

Background: Endurance training induces physiological cardiac hypertrophy and elevates adipose tissue lipolysis.
Results: Adipose-specific adipose triglyceride lipase (Atgl)-knock-out mice exhibit attenuated exercise-induced cardiac hypertrophy likely mediated by the lack of C16:1n7 palmitoleate actions on the heart.
Conclusion: Atgl-mediated adipose lipolysis regulates physiological cardiac hypertrophy.
Significance: Adipose-derived lipokines may serve as important molecular mediators of cardiac physiology and pathology.
Keywords: adipose tissue metabolism, adipose triglyceride lipase (ATGL), cardiac hypertrophy, cardiac metabolism, lipid signaling
Abstract
Endurance exercise training induces substantial adaptive cardiac modifications such as left ventricular hypertrophy (LVH). Simultaneously to the development of LVH, adipose tissue (AT) lipolysis becomes elevated upon endurance training to cope with enhanced energy demands. In this study, we investigated the impact of adipose tissue lipolysis on the development of exercise-induced cardiac hypertrophy. Mice deficient for adipose triglyceride lipase (Atgl) in AT (atATGL-KO) were challenged with chronic treadmill running. Exercise-induced AT lipolytic activity was significantly reduced in atATGL-KO mice accompanied by the absence of a plasma fatty acid (FA) increase. These processes were directly associated with a prominent attenuation of myocardial FA uptake in atATGL-KO and a significant reduction of the cardiac hypertrophic response to exercise. FA serum profiling revealed palmitoleic acid (C16:1n7) as a new molecular co-mediator of exercise-induced cardiac hypertrophy by inducing nonproliferative cardiomyocyte growth. In parallel, serum FA analysis and echocardiography were performed in 25 endurance athletes. In consonance, the serum C16:1n7 palmitoleate level exhibited a significantly positive correlation with diastolic interventricular septum thickness in those athletes. No correlation existed between linoleic acid (18:2n6) and diastolic interventricular septum thickness. Collectively, our data provide the first evidence that adipose tissue lipolysis directly promotes the development of exercise-induced cardiac hypertrophy involving the lipokine C16:1n7 palmitoleate as a molecular co-mediator. The identification of a lipokine involved in physiological cardiac growth may help to develop future lipid-based therapies for pathological LVH or heart failure.
Introduction
Intensive and prolonged physical exercise results in major cardiac adaptations leading to amplified cardiac output that meets increased peripheral oxygen and energy demands (1). Exercise-induced cardiac changes include enlarged left ventricular (LV)7 internal dimensions and augmented LV wall thickness in the presence of preserved cardiac function (1). These changes are accompanied by maintenance of FA oxidation as the predominant cardiac energy source. In contrast, the development of pathological or maladaptive LV hypertrophy during chronic hypertension or aortic valve disease results in augmented hypertrophic responses associated with LV dysfunction, cardiac fibrosis, and prevailing glucose oxidation (2). Improved knowledge about the mechanisms underlying exercise-induced/physiological cardiac hypertrophy is of clinical relevance because molecular effectors such as insulin-like growth factor 1 (Igf1) that induce beneficial cardiac growth are potential therapeutic targets for the treatment of maladaptive hypertrophy and heart failure (3).
New mechanisms underlying the development of LVH may arise from shifting the focus from the heart to accompanied peripheral exercise-induced physiological responses. To provide the heart and skeletal muscle with a sufficient amount of energy during prolonged physical exercise, a switch in energy substrates occurs from an initial preference for glucose that later changes to fatty acids (FAs) (3). Increased catecholamine levels induce the hydrolysis of triacylglycerol (TG) in white adipose tissue (WAT) mediating the release of free fatty acids (FFAs) into the circulation that provide energy substrates for the heart and skeletal muscle (4). In addition to their role as biomolecules for energy production, FAs have been recently characterized as signaling molecules directly regulating cellular responses (5). Along this line, exercise-mediated stimulation of WAT lipolysis releases a broad variety of FAs into the bloodstream, some of which may directly act as molecular effectors on other organs, including the heart. To liberate FAs from WAT, TG hydrolysis is catalyzed by two major adipose tissue lipases, hormone-sensitive lipase (Hsl) and adipose triglyceride lipase (Atgl), under hormonal control (6).
In this study, we aimed to identify new FA-based molecular effectors from WAT for exercise-induced nonproliferative cardiac growth. For this, we studied adipose tissue-specific Atgl-deficient mice (atATGL-KO) with attenuated WAT lipolysis and reduced FA release during chronic exercise. We then performed a serum FA profiling using HPLC/triple quad mass spectrometer technology to identify individual FAs involved in the development of physiological cardiac hypertrophy. Finally, to translate these results into the human situation, we studied endurance athletes by echocardiography and performed serum FA profiling to correlate individual FA levels with the degree of training-induced LVH.
Experimental Procedures
Animals
All animal procedures were performed in accordance with the guidelines of the German Law on the Protection of Animals. Fat tissue-specific Atgl-deficient mice (atATGL-KO) were generated by crossing B6.129-Pnpla2tm1Eek (atgl-flox) mice (7) with B6.Cg-Tg(fabp4-cre)1Rev/J mice. The highest expression of Atgl in mice has been determined in white/brown adipose tissue, in heart, and testis (8). To exclude the effects of Atgl deficiency in tissues other than adipose tissue, we analyzed Atgl expression in WAT, heart, and macrophages (Fig. 1A). Mice were housed in a facility with a 12-h light/dark cycle (25 °C) and fed at libitum with a standard diet (9). 5-Week-old female atgl-flox/flox Cre−/+ (atATGL-KO) mice and control littermate atgl-flox/flox Cre−/− animals (WT) were randomized to a treadmill-trained group (run, n = 10 atATGL-KO, and n = 10 WT mice) and control-sedentary animals (n = 10 atATGL-KO, and n = 10 WT mice). Run mice were adapted to treadmill training (Treadmill, TSE Sytems) by a gradual increase of the training intensity (0.05–0.25 m/s, 7° slope, 15–90 min/day/mouse, for 3 weeks). After the adaptation phase, mice were trained over 4 weeks using the following protocol: 0.25 m/s, 7° slope, 90 min/day/mouse, as described previously (9). To avoid differences in nutritional status of the mice, all animals were trained in the fed-state. At the end of the training regime, mice underwent echocardiographic analysis (10). Blood samples were collected before/after 45 min of training in fed mice for analysis of serum glucose and FFA (9). Body composition was determined by NMR (Bruker's MiniSpec MQ10). The respiratory quotient during exercise was determined using indirect calorimetric analysis combined with treadmill training (TSE Systems) (9).
FIGURE 1.

Phenotypic characterization of atATGL-KO mice. A, RT-PCR analysis of Atgl expression, measured in WAT, the left ventricle of the heart, and bone marrow-derived macrophages (BMdMφ). B, representative images of gonadal fat pads isolated from WT and atATGL-KO mice. Exercise-mediated metabolic changes. C, ex vivo lipolysis assay in murine gonadal adipose tissue explants collected from trained mice immediately following their final running session (n = 3–4/group). FFA release assays were performed using perigonadal white adipose tissue pads (n = 3–4/mouse). *, p < 0.05 versus WT mice (unpaired t test). D, change in plasma FFAs; F, glucose of trained mice before (pre) and directly after (post) their final running session. *, p < 0.05 versus pre/post difference in WT mice; °, p < 0.05 versus WT (pre); #, p < 0.05 versus WT (post) (n = 9; two-way ANOVA with repeated measures (Bonferroni post-test)). E, respiratory quotient (RQ) from trained mice during their final running sessions using indirect calorimetry integrated into the treadmill system (CaloTreadmill; TSE System). *, p < 0.05 versus WT (n = 4; two-way ANOVA with repeated measures (Bonferroni post-test)). G, profile of selected cardiac ceramides. Results represent samples obtained from WT and atATGL-KO mice (n = 3), analyzed using rapid resolution HPLC/tandem MS.
In an additional set of experiments, atATGL-KO and WT mice were supplemented orally with C16:1n7 or C18:1 n7 (300 mg/kg/day) (11) during 4 weeks of the main training period. Afterward, the mice underwent echocardiographic analysis.
Small Animal PET
To assess glucose metabolism, the animals were injected after an overnight rest with ∼7 MBq of 2-[18F]fluorodeoxyglucose and imaged using a small animal PET (Inveon dedicated PET, Siemens) (9). To evaluate FA metabolism after a resting period and overnight starvation, ∼2 MBq of [18F]fluoro-4-thia-palmitate were injected (12). Data were reconstructed using three-dimensional Ordered Subset Expectation Maximization (OSEM); image analysis was performed using the image analysis software AMIDE (13).
mRNA Analysis
Total RNA was isolated using the RNeasy micro kit (Qiagen). RNA samples were reverse-transcribed (Promega) and used in quantitative PCRs in the presence of a fluorescent dye (SYBR Green, Life Sciences) (9).
The expression analysis of bone marrow-derived cells was performed as described previously (14). Briefly, isolated primary bone marrow-derived cells from female atATGL-KO mice and their control littermates were differentiated in vitro into macrophages using 10% L929-conditioned medium. After 7 days of differentiation, bone marrow-derived macrophages were harvested, and total RNA was isolated, as described above.
Ex Vivo Lipolysis Assay in Gonadal Adipose Tissue Explants
The ex vivo lipolysis assay was described previously. Gonadal fat pads isolated from the mice directly after training (n = 3–4/group) were incubated in DMEM containing 2% FA-free BSA (basal lipolysis) and forskolin (10 μm) (stimulated lipolysis) for 1 h at 37 °C. FFA content was quantified using the HR-NEFA (Wako Diagnostics) (9).
FA Profiling of Serum Samples and HL-1 Cell Fractions
100 μl of serum were hydrolyzed under alkaline-methanolic conditions for 60 min at 80 °C. The samples were neutralized and diluted 1:10 with methanol containing the following internal standards: C15:0, C21:0 50 μg; C20:4-d8, C18:2-d4 5 μg; and C20:5-d5, C22:6-d5 1 μg. HPLC measurement was performed using an Agilent 1200 HPLC system with binary pump, autosampler, and column thermostat equipped with a Phenomenex Kinetex-C18 column 2.6 μm, 2.1 × 150-mm column using a solvent system of aqueous formic acid (0.1%) and acetonitrile. The solvent gradient started at 30% acetonitrile and was increased to 98% within 10 min with a flow rate of 0.4 ml/min and a 5-μl injection volume. The HPLC was coupled with an Agilent 6460 triple quad mass spectrometer with electrospray ionization source operated in negative selected ion mode.
Measurement of Ceramides in Heart Muscles
About 30-mg tissue samples were homogenized with liquid nitrogen. Next, an internal standard consisting of 10 ng of ceramide 17:0 (Avanti Polar Lipids, AL) in 2 ml of ethyl acetate/isopropyl alcohol/water (60:30:10) was added. The mixture was vortexed for 15 s and then ultrasonicated for 30 s. This was repeated three times followed by centrifugation for 10 min at 5000 rpm. The clear supernatant was taken and evaporated to dryness under a stream of nitrogen at 50 °C. The residues were dissolved in 150 μl of methanol and analyzed using an Agilent 1290 HPLC system with binary pump, autosampler, and column thermostat with a Kinetex C-18, 2.1 × 150 mm, 2.6 μm (Phenomenex, Aschaffenburg, Germany) column using a solvent system of aqueous formic acid (0.1%) and methanol. The elution gradient was started at 95% methanol, which was increased within 10 min to 99% and held there for 2 min. The flow rate was set at 0.4 ml/min. The injection volume was 1 μl. The HPLC was coupled with an Agilent 6490 Triple quad mass spectrometer (Agilent Technologies, Santa Clara, CA) with electrospray ionization source. The source parameters were as follows: Drying gas, 140 °C/14 liters/min; sheath gas, 380 °C/10 liters/min; capillary voltage, 5500 V; Nozzel voltage, 2000 V, and nebulizer pressure, 30 p.s.i. Analysis was performed in multiple reaction monitoring in a positive mode. The results were calculated compared with ceramide 17:0 as internal standard.
Cell Culture Experiments
Mouse HL-1 cardiomyocytes, kindly provided by W. C. Claycomb (Louisiana State University, LA), were cultivated as described previously (15). Human (GATA-4+/α-sarcomeric actin+) primary cardiomyocytes (primary human cardiomyocytes) were purchased from Promocell and cultivated accordingly to the manufacturer's instructions, as described previously (16). After a 24-h starvation period (0.5% FBS), cells were stimulated with 100 nm endothelin 1, a mix of FFAs (FA mix) dissolved in 10% FFA-free BSA (C16:0, C16:1n7, and C14:0), (C16:0, C14:0), (C16:0, C18:1, and C18:2), or C16:1n7 alone for 6 h (mRNA expression) or 30 min (protein phosphorylation). FFAs were used in equimolar serum concentrations estimated by FA profiling, C16:1 low was 10 times lower as serum concentration.
HL-1 Cell Fractionation
For cell fractionation experiments, the membrane fractionation kit (Ab 139409, Abcam) was used according to the manufacturer's protocol.
Western Immunoblotting
For Western blot analysis, HL-1 and primary HC were lysed in RIPA buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 1% Nonidet P-40, 2.5% glycerol, 1 mm EGTA, 50 mm NaF, 1 mm Na3VO4, 10 mm Na4P2O7, 100 μm phenylmethylsulfonyl fluoride, and complete protease/phosphatase inhibitor mixture (Phos-stop and Complete Mini, Roche Diagnostics)). Lysates were analyzed by immunoblotting using antibodies raised against pS473-Akt and total-Akt (4060 and 9272 from Cell Signaling Technologies, dilution 1:2000) and secondary horseradish-conjugated antibodies (Jackson ImmunoResearch, dilution 1:10,000). For detection, enhanced chemiluminescent reagents (ECL kit, Thermo Scientific) were used.
Cell fractions were analyzed using the membrane fractionation WB mixture (Ab 140365, Abcam) providing the mix of Ab specific for different cellular fractions as follows: anti-sodium potassium ATPase (plasma membrane marker); anti-Grp78 (endoplasmic reticulum marker) and anti-Atp5a (mitochondrial membrane marker); anti-Gapdh (cytosolic marker) and anti-histone H3 (nuclear marker). WB analysis was performed according to the manufacturer's protocol.
Immunostaining and Cell Size Quantification
Cells were fixed with formaldehyde (3.7%) in phosphate-buffered saline (PBS), permeabilized for 10 min with 0.5% Triton X-100 in PBS, and blocked for 1 h with PBS containing 0.1% Triton X-100 and goat serum (10%). Cells were incubated with primary antibody for sarcomeric α-Actinin (mouse monoclonal antibody, dilution 1:200, Sigma) for 1 h in the same solution as blocking solution. Cells were washed three times with PBS, incubated with secondary AlexaFluor® 488-conjugated antibody (goat anti-rabbit polyclonal IgG, dilution 1:400, Molecular Probes; green) and washed three times with PBS. Next, nuclei were stained for 5 min with DAPI (dilution 1:1000, Thermo Scientific, blue), washed three times with PBS, and mounted with mounting solution (Dako). Proteins were visualized with an inverted fluorescence phase-contrast microscope (BZ-9000E, All-in-One fluorescence microscope, Keyence) at a ×20 and ×60 magnification, and images were captured using a digital camera CFI60 (Nikon). Cell size was determined using BZ-II analyzer software and Dynamic Cell Count Tool (Keyence).
Histology
Cardiac tissue isolated from mice were formalin-fixed, paraffin-embedded, and stained with hematoxylin and eosin (H&E). For evaluation of LV cardiomyocyte size, H&E-stained cross-sections were analyzed using Analysis software (Olympus). For this, 50 randomly selected cells/LV were quantified in at least three random fields from n = 3 mice (×100 magnification). Picrosirius red staining was performed according to the manufacturer's instructions (Morphisto, Germany) with a small modification; the cardiac tissue was incubated two times in 6% acetic acid for 20 min prior staining.
Athletes and Echocardiography
25 male endurance athletes from the German cross-country skiing (n = 9)/biathlon (n = 16) national team underwent physical examinations, two-dimensional echocardiography, and serum sampling. The cohort was between 18 and 28 years old. All participants provided written consent, and ethical approval was obtained from the ethics committee (University Hospital Klinikum rechts der Isar, Munich, Germany). Serum samples were taken in the early morning prior to breakfast and subjected to lipid profiling (100 μl).
Two-dimensional echocardiography at rest was performed by an experienced echocardiography specialist according to standard procedures and measurements recommended previously (17). An IE 33 system with a 3.5 MHz transducer (Philips Healthcare, Hamburg, Germany) was used for all investigations. The examination included the documentation of standard parasternal and apical views and the use of continuous pulsed wave and colored Doppler techniques. Left ventricular mass was calculated using the formula established by Devereux et al. (18).
Statistical Analysis
Comparison of mean values between groups was evaluated by two-way ANOVA (Bonferroni post-test), two-way ANOVA with repeated measures (Bonferroni post-test), one-way ANOVA (Tukey's or Bonferroni multiple comparison test), or unpaired t tests, as appropriate. Exact value of n is provided for each type of experiments. Correlation analyses in the clinical study were performed using Pearson's test. Statistical significance was assumed at p < 0.05. Vertical lines in the histograms indicate standard error of the mean (S.E.).
Results
The atATGL-KO mice showed complete deletion of Atgl expression in WAT but not in the heart or bone marrow-derived macrophages and an enlargement of WAT depots, increased fat pad weights, and increased overall fat mass (Fig. 1, A and B, and Table 1). Moreover, WAT lipolysis was significantly diminished in atATGL-KO mice, when compared with WT littermates (Fig. 1C). In accordance with the importance of adipose ATGL for exercise-induced WAT lipolysis, circulating FFA level, measured before and after training, did significantly increase after training in WT mice but not in atATGL-KO mice (Fig. 1D). Consistently, atATGL-KO mice exhibited a significantly higher peri-exercise respiratory quotient confirming attenuated systemic lipid oxidation in the absence of adipose ATGL (Fig. 1E). Plasma glucose levels did not differ between the genotypes (Fig. 1F). Previously, ATGL deficiency in cardiomyocytes resulted in increased cardiac ceramide levels (19). However, deletion of ATGL in WAT did not affect cardiac ceramide levels (Fig. 1G). Taken together, atATGL-KO mice exhibited impaired WAT lipolysis associated with reduced exercise-induced plasma FFA appearance and systemic lipid oxidation.
TABLE 1.
Metabolic phenotyping
All measurements were performed after 4 weeks of training. Values are shown as means ± S.E. of the mean. BW is body weight.
| WT sedentary | WT run | atATGL-KO sedentary | atATGL-KO run | |
|---|---|---|---|---|
| Glucose (mg/dl) | 130.9 ± 7.61 | 135.1 ± 9.17 | 145.6 ± 11.45 | 132.3 ± 5.98 |
| BW (g) | 23.2 ± 0.4 | 22.8 ± 0.5 | 24.5 ± 0.7 | 24.4 ± 0.4 |
| Lean mass (NMR) | 17.19 ± 0.39 | 16.88 ± 0.34 | 16.79 ± 0.34 | 16.94 ± 0.37 |
| Fat mass (NMR) | 3.6 ± 0.3 | 3.3 ± 0.3 | 5.1 ± 0.3a | 4.8 ± 0.3a |
| WAT perirenal (g) | 0.1 ± 0.01 | 0.1 ± 0.01 | 0.2 ± 0.03a | 0.2 ± 0.02a |
| WAT gonadal (g) | 0.4 ± 0.05 | 0.3 ± 0.03 | 0.5 ± 0.06 | 0.4 ± 0.04 |
a p < 0.01 versus WT run; two-way ANOVA (Bonferroni post-test).
Subsequently, small animal PET was performed in sedentary and trained mice to assess cardiac FA and glucose uptake. Trained mice were examined at the end of the 4-week training after an overnight starving/resting period. As depicted in Fig. 2A, trained WT mice markedly increased cardiac FA uptake when compared with sedentary controls. Accordingly, cardiac glucose uptake was reduced in exercising WT mice. In consonance with the reduced FA mobilization in trained atATGL-KO mice, exercise-mediated cardiac FA uptake was completely abolished and even lower than sedentary controls, accompanied by an increase in cardiac glucose uptake (Fig. 2A). Reduced cardiac FA uptake in atATGL-KO mice did not result from diminished expression of cardiac FA transporters (Fig. 2B). In line with a reduction of cardiac FA uptake and utilization, cardiac expression of key enzymes involved in downstream FA metabolism, such as long chain acyl-CoA synthetase 1 (Acsl1), was reduced or tended to decrease (peroxisome proliferator-activated receptor α (Ppar-α)) in atATGL-KO mice (Fig. 2B). In association with changes in cardiac metabolism, chronic exercise leads to the development of physiological cardiac hypertrophy. LV mass assessed by echocardiography was significantly increased in WT mice challenged with endurance training compared with sedentary mice (Table 2). This response was significantly attenuated in atATGL-KO mice (Fig. 2C and Table 2). Histological analysis of myocyte size from LV samples (H&E staining) revealed a moderate increase of cardiomyocyte size in trained WT mice when compared with sedentary WT mice (LV myocyte cross-sectional area: sedentary WT, 258.6 ± 6.9 μm2 versus trained WT, 291.0 ± 7.1 μm2, p < 0.01) (Fig. 2D). This regulation was not visible in atATGL-KO mice (LV myocyte cross-sectional area: sedentary-KO, 273.3 ± 7.5 μm2 vs. trained-KO, 277.1 ± 6.8 μm2, p = non-significant) (Fig. 2D). Together these data suggest that FA mobilization from adipose tissue and peripheral FA availability may determine cardiac morphology during chronic exercise. During the development of pathological cardiac hypertrophy, inflammatory processes such as macrophage invasion and fibrotic remodeling occur. In contrast, exercised-induced LV mass increase was neither associated with cardiac fibrosis (Fig. 2E) nor with macrophage invasion assessed by expression of the macrophage marker Cd68 (Fig. 2F). More importantly, deletion of Atgl in WAT did not affect these processes (Fig. 2, E and F).
FIGURE 2.

Exercise-mediated changes in cardiac morphology/metabolism. A, representative images of the myocardial FA uptake ([18F]fluoro-4-thia-palmitate (18F-FTP)) (top 4 panels) and glucose uptake (2-[18F]fluorodeoxyglucose (18F-FDG) (bottom 4 panels), measured using small animal PET. Quantification of [18F]fluoro-4-thia-palmitate and 2-[18F]fluorodeoxyglucose PET, shown as percent change of mean sedentary myocardial uptake, was calculated as injection dose (%ID) relative to LVM (%ID/LVM) (n = 5; *, p < 0.05 versus WT (unpaired t test)). B, analysis of mRNA expression in LV tissue collected from trained mice immediately following their final running session, as a marker of lipid uptake and mitochondrial fatty acid oxidation. Quantitative RT-PCR studies were carried out using total RNA isolated from LV tissue. Data are presented as x-fold over WT mice. Cd36, Cluster of Differentiation 36; Fatp1, fatty acid transport protein 1; Acsl, acyl-CoA synthetase-1; Ppar-α, peroxisome proliferator-activated receptor α. *, p < 0.05 versus WT mice (n = 8; unpaired t test). C, cardiac adaptation expressed as percent change from mean sedentary LVM, as described previously (37). *, p < 0.05 versus WT mice (n = 9; unpaired t test). D, H&E -stained heart sections from WT and atATGL-KO mice. E, Picrosirius red-stained heart sections from WT and atATGL-KO mice (fibrosis), positive control (right image). LV samples were from mice with transverse aortic constriction (TAC) resulting in pathological cardiac hypertrophy. F, quantitative RT-PCR analysis of Cd68 mRNA expression levels measured in LVs of WT and atATGL-KO mice, as indicated.
TABLE 2.
Echocardiographic analysis of the mice
Echocardiographic analysis was performed after 4 weeks of training. Values are shown as means ± S.E. of the mean. LVIDd, LV internal diameter during diastole; HR, heart rate, EF, ejection fraction; sed, sedentary.
| WT sed | WT run | atATGL-KO sed | atATGL-KO run | |
|---|---|---|---|---|
| PWd, mm | 0.52 ± 0.007 | 0.58 ± 0.005a | 0.54 ± 0.007 | 0.56 ± 0.004 |
| IVSd, mm | 0.52 ± 0.007 | 0.59 ± 0.006b | 0.54 ± 0.007 | 0.59 ± 0.003c |
| LVIDd, mm | 4.20 ± 0.14 | 4.43 ± 0.07 | 4.08 ± 0.05 | 4.20 ± 0.02 |
| HR (beats/min) | 409 ± 10.1 | 394 ± 10.2 | 433 ± 11.2 | 440 ± 10.0d |
| EF% | 47 ± 2.7 | 43 ± 2.2 | 46 ± 1.6 | 45 ± 1.3 |
| LVM (mg) | 74.4 ± 4.5 | 92.9 ± 3.5b | 72.8 ± 2.1 | 81.2 ± 0.9d |
| LV/BW (mg/g) | 3.2 ± 0.17 | 4.2 ± 0.15b | 3.0 ± 0.05 | 3.4 ± 0.06e,f |
a p < 0.001 versus sedentary.
b p < 0.01 versus WT sedentary.
c p < 0.01 versus atATGL-KO sedentary.
d p < 0.05 versus WT run.
e p < 0.01 versus WT run.
f p < 0.05 versus atATGL-KO sedentary; two-way ANOVA (Bonferroni post test).
Recently published data from Riquelme et al. (5) indicated a prohypertrophic action of several FA species. The lack of an exercise-induced increase of circulating FA levels in atATGL-KO mice associated with the attenuated cardiac hypertrophic response suggests also a potential role of FAs as prohypertrophic mediators in our model. To investigate the role of FAs in nonproliferative cardiomyocyte growth, we next stimulated HL-1 cells with the FA mixture containing C14:0, C16:0, and C16:1n7, as described previously (5), and analyzed cellular hypertrophy (Fig. 3, A and B). The FA mixture, effectively induced cardiomyocyte hypertrophy in exercise-relevant serum concentrations (Fig. 3, A and B). Lack of C16:1n7 in this mixture resulted in the absence of pro-hypertrophic effects (Fig. 3, A and B). To identify the putative FA effector of hypertrophy development in our model, we performed a comprehensive serum analysis of circulating FAs using HPLC/MS measurements (Fig. 3C). Among the prohypertrophic FAs, only C16:1n7 serum levels followed a pattern corresponding to the observed cardiac phenotype (Fig. 3C). C16:1n7 levels increased significantly in WT mice after training but not in atATGL-KO mice (Fig. 3C). In contrast, C14:0 and C16:0 also increased in atATGL-KO mice (Fig. 3C). Despite the overall low serum concentration of C16:1n7 compared with other FAs, C16:1n7 has recently been identified as an adipose-derived hormone regulating biochemical and physiological processes in other organs such as skeletal muscle and liver (20). Thus, we hypothesized that C16:1n7 might be a crucial molecular co-mediator of cardiac hypertrophy in our model. To prove this, we next treated HL-1 cells with C16:1n7 alone or a FA mixture reflecting the serum FA composition (C16:0, C18:1, and C18:2), and we analyzed cellular hypertrophy by assessing the cell area (Fig. 3, D–F) and cellular protein content (Fig. 3G). C16:1n7 alone effectively induced nonproliferative cardiomyocyte growth in exercise-relevant serum concentrations, whereas this effect was absent in HL-1 cells stimulated with the FA mixture (Fig. 3, D and F). These data suggest that C16:1n7 liberated from adipose tissue may support the development of training-induced LVH. Moreover, the mRNA expression of the atrial natriuretic factor (Anf) and β-cardiac myosin heavy chain isogene (β-Mhch, both markers of pathological hypertrophy, was not increased under C16:1n7 stimulation indicating the induction of a physiological response (Fig. 3H).
FIGURE 3.

Hypertrophic response to C16:1n7. A, hypertrophic response of murine cardiac HL-1 cells to FAs; representative immunofluorescence images of HL-1 cells, treated with 100 nm endothelin 1 (Et-1) (B), FA mix: C14:0, C16:0, and C16:1n7 (D), or C14:0 and C16:0 (F), or vehicle control (A, C, and E) for 6 h (left panel). B, cell surface area calculated from the images of HL-1 cells, treated with the indicated compounds for 6 h. Data are presented as x-fold over control; ***, p < 0.001 versus control, one-way ANOVA (Bonferroni post-test) or unpaired t test. C, profile of selected serum lipids. Results represent serum samples obtained from sedentary (sed) and trained mice (n = 5), analyzed using rapid resolution HPLC/tandem MS (*, p < 0.05 versus WT sedentary; ***, p < 0.001 versus WT sedentary; °, p < 0.05) versus WT run (n = 5; two-way ANOVA (Bonferroni post-test)). D, hypertrophic response of murine cardiac HL-1 cells to C16:1n7; representative immunofluorescence images of HL-1 cells, treated with 100 nm endothelin 1 (B), C16:1n7 low (D)/high (F) concentration, C16:0; C18:1, and C18:2 (H), or vehicle controls (A, C, E, and G) for 6 h. E and F, cell surface area calculated from the images of HL-1 cells, treated with the indicated compounds for 6 h. Data are presented as x-fold over control (***, p < 0.001 versus control, one-way ANOVA (Bonferroni post-test) (E) or unpaired t test (F)). G, ratio of protein/DNA, n = 8; *, p < 0.05 versus control. H, analysis of Anf and β-Mhch chain isogene mRNA expression in cardiac HL-1 cells, treated with 100 nm endothelin 1, C16:1n7 low/high concentration, or vehicle control for 6 h. Anf, atrial natriuretic factor; β-Mhch, β-cardiac myosin heavy chain isogene. *, p < 0.05 versus control (n = 3; one-way ANOVA (Tukey's or Bonferroni post-test)).
A hallmark of the development of physiological cardiac hypertrophy is the short term activation of the cardiomyocytic serine/threonine protein kinase Akt. Short term treatment of HL-1 cells and primary human cardiomyocytes with C16:1n7 markedly stimulated Akt phosphorylation supporting the induction of an adaptive physiological hypertrophic response by C16:1n7 (Fig. 4, A and B). To further elicit the mechanism of action of C16:1n7, we analyzed cellular C16:1n7 appearance in HL-1 whole cells and in subcellular fractions (membrane, cytosolic, and nuclear) 6 h after C16:1n7 stimulation by using cell fractionation followed by HPLC/MS measurements (Fig. 4, C–E). Cells were washed multiple times after C16:1n7 stimulation to exclude contamination of cell pellets with the FA. As depicted in Fig. 4C, C16:1n7 stimulation led to a significant cellular accumulation of C16:1n7. Interestingly, C16:1n7 predominantly occurred in nuclear fractions, and modest induction of its occurrence was observed in cell membrane fractions (Fig. 4D). Together, these results suggest that C16:1n7 likely acts via Akt signaling and via an intracellular/nuclear mechanism.
FIGURE 4.

Cellular actions of C16:1n7. A, WB analysis of P-Akt and Akt of HL-1 cells; B, primary human cardiac myocytes stimulated with C16:1n7 for 30 min, as indicated. Right panels, pAkt/Akt ratio calculated based on 3–4 independent experiments using ImageJ software. *, p < 0.05 versus control; ***, p < 0.01 versus control, unpaired t test. C, C16:1n7 levels measured in HL-1 cells stimulated with C16:1 or vehicle for 6 h. Results represent cell pellet samples (n = 3), analyzed using rapid resolution HPLC/tandem MS; ***, p < 0.001 versus control, unpaired t test; D, C16:1n7 levels measured in the different cellular fractions of HL-1 cells, stimulated with C16:1 or vehicle for 6 h. Results represent fractionation samples: membrane (M), cytosol (C), and nuclear (N) fractions (n = 3), analyzed using rapid resolution HPLC/tandem MS. *, p < 0.05 versus control; unpaired t test. E, WB analysis of different cellular fractions isolated from HL-1 cells treated with C16n7 or vehicle for 6 h. MM-Marker, mitochondrial membrane marker; PM-Marker, plasma membrane marker; ER-Marker, endoplasmic reticulum marker. For details see under “Experimental Procedures.”
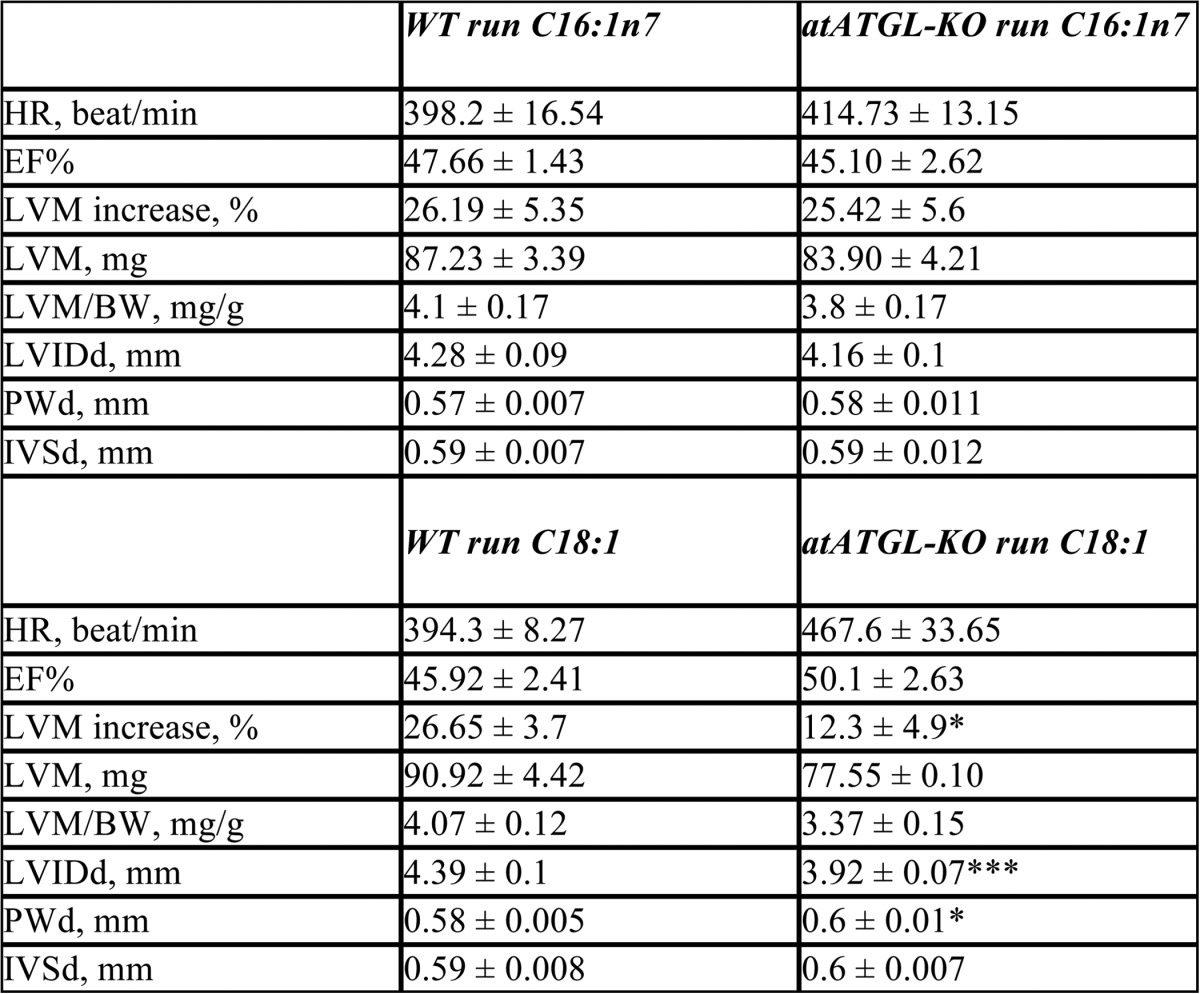
To further support a role of C16:1n7 as a co-regulator of training-induced cardiac hypertrophy in vivo, we assessed WT and atATGL-KO mice who either received dietary enrichment of C16:1n7 or C18:1 during their 4-week training regimen. Only dietary supplementation with C16:1n7 rescued the impaired exercise-induced cardiac hypertrophy in atATGL-KO mice to levels comparable with WT mice, whereas C18:1 supplementation did not affect the hypertrophic response in atATGL-KO mice (Fig. 5A and Table 3). These data suggest that the lack of C16:1n7 contributes to the cardiac phenotype observed in atATGL-KO mice after exercise. Importantly, C16:1n7 supplementation led to a consistent increase of C16:1n7 plasma levels during the training period in WT and atATGL-KO mice, which were not observed with C18:1 supplementation (Fig. 5B). We also analyzed AKT- phosphorylation in LV samples from supplemented, trained mice. Cardiac Akt phosphorylation in vivo was not regulated by FA supplementation in trained mice (data not shown).
FIGURE 5.
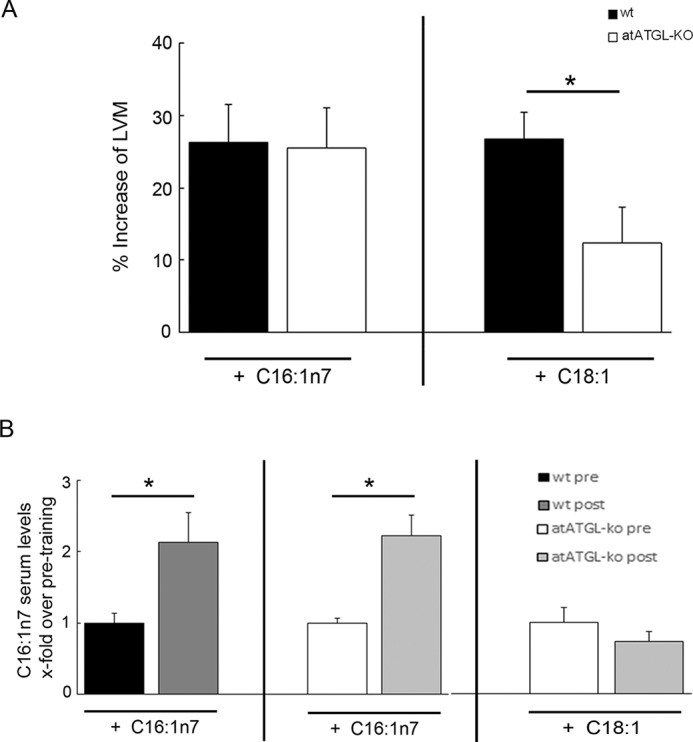
C16:1n7 supplementation in vivo. A, cardiac adaptation of mice supplemented with C16:1n7 or C18:1 during the exercise period. Cardiac adaptation is expressed as percent change from mean sedentary LVM, as described previously (37) (n = 4–5; *, p < 0.05 versus WT mice, unpaired t test). B, C16:1n7 serum levels in trained mice supplemented with C16:1n7 or C18:1 obtained before (pre) and (post) their final running sessions. Results represent serum samples (n = 5), analyzed using rapid resolution HPLC/tandem MS (*, p < 0.05 versus samples obtained before training (pre), unpaired t test).
TABLE 3.
Echocardiographic analysis of the mice supplemented with C16:1n7
Echocardiographic analysis was performed after 4 weeks of training. Values are shown as means ± S.E. of the mean; LVM increase was calculated as described for Fig. 2. LVIDd, LV internal diameter during diastole.
*p < 0.05 versus WT run.
***p < 0.001 versus WT run, unpaired t test.
For translation of our results to human physiology, we performed echocardiography and serum FA analysis (C16:1n7 and C18:2n6) in 25 male highly trained endurance athletes, and we analyzed the parameters of LVH, including diastolic interventricular septum thickness (IVSd), diastolic posterior wall thickness (PWd), end-diastolic left ventricle diameter, and LVM. Mean IVSd, PWd, end-diastolic left ventricle diameter, and LVM values (Table 4) indicated training-induced LVH in endurance athletes compared with previously studied control subjects (21). We observed a highly significant linear correlation between C16:1n7 serum level and IVSd (Fig. 6, upper left). The C16:1n7 level did not correlate with the PWd level (Fig. 6, middle left). A positive correlation between C16:1n7 and LVM could be detected but did not reach statistical significance (Fig. 6, lower left). To exclude that this correlation is a result from an increase in serum FA levels per se, we also studied C18:2n6 serum levels, a FA with highest serum concentrations in human serum samples. No correlation could be detected between C18:2n6 level and echocardiographic LVH parameters (Fig. 6, right) suggesting a specific association between C16:1n7 and IVSd.
TABLE 4.
Echocardiographic analysis and fatty acid levels measured in athletes
BSA is body surface area; IVSd is septum thickness during diastole. Values are shown as mean and Minimum-maximum values.
| Parameters | Mean | Minimum-maximum |
|---|---|---|
| n = 25 | ||
| Age (years) | 23 | 18–28 |
| Height (cm) | 181.5 | 170.4–188.5 |
| Weight (kg) | 76.7 | 61.4–88.3 |
| LVM (g) | 221.6 | 139.0–300.7 |
| LVM/BSA (g/m2) | 112.4 | 72.5–145.1 |
| IVSd (mm) | 9.7 | 7.0–12.5 |
| PWd (mm | 10.1 | 8.0–12.0 |
| LVEDd (mm) | 54.4 | 46.0–67.0 |
| C16:1n7 (palmitoleic acid) (μg/ml) | 27.1 | 13.8–49.7 |
| C18:2n6 (linoleic acid) (μg/ml) | 687.4 | 513.4–969.0 |
FIGURE 6.

LVH and serum FA level in endurance athletes. Echocardiographic analysis, including IVSd, PWd, and LVM, was performed in 25 endurance athletes. Echocardiography data were correlated to serum FA levels of C16:1n7 (left panel) and C18:2n6 (right panel), measured in the serum of athletes. r = Pearson's correlation coefficient.
Discussion
This study demonstrates for the first time that adipose tissue lipolysis regulated by adipose Atgl and associated with lipokine liberation (C16:1n7) promotes exercise-induced cardiac hypertrophy. The relevance of C16:1n7 as a pro-hypertrophic cofactor during training is further supported by a positive correlation between C16:1n7 serum level and echocardiographic LVH parameters in endurance athletes.
In consonance with the work of Ahmadian et al. (22), an impairment of Atgl-mediated lipolysis in WAT led to AT hypertrophy. Moreover, when subjected to chronic training, atATGL-KO mice exhibited an impaired AT lipolysis associated with the absence of exercise-induced plasma FFA appearance and reduced systemic lipid oxidation. Absence of adaptation of circulating FFAs to increased energy demands during exercise has been previously described in two other models of Atgl deficiency (23, 24). Schoiswohl et al. (24) demonstrated in an acute exercise model that mice overexpressing Atgl specifically in the heart, while lacking Atgl in all other tissues, exhibit significantly reduced appearance of plasma FFAs and glycerol after training. In contrast to our results, these authors also observed a significant decrease of plasma glucose in Atgl-deficient mice after training, likely resulting from the relatively short and acute form of exercise. Likewise, in a separate study, the global Atgl-KO also resulted in a reduction of maximal running velocity and endurance capacity underscoring the importance of this enzyme for regular substrate metabolism during exercise (23).
The effect of adipocyte-specific Atgl deletion on adaptive responses to chronic exercise training has not yet been previously examined. In this study, chronically trained atATGLKO mice exhibited diminished development of adaptive LVH and reduced capability of cardiac FA uptake. Thus, this is the first report investigating the lack of Atgl in AT and its impact on cardiac metabolism/function. In contrast, previous studies mainly focused on the role of myocardial Atgl. Kienesberger et al. (25) demonstrated that cardiomyocyte-specific overexpression of Atgl results in reduced myocardial TG content and an improved systolic function. These data are in line with Atgl loss-of-function studies showing a marked increase of cardiac TG accumulation and impairment of LV systolic function (26–28). In addition, Gao et al. (19) demonstrated that Atgl deficiency in neonatal rat cardiomyocytes leads to accumulation of ceramides, a process not observed in our model. More importantly, Haemmerle et al. (26) also investigated transgenic mice with Atgl exclusively expressed in cardiac muscle. The authors show that plasma FA and TG concentrations were much lower in those mice accompanied by an increase in cardiac Ppar-α and Ppar-δ target gene expression, suggesting that plasma lipid concentrations do not correlate with the activity level of the myocardial Ppar pathway and the severity of the cardiac phenotype. This is in contrast to our data clearly indicating a close relationship between plasma lipid fuel availability, cardiac uptake, phenotype, and gene expression. Accordingly, cardiac expression of the Ppar-α target gene Acsl1 (29) was down-regulated in atATGL-KO mice. WAT-Atgl mediates the release of systemic FA ligands for Ppar-α activation in other organs (22). Thus, suppression of WAT lipolysis in atATGL-KO mice results in reduced cardiac FA availability, which may attenuate cardiac Ppar-α activity and subsequent gene regulation. The discrepancy between observations by Haemmerle et al. (26) and our data may result from distinct experimental settings. Cardiac Atgl overexpression may induce a distinct cardiac metabolic program, which differs from hearts with physiological Atgl levels, as in case of the mice used in our study.
We identified C16:1n7, liberated by Atgl-mediated adipocyte lipolysis in response to training, as a molecular co-mediator of training-induced cardiac hypertrophy. In consonance with our data, Riquelme et al. (5) recently identified similar FAs as prohypertrophic factors in a different model of physiological postprandial cardiac hypertrophy. This study was not designed to prove that C16:1n7 alone is sufficient to directly induce cardiac hypertrophy. Rather our data support the notion that FAs may act as co-regulators of physiological responses in concert with other mediators in vivo (3).
C16:1n7 has been recently characterized as an adipose-derived lipid hormone regulating muscular insulin sensitivity and hepatic lipid metabolism (20). In this regard, C16:1n7 markedly potentiated Akt signaling in skeletal muscle (20). Cardiac Akt phosphorylation was not regulated by C16:1n7 during chronic administration in vivo. However, we show that the C16:1n7-mediated stimulation of cardiac cellular hypertrophy is accompanied by an induction of Akt phosphorylation in murine HL-1 cardiomyocytes and in primary human cardiomyocytes. Akt phosphorylation in vivo likely follows an intermittent pattern according to the training period (3). These characteristics make it difficult to analyze the Akt signal in vivo during chronic FA administration.
The upstream signaling pathway of C16:1n7-induced Akt phosphorylation and subsequent cardiomyocyte hypertrophy remains to be determined. Potential candidates include the G-protein-coupled receptor Gpr120, which signals through a Gαq-dependent pathway after C16:1n7 binding, or porcupine, a membrane-bound O-acyltransferase involved in the attachment of C16:1n7 to serine residues (30, 31). Lipid modification of serine residues has been recently identified as a pivotal processes in Wnt signaling, a pathway upstream of Akt, and involved in cardiac remodeling (31, 32). In addition, the marked occurrence of C16:1n7 in the cell nucleus 6 h after stimulation points toward a nuclear action of the FA. Palmitoleic acid has been shown to induce Ppar-δ transcriptional activity (33). Because cardiomyocytic Ppar-δ is an established regulator of cardiomyocyte morphology and function, this nuclear receptor might be a potential target of action of the C16:1n7 in the cell nucleus (34).
To prove the relevance of our results for human physiology, we demonstrated that the C16:1n7 serum level significantly correlates with echocardiographic LVH parameters in endurance athletes. Increased serum FFA levels have been previously documented during exercise (35). To exclude that LVH parameters in athletes correlate with increased FFA levels per se, we analyzed C18:2n6 showing no correlation to LVH. Chiefly, liberation of FFAs during prolonged exercise occurs for the maintenance of enhanced skeletal muscle and cardiac energy demands. Compared with C18:2n6 serum concentrations, C16:1n7 levels are very low making it unlikely that C16:1n7 serves as a major cardiac energy source. On the contrary, our data point toward an involvement of C16:1n7 in cardiac cellular growth, likely in concert with other mediators (3). We are aware that the observed correlation does not allow any causative conclusions; however, these data, at least in part, support the potential action of C16:1n7 on LV wall thickening.
Recently published studies pointed out that molecular effectors involved in beneficial adaptive cardiac growth may be used as therapeutic targets for maladaptive cardiovascular disease (3). Along this line, one may speculate about a potential therapeutic role of lipid-derived, e.g. C16:1n7-based, interventions. This is further supported by studies demonstrating a beneficial role of food-derived or supplemented other FAs such as ω-3 FAs in primary and/or secondary prevention of cardiovascular disease (36). Future studies will be required to fully understand the therapeutic options of C16:1n7 itself or key hepatic enzymes involved in de novo C16:1n7 synthesis and to develop a lipid-based therapy for cardiovascular diseases.
Collectively, our data provide the first evidence that adipose tissue lipolysis directly promotes the development of exercise-induced cardiac hypertrophy involving the lipokine C16:1n7 palmitoleate. The identification of a lipokine mediating physiological cardiac growth may help to develop future lipid-based therapies for pathological LVH or chronic heart failure.
Author Contributions
A. F. L. substantially contributed to conception and design of the study, acquisition of data, data analysis and interpretation, drafted the article, and revised the article critically for important intellectual content. M. C. K., M. R., and R. K. substantially contributed to conception of the study, acquisition of data, and data analysis and interpretation (M. C. K., PET analysis; M. R., HPLC/MS analysis; R. K., histological analysis). V. B., in vivo experiments; S. B., in vivo/in vitro experiments; E. S., in vitro experiments; Z. B., in vitro experiments; E. J., in vitro experiments; J.S., in vivo experiments; J. G., in vivo experiments; A. K. S., in vitro experiments; A. B., in vitro experiments; A. S., PET analysis; and K. B., human studies substantially contributed to acquisition of data, data analysis, and interpretation. M. H. and B. W. substantially contributed to conception of the human study and revised the article critically for important intellectual content. E. E. K. substantially contributed to conception and design of the study (in vivo experiments with atATGL-KO mice), data interpretation, and revised the article critically for important intellectual content. U. K. substantially contributed to acquisition of funding, conception, and design of the study, data analysis and interpretation, drafted the article, and revised the article critically for important intellectual content. All authors finally approved the version to be published.
Acknowledgments
We thank Christiane Sprang, Beata Hoeft, and Manuela Sommerfeld (Institute of Pharmacology, Charité-Universitaetsmedizin Berlin, CCR, Berlin, Germany) for their excellent technical support; Xiang Li, Katja Hirsch (Dept. of Nuclear Medicine, University Hospital Wuerzburg, Wuerzburg, Germany), and Ina Israel for the help with PET experiments; and Stefan Anker (Charité-Universitaetsmedizin Berlin, CCR, Germany) for using the NMR device (Brucker's MiniSpec MQ10).
This work was supported by Deutsche Forschungsgemeinschaft Grant FG 1054/2 (Ki712/5-2). This work was also supported in part by National Institutes of Health Grant R01DK090166 (to E. E. K.). M. R. is an employee of Lipidomix GmbH, Berlin, Germany.
- LV
- left ventricle
- LVM
- left ventricular mass
- FA
- fatty acid
- PWd
- posterior wall thickness
- LVH
- left ventricular hypertrophy
- ATGL
- adipose-specific adipose triglyceride lipase
- AT
- adipose tissue
- WAT
- white adipose tissue
- FFA
- free fatty acid
- TG
- triacylglycerol
- PPAR
- peroxisome proliferator-activated receptor
- ANOVA
- analysis of variance
- IVSd
- diastolic interventricular septum thickness
- WB
- Western blot
- Ab
- antibody.
References
- 1. Rawlins J., Bhan A., Sharma S. (2009) Left ventricular hypertrophy in athletes. Eur. J. Echocardiogr. 10, 350–356 [DOI] [PubMed] [Google Scholar]
- 2. Heineke J., Molkentin J. D. (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600 [DOI] [PubMed] [Google Scholar]
- 3. Maillet M., van Berlo J. H., Molkentin J. D. (2013) Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 14, 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson D., Karpe F., Lafontan M., Frayn K. (2012) Physical activity and exercise in the regulation of human adipose tissue physiology. Physiol. Rev. 92, 157–191 [DOI] [PubMed] [Google Scholar]
- 5. Riquelme C. A., Magida J. A., Harrison B. C., Wall C. E., Marr T. G., Secor S. M., Leinwand L. A. (2011) Fatty acids identified in the Burmese python promote beneficial cardiac growth. Science 334, 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Young S. G., Zechner R. (2013) Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev. 27, 459–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sitnick M. T., Basantani M. K., Cai L., Schoiswohl G., Yazbeck C. F., Distefano G., Ritov V., DeLany J. P., Schreiber R., Stolz D. B., Gardner N. P., Kienesberger P. C., Pulinilkunnil T., Zechner R., Goodpaster B. H., et al. (2013) Skeletal muscle triacylglycerol hydrolysis does not influence metabolic complications of obesity. Diabetes 62, 3350–3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kershaw E. E., Hamm J. K., Verhagen L. A. W., Peroni O., Katic M., Flier J. S. (2006) Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes 55, 148–157 [PMC free article] [PubMed] [Google Scholar]
- 9. Foryst-Ludwig A., Kreissl M. C., Sprang C., Thalke B., Böhm C., Benz V., Gürgen D., Dragun D., Schubert C., Mai K., Stawowy P., Spranger J., Regitz-Zagrosek V., Unger T., Kintscher U. (2011) Sex differences in physiological cardiac hypertrophy are associated with exercise-mediated changes in energy substrate availability. Am. J. Physiol. Heart Circ. Physiol. 301, H115–H122 [DOI] [PubMed] [Google Scholar]
- 10. Fliegner D., Schubert C., Penkalla A., Witt H., Kararigas G., Kararigas G., Dworatzek E., Staub E., Martus P., Ruiz Noppinger P., Kintscher U., Gustafsson J. A., Regitz-Zagrosek V. (2010) Female sex and estrogen receptor-β attenuate cardiac remodeling and apoptosis in pressure overload. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1597–R1606 [DOI] [PubMed] [Google Scholar]
- 11. Bolsoni-Lopes A., Festuccia W. T., Farias T. S., Chimin P., Torres-Leal F. L., Derogis P. B., de Andrade P. B., Miyamoto S., Lima F. B., Curi R., Alonso-Vale M. I. (2013) Palmitoleic acid (n-7) increases white adipocyte lipolysis and lipase content in a PPARα-dependent manner. Am. J. Physiol. Endocrinol. Metab. 305, E1093–E1102 [DOI] [PubMed] [Google Scholar]
- 12. DeGrado T. R., Kitapci M. T., Wang S., Ying J., Lopaschuk G. D. (2006) Validation of 18F -fluoro-4-thia-palmitate as a PET probe for myocardial fatty acid oxidation: effects of hypoxia and composition of exogenous fatty acids. J. Nucl. Med. 47, 173–181 [PubMed] [Google Scholar]
- 13. Loening A. M., Gambhir S. S. (2003) AMIDE: a free software tool for multimodality medical image analysis. Mol. Imaging 2, 131–137 [DOI] [PubMed] [Google Scholar]
- 14. Weischenfeldt J., Porse B. (2008) Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. 2008, pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- 15. Claycomb W. C., Lanson N. A. Jr., Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., Izzo N. J. Jr. (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 95, 2979–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boon R. A., Iekushi K., Lechner S., Seeger T., Fischer A., Heydt S., Kaluza D., Tréguer K., Carmona G., Bonauer A., Horrevoets A. J., Didier N., Girmatsion Z., Biliczki P., Ehrlich J. R., et al. (2013) MicroRNA-34a regulates cardiac ageing and function. Nature 495, 107–110 [DOI] [PubMed] [Google Scholar]
- 17. Lang R. M., Bierig M., Devereux R. B., Flachskampf F. A., Foster E., Pellikka P. A., Picard M. H., Roman M. J., Seward J., Shanewise J. S., Solomon S. D., Spencer K. T., Sutton M. S., Stewart W. J. (2005) Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J. Am. Soc. Echocardiogr. 18, 1440–1463 [DOI] [PubMed] [Google Scholar]
- 18. Devereux R. B., Alonso D. R., Lutas E. M., Gottlieb G. J., Campo E., Sachs I., Reichek N. (1986) Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am. J. Cardiol. 57, 450–458 [DOI] [PubMed] [Google Scholar]
- 19. Gao H., Feng X. J., Li Z. M., Li M., Gao S., He Y. H., Wang J. J., Zeng S. Y., Liu X. P., Huang X. Y., Chen S. R., Liu P. Q. (2015) Downregulation of adipose triglyceride lipase promotes cardiomyocyte hypertrophy by triggering the accumulation of ceramides. Arch. Biochem. Biophys. 565, 76–88 [DOI] [PubMed] [Google Scholar]
- 20. Cao H., Gerhold K., Mayers J. R., Wiest M. M., Watkins S. M., Hotamisligil G. S. (2008) Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 134, 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pluim B. M., Zwinderman A. H., van der Laarse A., van der Wall E. E. (2000) The athlete's heart. A meta-analysis of cardiac structure and function. Circulation 101, 336–344 [DOI] [PubMed] [Google Scholar]
- 22. Ahmadian M., Abbott M. J., Tang T., Hudak C. S., Kim Y., Bruss M., Hellerstein M. K., Lee H. Y., Samuel V. T., Shulman G. I., Wang Y., Duncan R. E., Kang C., Sul H. S. (2011) Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 13, 739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huijsman E., van de Par C., Economou C., van der Poel C., Lynch G. S., Schoiswohl G., Haemmerle G., Zechner R., Watt M. J. (2009) Adipose triacylglycerol lipase deletion alters whole body energy metabolism and impairs exercise performance in mice. Am. J. Physiol. Endocrinol. Metab. 297, E505–E513 [DOI] [PubMed] [Google Scholar]
- 24. Schoiswohl G., Schweiger M., Schreiber R., Gorkiewicz G., Preiss-Landl K., Taschler U., Zierler K. A., Radner F. P., Eichmann T. O., Kienesberger P. C., Eder S., Lass A., Haemmerle G., Alsted T. J., Kiens B., et al. (2010) Adipose triglyceride lipase plays a key role in the supply of the working muscle with fatty acids. J. Lipid Res. 51, 490–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kienesberger P. C., Pulinilkunnil T., Sung M. M., Nagendran J., Haemmerle G., Kershaw E. E., Young M. E., Light P. E., Oudit G. Y., Zechner R., Dyck J. R. (2012) Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol. Cell. Biol. 32, 740–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haemmerle G., Moustafa T., Woelkart G., Büttner S., Schmidt A., van de Weijer T., Hesselink M., Jaeger D., Kienesberger P. C., Zierler K., Schreiber R., Eichmann T., Kolb D., Kotzbeck P., Schweiger M., et al. (2011) ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 17, 1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zierler K. A., Jaeger D., Pollak N. M., Eder S., Rechberger G. N., Radner F. P., Woelkart G., Kolb D., Schmidt A., Kumari M., Preiss-Landl K., Pieske B., Mayer B., Zimmermann R., Lass A., et al. (2013) Functional cardiac lipolysis in mice critically depends on comparative gene identification-58. J. Biol. Chem. 288, 9892–9904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kienesberger P. C., Pulinilkunnil T., Nagendran J., Young M. E., Bogner-Strauss J. G., Hackl H., Khadour R., Heydari E., Haemmerle G., Zechner R., Kershaw E. E., Dyck J. R. (2013) Early structural and metabolic cardiac remodelling in response to inducible adipose triglyceride lipase ablation. Cardiovasc. Res. 99, 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schoonjans K., Watanabe M., Suzuki H., Mahfoudi A., Krey G., Wahli W., Grimaldi P., Staels B., Yamamoto T., Auwerx J. (1995) Induction of the acyl-coenzyme A synthetase gene by fibrates and fatty acids is mediated by a peroxisome proliferator response element in the C promoter. J. Biol. Chem. 270, 19269–19276 [DOI] [PubMed] [Google Scholar]
- 30. Glass C. K., Olefsky J. M. (2012) Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 15, 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takada R., Satomi Y., Kurata T., Ueno N., Norioka S., Kondoh H., Takao T., Takada S. (2006) Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev. Cell 11, 791–801 [DOI] [PubMed] [Google Scholar]
- 32. Bergmann M. W. (2010) WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ. Res. 107, 1198–1208 [DOI] [PubMed] [Google Scholar]
- 33. Brown J. D., Oligino E., Rader D. J., Saghatelian A., Plutzky J. (2011) VLDL hydrolysis by hepatic lipase regulates PPARδ transcriptional responses. PLoS ONE 6, e21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng L., Ding G., Qin Q., Huang Y., Lewis W., He N., Evans R. M., Schneider M. D., Brako F. A., Xiao Y., Chen Y. E., Yang Q. (2004) Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 10, 1245–1250 [DOI] [PubMed] [Google Scholar]
- 35. Mittendorfer B., Horowitz J. F., Klein S. (2002) Effect of gender on lipid kinetics during endurance exercise of moderate intensity in untrained subjects. Am. J. Physiol. Endocrinol. Metab. 283, E58–E65 [DOI] [PubMed] [Google Scholar]
- 36. Kromhout D., de Goede J. (2014) Update on cardiometabolic health effects of ω-3 fatty acids. Curr. Opin. Lipidol. 25, 85–90 [DOI] [PubMed] [Google Scholar]
- 37. Konhilas J. P., Maass A. H., Luckey S. W., Stauffer B. L., Olson E. N., Leinwand L. A. (2004) Sex modifies exercise and cardiac adaptation in mice. Am. J. Physiol. Heart Circ. Physiol. 287, H2768–H2776 [DOI] [PMC free article] [PubMed] [Google Scholar]