant-ATP to A-CAT was most severely affected by the mutation of three residues (Leu-716, Lys-645, and Glu-713) that directly contact the adenine ring (Fig. 4).
The invariant Lys-645 residue in the β7 strand and the invariant Glu-713 residue in the hinge connecting the N- and C-lobes participate in a salt bridge interaction. This interaction positions the side chain of Lys-645 to interact with the N2 atom of the adenine base and the α-phosphoryl group. In contrast, the equivalent residue in PKA, Lys-72, interacts electrostatically with Glu-91 in the αC-helix and, as a result, is positioned between the α- and β-phosphoryl groups of ATP (Fig. 6) (43). Mutation of Lys-72 to alanine in yeast PKA or to arginine in ERK2 and Lck strongly inhibits kinase activity but has little or no effect on the Km value for ATP (44–46). The invariant N-lobe lysine residue therefore performs different functions in A-CAT and PKA, as might be predicted based on their distinct interactions with the nucleotide. Lys-645 in A-CAT plays a critical role in binding the nucleotide, whereas Lys-72 in PKA plays an essential role in the phosphoryl transfer reaction. There is no residue equivalent to Glu-713 in PKA. Glu-713 is likely to play a central role in defining the nucleotide specificity of A-CAT via a hydrogen bond interaction with the adenine N6 amino group.
The side chain of Phe-720 lies below the adenine ring and ribose moiety (Fig. 4). A Phe-720 to serine mutation did not impair nucleotide binding but did strongly inhibit catalytic activity. Phe-720 is conserved in the α-kinases in terms of hydrophobicity but not in terms of exact residue identity. The side chain of Phe-720 extends inward to make a hydrophobic interaction with the side chain methyl group of the conserved Thr-765 residue in the innermost catalytic loop, suggesting that its primary function is to anchor and properly position the three catalytic loops.
Chelation of Mg1 by Gln-758 Is Essential for Catalysis
Mg2+ enhanced the binding of ATP and ADP, but not AMP or adenosine, to A-CAT. The Mg2+ independence of AMP binding is at odds with the observation that the A-CAT·AMP structure, like the A-CAT·ADP and A-CAT-D766A·ATP structures, contains a Mg2+ ion (Mg1) ligated by the ribose 3′-hydroxyl group, the α-phosphoryl group, and the highly conserved Gln-758 residue (19). This result can be reconciled if Mg1 does not play an important role in promoting the binding of nucleotide to A-CAT but, instead, functions primarily in catalysis. Strong support for this interpretation comes from the finding that the mutation of Gln-758 to alanine decreased only slightly the binding affinity for mant-ATP but severely impaired catalytic activity. Chelation of Mg1 by Gln-758 is therefore likely to play an essential role in orienting the nucleotide phosphoryl groups for catalysis. The function of Gln-758 differs from its counterpart in PKA, Asn-171, which coordinates a nonessential Mg2+ ion that bridges the α- and γ-phosphates (Fig. 6) (4).
Mg1 is also coordinated by the ribose 3′-hydroxyl group. Interestingly, d3′ATP, which lacks the 3′-hydroxyl group, was hydrolyzed at a rate only 3-fold lower than ATP. Thus, the absence of the 3′-hydroxyl group does not have nearly as great an effect on catalytic activity as the Q758A mutation. We interpret this result to show that Gln-758, which provides the sole protein ligand, has a much more critical role in the chelation of Mg1, and hence in the proper orientation of the nucleotide phosphates, than the ribose 3′-hydroxyl group. Indeed, ligation of Mg1 by the ribose 3′-hydroxyl group bends the AMPPCP phosphoryl groups into a U-shaped conformation that is not suitable for catalysis (19).
The side chain of the invariant Lys-722 residue is positioned to interact with the ATP γ-phosphate (Fig. 4). A K722N mutation reduced mant-ATP binding by less than 2-fold, increased the Km values for ATP by 2–3-fold, and reduced kcat by 6-fold. The equivalent residue in PKA is Lys-168 which, when mutated to alanine, increases the Km value for ATP by 2.5-fold and reduces kcat by 50-fold (46). Theoretical analysis indicates that Lys-168 interacts with the transferring phosphoryl group throughout the reaction process and contributes to the stabilization of the product state (47). Lys-722 clearly plays a less important role in the A-CAT phosphotransferase reaction than does Lys-168 in PKA, likely due to differences in the catalytic mechanisms employed by the two kinases.
In summary, the results presented here provide additional evidence that A-CAT, and by extension other α-kinases, exhibits catalytic properties that differ in important respects from those described for conventional ePKs. These include differences in the reactions catalyzed and the chemical mechanism of phosphoryl transfer, which in turn are reflected in altered functions for a number of key active site residues. Further work is required to understand how the unique catalytic properties of the α-kinases might influence their ability to regulate cellular processes such as cell motility, protein translation, and gene expression.
Author Contributions
G. C. designed the study and wrote the paper. Z. J. helped design experiments and analyze the data. Y. Y. constructed vectors, expressed and purified proteins, and characterized their functional properties and helped write the paper. Q. Y. crystallized proteins and solved the x-ray structures. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Kim Munro of the Queen's University Protein Function Discovery Laboratory for help with the fluorescence measurements and Dr. Jiaxi Wang of the Queen's University Department of Chemistry Mass Spectrometry Facility for peptide analysis. Structural data were collected using Beamline X4A at the National Synchrotron Light Source, Brookhaven National Laboratory, a United States Department of Energy facility, supported by the New York Structural Biology Consortium.
This work was supported by Natural Science and Engineering Research Council of Canada Discovery Grant RGPIN 391522 (to G. P. C.) and Grant RGPIN 203705 (to Z. J). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4ZME and 4ZMF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ePK
- eukaryotic protein kinase
- MHCK
- Dictyostelium myosin-II heavy chain kinase
- A-CAT
- the α-kinase domain of MHCK A
- PKL
- protein kinase-like
- MBP
- myelin basic protein
- mant
- methyl-anthraniloyl
- AMPPCP
- adenosine 5′-(β,γ-methylenetriphosphate)
- TES
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate
- d3′ATP
- 3′-deoxy-ATP
- TRPM7
- transient receptor potential melastatin-like 7
- eEF2
- eukaryotic elongation factor 2
- ADN
- adenosine.
References
- 1. Kannan N., Taylor S. S., Zhai Y., Venter J. C., Manning G. (2007) Structural and functional diversity of the microbial kinome. PLoS Biol. 5, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 3. Scheeff E. D., Bourne P. E. (2005) Structural evolution of the protein kinase-like superfamily. PLoS Comput. Biol. 1, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor S. S., Kornev A. P. (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Middelbeek J., Clark K., Venselaar H., Huynen M. A., van Leeuwen F. N. (2010) The α-kinase family: an exceptional branch on the protein kinase tree. Cell. Mol. Life Sci. 67, 875–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ryazanov A. G. (2002) Elongation factor-2 kinase and its newly discovered relatives. FEBS Lett. 514, 26–29 [DOI] [PubMed] [Google Scholar]
- 7. Crawley S. W., Côté G. P. (2008) Determinants for substrate phosphorylation by Dictyostelium myosin II heavy chain kinases A and B and eukaryotic elongation factor-2 kinase Biochim. Biophys. Acta 1784, 908–915 [DOI] [PubMed] [Google Scholar]
- 8. Luo X., Crawley S. W., Steimle P. A., Egelhoff T. T., Cote G. P. (2001) Specific phosphorylation of threonine by the Dictyostelium myosin II heavy chain kinase family. J. Biol. Chem. 276, 17836–17843 [DOI] [PubMed] [Google Scholar]
- 9. Futey L. M., Medley Q. G., Côté G. P., Egelhoff T. T. (1995) Structural analysis of myosin heavy chain kinase A from Dictyostelium. Evidence for a highly divergent protein kinase domain, an amino-terminal coiled-coil domain, and a domain homologous to the b-subunit of heterotrimeric G proteins J. Biol. Chem. 270, 523–529 [DOI] [PubMed] [Google Scholar]
- 10. Yumura S., Yoshida M., Betapudi V., Licate L. S., Iwadate Y., Nagasaki A., Uyeda T. Q., Egelhoff T. T. (2005) Multiple myosin II heavy chain kinases: roles in filament assembly control and proper cytokinesis in Dictyostelium. Mol. Biol. Cell 16, 4256–4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Côte G. P., Bukiejko U. (1987) Purification and characterization of a myosin heavy chain kinase from Dictyostelium discoideum. J. Biol. Chem. 262, 1065–1072 [PubMed] [Google Scholar]
- 12. Egelhoff T. T., Lee R. J., Spudich J. A. (1993) Dictyostelium myosin heavy chain phosphorylation sites regulate myosin filament assembly and localization in vivo. Cell 75, 363–371 [DOI] [PubMed] [Google Scholar]
- 13. Ryazanov A. G., Shestakova E. A., Natapov P. G. (1988) Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 334, 170–173 [DOI] [PubMed] [Google Scholar]
- 14. Kenney J. W., Moore C. E., Wang X., Proud C. G. (2014) Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 55, 15–27 [DOI] [PubMed] [Google Scholar]
- 15. Faller W. J., Jackson T. J., Knight J. R., Ridgway R. A., Jamieson T., Karim S. A., Jones C., Radulescu S., Huels D. J., Myant K. B., Dudek K. M., Casey H. A., Scopelliti A., Cordero J. B., Vidal M., et al. (2015) mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517, 497–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark K., Langeslag M., van Leeuwen B., Ran L., Ryazanov A. G., Figdor C. G., Moolenaar W. H., Jalink K., van Leeuwen F. N. (2006) TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 25, 290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clark K., Middelbeek J., Lasonder E., Dulyaninova N. G., Morrice N. A., Ryazanov A. G., Bresnick A. R., Figdor C. G., van Leeuwen F. N. (2008) TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J. Mol. Biol. 378, 790–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krapivinsky G., Krapivinsky L., Manasian Y., Clapham D. E. (2014) The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell 157, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ye Q., Crawley S. W., Yang Y., Côté G. P., Jia Z. (2010) Crystal structure of the α-kinase domain of Dictyostelium myosin heavy chain kinase A. Sci. Signal. 3, ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamaguchi H., Matsushita M., Nairn A. C., Kuriyan J. (2001) Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol. Cell 7, 1047–1057 [DOI] [PubMed] [Google Scholar]
- 21. Endicott J. A., Noble M. E., Johnson L. N. (2012) The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613 [DOI] [PubMed] [Google Scholar]
- 22. Moore C. E., Regufe da Mota S., Mikolajek H., Proud C. G. (2014) A conserved loop in the catalytic domain of Eukaryotic elongation factor 2 kinase plays a key role in its substrate Specificity. Mol. Cell. Biol. 34, 2294–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adams J. A. (2001) Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 101, 2271–2290 [DOI] [PubMed] [Google Scholar]
- 24. Madhusudan, Akamine P., Xuong N. H., Taylor S. S. (2002) Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat. Struct. Biol. 9, 273–277 [DOI] [PubMed] [Google Scholar]
- 25. Ferreira-Cerca S., Sagar V., Schäfer T., Diop M., Wesseling A. M., Lu H., Chai E., Hurt E., LaRonde-LeBlanc N. (2012) ATPase-dependent role of the atypical kinase Rio2 on the evolving pre-40S ribosomal subunit. Nat. Struct. Mol. Biol. 19, 1316–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferreira-Cerca S., Kiburu I., Thomson E., LaRonde N., Hurt E. (2014) Dominant Rio1 kinase/ATPase catalytic mutant induces trapping of late pre-40S biogenesis factors in 80S-like ribosomes. Nucleic Acids Res. 42, 8635–8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. LaRonde N. A. (2014) The ancient microbial RIO kinases. J. Biol. Chem. 289, 9488–9492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Côté G. P., Luo X., Murphy M. B., Egelhoff T. T. (1997) Mapping of the novel protein kinase catalytic domain of Dictyostelium myosin II heavy chain kinase A. J. Biol. Chem. 272, 6846–6849 [DOI] [PubMed] [Google Scholar]
- 29. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 33. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK–a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 36. Crawley S. W., Gharaei M. S., Ye Q., Yang Y., Raveh B., London N., Schueler-Furman O., Jia Z., Côté G. P. (2011) Autophosphorylation activates Dictyostelium myosin II heavy chain kinase A by providing a ligand for an allosteric binding site in the α-kinase domain. J. Biol. Chem. 286, 2607–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 38. Ni Q., Shaffer J., Adams J. A. (2000) Insights into nucleotide binding in protein kinase A using fluorescent adenosine derivatives Protein Sci. 9, 1818–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laronde-Leblanc N., Guszczynski T., Copeland T., Wlodawer A. (2005) Structure and activity of the atypical serine kinase Rio1. FEBS J. 272, 3698–3713 [DOI] [PubMed] [Google Scholar]
- 40. Chen C., Ha B. H., Thévenin A. F., Lou H. J., Zhang R., Yip K. Y., Peterson J. R., Gerstein M., Kim P. M., Filippakopoulos P., Knapp S., Boggon T. J., Turk B. E. (2014) Identification of a major determinant for serine-threonine kinase phosphoacceptor specificity. Mol. Cell 53, 140–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taylor S. S., Yang J., Wu J., Haste N. M., Radzio-Andzelm E., Anand G. (2004) PKA: a portrait of protein kinase dynamics. Biochim. Biophys. Acta 1697, 259–269 [DOI] [PubMed] [Google Scholar]
- 42. Bhatnagar D., Roskoski R. Jr., Rosendahl M. S., Leonard N. J. (1983) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: a new fluorescence displacement titration technique for characterizing the nucleotide binding site on the catalytic subunit. Biochemistry 22, 6310–6317 [DOI] [PubMed] [Google Scholar]
- 43. Cheng Y., Zhang Y., McCammon J. A. (2005) How does the cAMP-dependent protein kinase catalyze the phosphorylation reaction: an ab initio QM/MM study. J. Am. Chem. Soc. 127, 1553–1562 [DOI] [PubMed] [Google Scholar]
- 44. Robinson M. J., Harkins P. C., Zhang J., Baer R., Haycock J. W., Cobb M. H., Goldsmith E. J. (1996) Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry 35, 5641–5646 [DOI] [PubMed] [Google Scholar]
- 45. Carrera A. C., Alexandrov K., Roberts T. M. (1993) The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc. Natl. Acad. Sci. U.S.A. 90, 442–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gibbs C. S., Zoller M. J. (1991) Rational scanning mutagenesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. J. Biol. Chem. 266, 8923–8931 [PubMed] [Google Scholar]
- 47. Valiev M., Yang J., Adams J. A., Taylor S. S., Weare J. H. (2007) Phosphorylation reaction in cAPK protein kinase-free energy quantum mechanical/molecular mechanics simulations. J. Phys. Chem. B 111, 13455–13464 [DOI] [PubMed] [Google Scholar]