

Background: AICAR is a potent anti-proliferative compound, but the basis of its cytotoxicity is poorly understood.
Results: AICAR affects NTP homeostasis in a carbon source-dependent way, in both yeast and human cells.
Conclusion: AICAR balance with nucleotides triphosphate is critical for its in vivo effects.
Significance: AICAR is significantly more cytotoxic on glucose and thus potentially targets cells prone to Warburg effect.
Keywords: nucleoside/nucleotide analog, nucleoside/nucleotide metabolism, purine, yeast genetics, yeast metabolism
Abstract
5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside monophosphate (AICAR) is a natural metabolite with potent anti-proliferative and low energy mimetic properties. At high concentration, AICAR is toxic for yeast and mammalian cells, but the molecular basis of this toxicity is poorly understood. Here, we report the identification of yeast purine salvage pathway mutants that are synthetically lethal with AICAR accumulation. Genetic suppression revealed that this synthetic lethality is in part due to low expression of adenine phosphoribosyl transferase under high AICAR conditions. In addition, metabolite profiling points to the AICAR/NTP balance as crucial for optimal utilization of glucose as a carbon source. Indeed, we found that AICAR toxicity in yeast and human cells is alleviated when glucose is replaced by an alternative carbon source. Together, our metabolic analyses unveil the AICAR/NTP balance as a major factor of AICAR antiproliferative effects.
Introduction
Purine nucleotides are key metabolites that are efficiently interconverted resulting in fine-tuned balances between adenylic and guanylic derivatives but also between mono-, di-, and triphosphate species. In particular, the AMP to ATP ratio is a major metabolic signal informing the cell about its energy state. When energy is required, ATP is consumed, and the resulting ADP is used to replenish the ATP pool in part via the adenylate kinase reaction that generates AMP and ATP from two molecules of ADP. This reaction does not require exogenous energy and can thus rapidly refill the ATP pool upon demand. However, this reaction is limited by the size of the AXP pool and rapidly leads to ADP depletion and AMP accumulation. Increased AMP concentration is perceived as a low energy signal and contributes to redirect the metabolism to catabolism (energy providing reactions) notably via activation of the AMP-kinase (AMPK).4 Metabolic transitions can therefore be experimentally triggered by modifying the AMP/ATP ratio. However, AMP accumulation is difficult to control experimentally, and an AMP mimic molecule named AICAR (Fig. 1A), which can be provided as a riboside precursor form (AICAr; Fig. 1A) in the growth medium, is often used as low energy mimetic. AICAR clearly activates AMPK in vitro and in vivo but also shows many effects that are not AMPK-dependent (1, 2). Whether these effects are the result of AMP mimicry or not is unknown. The yeast AMPK ortholog Snf1 is activated by ADP rather than AMP (3), and consistently the yeast AICAR transcriptional signature did not include genes described as strongly regulated by Snf1p (4). Yeast therefore is a good experimental system to study the effects of AICAR that are AMPK-independent. Identification of AMPK-independent targets of AICAR is crucial because it was recently established that the highly promising antiproliferative properties of AICAR are largely AMPK-independent (5, 6). Importantly, AICAR cytotoxic effects were higher on trisomic mouse embryonic fibroblasts than on their euploid counterpart (7). This property makes AICAR a promising anti-tumor molecule because most solid tumor cells are aneuploid, i.e. have an incorrect number of chromosomes.
FIGURE 1.
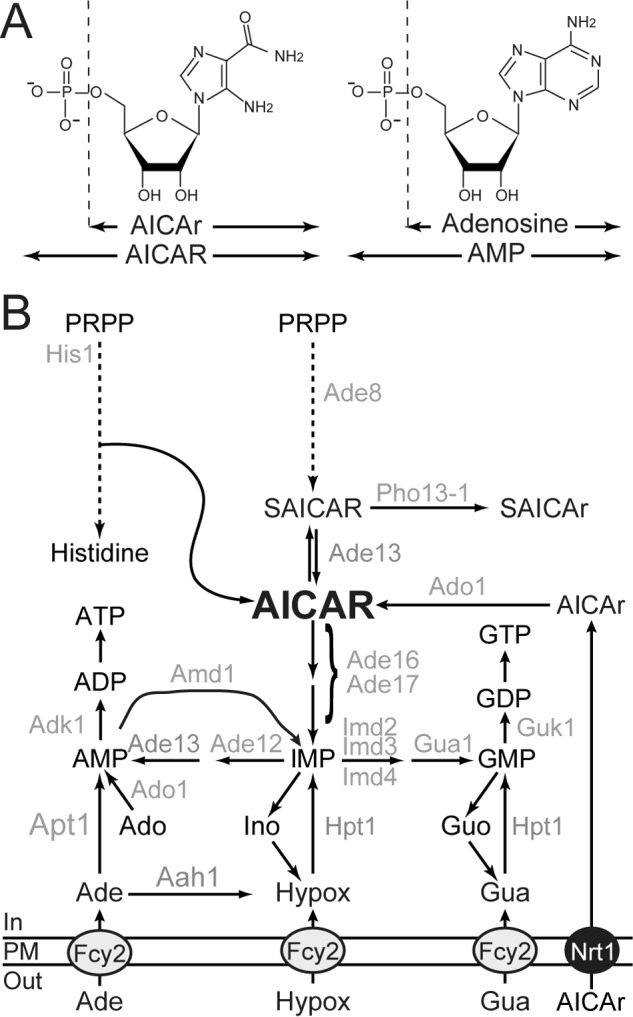
Schematic representation of purine and histidine pathways in yeast. A, chemical structures of AICAR, AMP, and their riboside derivatives. B, only the enzymes mentioned in the text are indicated. Ade, adenine; Ado, adenosine; Guo, guanosine; Hypox, hypoxanthine; Ino, inosine; PM, plasma membrane; PRPP, 5-phosphoribosyl-1-pyrophosphate; SAICAr, succinyl-AICAr; SAICAR, succinyl-AICAR; S-AMP, succinyl-AMP.
As for mammalian cells, AICAR can be cytotoxic for yeast cells (8). Understanding the molecular basis of AICAR toxicity in yeast can thus give important clues on its effects in other eukaryotic cells. However, it should be stressed that, by opposition to mammalian cells, simple addition of AICAr to the growth medium does not result in AICAR monophosphate accumulation in yeast unless the ADE16 ADE17 genes, encoding AICAR transformylase IMP-cyclohydrolase, are mutated (Fig. 1B) (5). This most probably reflects the fact that yeast AICAR transformylase IMP-cyclohydrolase very efficiently metabolizes AICAR in vivo as discussed in Ref. 5. Thus, studies of AICAR effects in yeast necessitate adequate genetic backgrounds affecting synthesis and/or utilization of AICAR. We have previously shown that AICAR can be detoxified in its riboside form (AICAr) in an adenosine kinase mutant (ado1) unable to resynthesize AICAR from AICAr (9) (Fig. 1B). We also identified a mutant form of the phosphatase Pho13, named Pho13-1, which can detoxify succinyl-AICAR to succinyl-AICAR riboside and thereby diminish intracellular AICAR concentration (9) (Fig. 1B). Together, these results established that the riboside forms of AICAR and succinyl-AICAR are not toxic for yeast cells, a situation highly similar to what happens in mammalian cells where the vast majority of AICAR effects are abolished by adenosine kinase inhibitors (1). Thus, for both yeast and mammalian cells, it was concluded that AICAr riboside had to be metabolized to the monophosphate form to become active and cytotoxic. Transcriptomic analyses in yeast (4, 9) did not give any obvious key for AICAR toxicity, and thus the molecular mechanisms are still to be uncovered. To address this question, we took advantage of yeast genetics to isolate mutants unable to grow under conditions where AICAR accumulates. Here, we describe such yeast mutants and connect their AICAR sensitivity to their metabolite profile. Our results point to the AICAR/NTP balance as a crucial feature of AICAR toxicity. Importantly, we established that AICAR toxicity is tightly linked to growth on glucose as a carbon source and can be largely alleviated by alternative carbon sources both in yeast and in mammalian cells. This glucose-specific toxicity of AICAR could potently contribute to its antitumor properties because most cancer cells exclusively rely on glucose metabolism for energy production.
Experimental Procedures
Yeast Media, Strains, and Plasmids
SD is a synthetic minimal medium containing 0.5% ammonium sulfate, 0.17% yeast nitrogen base (Difco), 2% glucose. SDcasaW is SD medium supplemented with 0.2% casamino acids (Difco) and tryptophan (0.2 mm). When indicated, adenine (0.3 mm), guanosine (1 mm), hypoxanthine (0.3 mm), inosine (1 mm), and/or uracil (0.3 mm) was added in SDcasaW medium, resulting in media named SDcasaWA (+ adenine), SDcasaWHypox (+ hypoxanthine), SDcasaWAU (+ adenine + uracil), and SDcasaWUHypox (+ hypoxanthine + uracil). Synthetic medium containing different carbon source correspond to SD medium in which glucose has been replaced by 2% of the indicated carbon source. SC medium was prepared as described by Sherman et al. (10). SC complete medium is SC medium supplemented with adenine (0.3 mm), uracil (0.3 mm), histidine (0.06 mm), leucine (0.4 mm), lysine (0.06 mm), and tryptophan (0.2 mm). YPD medium is a complete medium containing 1% yeast extract, 2% bactopeptone, and 2% glucose. Yeast strains (Table 1) belong to, or are derived from, a set of disrupted strains isogenic to BY4741 or BY4742 purchased from Euroscarf. Multimutant strains were obtained by crossing, sporulation, and micromanipulation of meiosis progeny. Plasmids allowing overexpression of AAH1 (p2088), ADE12 (p1032), ADE13 (p1971), ADK1 (p2151 (11)), AMD1 (p2479 (12)), APT1 (p2091 (12)), HPT1 (p2149 (13)), GUA1 (p2141), GUK1 (p1814), IMD2 (p2093), IMD3 (p2100), IMD4 (p2102), and PHO13-1 (p4291 (9)) were obtained by cloning in the pCM189 vector (tetProm, CEN, URA3) (14). Cloning details for the unpublished plasmids are available upon request.
TABLE 1.
List of yeast strains used in this study
| Strain name | Genotype |
|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 |
| Y178 | MATα his3Δ1 leu2Δ0 ura3Δ0 |
| Y1095 | MATα ade16::kanMX4 ade17::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y1127 | MATa hpt1::KanMX4 his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| Y1759 | MATα apt1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y2080 | MATa aah1::KanMX4 amd1::KanMX4 his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| Y2340 | MATa aah1::kanMX4 ade8::kanMX4 his3Δ1 leu2 Δura3Δ0 |
| Y2950 | MATα ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y3578 | MATα adk1::kanMX4 amd1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y4479 | MATα his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y4499 | MATa ade16::kanMX4 ade17::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y5864 | MATa hpt1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y5868 | MATα hpt1::kanMX4 ade16::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y5870 | MATα ade16::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y5873 | MATα hpt1::kanMX4 ade17::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y5876 | MATα ade17::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y6750 | MATa aah1::kanMX4 his3Δ1 leu2Δ ra3Δ0 |
| Y6986 | MATα ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y7205 | MATα aah1–308 ade16::kanMX4 ade17::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 [tet-PHO13–1] |
| Y7314 | MATα pdc2 (L234S) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y7320 | MATα ade13 (G166S) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y7321 | MATα thi3 (S402F)ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y8480 | MATa ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y8793 | MATa ade16::kanMX4 ade17::kanMX4 aah1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y8845 | MATα thi80 (L90P) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y9303 | MATα aah1–308 ade16::kanMX4 ade17::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y9798 | MATα aah1::kanMX4 ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y10314 | MATa APT1-GFP-HIS3 his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 |
| Y10351 | MATa ade16::kanMX4 ade17::kanMX4 APT1-GFP-HIS3 his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 |
| Y10399 | MATa ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 kcs1:: kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
Isolation of AICAR-sensitive Mutants
To obtain AICAR-sensitive mutants, an ade16 ade17 strain (Y4999), transformed with a plasmid expressing the PHO13-1 dominant allele under the control of a tetracycline-repressible promoter, was plated on SDcasaWA medium, mutagenized with UV-light for 40 s, and then grown for 48 h at 30 °C. The resulting clones were then transferred by replica-plating on the same medium alone or medium containing doxycycline (10 mg/liter) to repress PHO13-1 expression. AICAR-sensitive mutants were isolated as clones unable to grow in the presence of doxycycline but growing in the absence of the antibiotic.
Metabolite Extraction and Separation by Liquid Chromatography
Extraction of yeast and mammalian cells metabolites was performed as described (5), and metabolite separation was performed on an ICS3000 chromatography station (Dionex, Sunnyvale, CA) using a carbopac PA1 column (250 × 2 mm; Dionex) with the 50–800 mm acetate gradient in 50 mm NaOH described in Ref. 15. Peaks were identified by their retention time, as well as by co-injection with standards and/or their UV spectrum signature.
Mammalian Cells
HeLa cells (ATCC, CCL-2) were propagated in DMEM, 4.5 g/liter glucose, 10% FCS, l-glutamine, and penicillin/streptomycin. Cells were then trypsinized and washed twice by centrifugation in glucose-free DMEM (Gibco Life Technologies catalog no. 11966-025) containing 10% dialyzed FCS and 1 mm pyruvate. They were then seeded at the density of 30,000 cells/cm2 in culture dishes that have been precoated for 2 h with 5 μg/ml fibronectin (Sigma catalog no. F4759) in PBS. The growth medium consisted of DMEM without glucose supplemented either with 5 mm glucose or 5 mm galactose and containing 10% dialyzed FCS, 1 mm pyruvate, 5 μg/ml insulin, 10 ng/ml EGF, 2 ng/ml FGF-2, and penicillin/streptomycin. After 24 h, AICAr was added to the culture medium, and the cells were further allowed to grow for 48 h. AICAr toxicity was determined by counting cells with a Multisizer 4 Coulter counter (Beckman).
Enzymatic Activities
For enzymatic activities, all measurements were done in initial rate conditions that were stable for at least 3 min at 30 °C for all enzymes tested. A potential effect of AICAR on enzymatic activities was tested by preincubating or not the protein extracts with AICAR (1 mm) for 2 min before substrate addition. Finally, all enzymatic reactions were stopped by boiling (3 min at 80 °C) a 100-μl aliquot of the reaction mix in 900 μl of ethanol/10 mm Hepes, pH 7 (4/1, v/v). Quantification of the reaction product(s) was done by liquid chromatography in the conditions described for intracellular metabolites determination. AMP deaminase enzymatic activity was measured by a method adapted from (16). Briefly, a total yeast protein extract was obtained from adk1 amd1 mutant cells (Y3578) overexpressing (p2479) or not (pCM189) AMD1 and grown in SDcasaWA medium (100 ml, 5 × 107 cells/ml). After harvesting, cells were resuspended in 500 μl of buffer A (50 mm triethanolamine, pH 7, 0.1 m KCl, and 100 μm dithiothreitol) and were disrupted by vortexing with glass beads (1 min, three times). The extract was then dialyzed twice against 2 liters of buffer A containing 10% (m/v) glycerol. Enzymatic activity was measured in 500 μl of buffer A containing AMP (100 μm), ATP (100 μm), and the protein extract (250 μg, added at time 0). AMP deaminase specific activity corresponds to nmol IMP synthetized/min/mg of protein. No IMP synthesis was detected in a protein extract obtained from the strain containing the pCM189 empty vector. Adenine phosphoribosyl transferase activity was determined using a total protein extract from an aah1 amd1 mutant strain (Y2080) containing a plasmid allowing overexpression of APT1 (p2091), as described in Ref. 17 but with modifications. Yeast cells grown in SDcasaW medium were harvested (100 ml; 5 × 107 cells/ml) and disrupted in 500 μl of 50 mm Tris/HCl, pH 7.5, containing 5 mm MgCl2, 20 mm KCl, and 10% glycerol (buffer B). The suspension was then centrifuged for 1 h at 21,000 × g and 4 °C, and the supernatant was dialyzed twice against 2 liters of buffer B. Enzymatic activity was measured in 500 μl of 50 mm Tris/HCl, pH 7.4, containing 5 mm MgCl2, 0.5 mg/ml BSA, 100 μm phosphoribosyl pyrophosphate, 10, 50 or 100 μm adenine, 0 or 1 mm AICAR, and 12 μg of protein extract (added at time 0). Adenine phosphoribosyl transferase specific activity was defined as nmol of AMP synthetized/min/mg of protein. No AMP synthesis was detected in the absence of phosphoribosyl pyrophosphate. Adenylate kinase (Adk) activity was measured on Saccharomyces cerevisiae purified protein (Merck Millipore catalog no. 474951) with a method derived from Ref. 18. The reaction mixture (500 μl) contained 100 mm Tris/HCl, pH 7.5, 0.1 m KCl, 5 mm MgCl2, 2 mm ADP, and 10 ng of purified Adk. Specific activity was defined as the amount of ATP synthetized/min/mg of purified protein.
Miscellaneous Methods
Yeast growth drop test were performed as described in Ref. 12.
Results
Mutations in the AAH1 Gene Are Synthetically Lethal with ade16 ade17 Mutations Resulting in AICAR Accumulation in Vivo
To isolate mutants which are affected by AICAR accumulation, we started from an ade16 ade17 double mutant that constitutively accumulates AICAR (∼ 1 mm compared with ∼1 μm in the wild-type strain (4)). In this double mutant strain, we introduced a plasmid carrying the PHO13-1 gene under control of a doxycycline repressible promoter. When expressed, PHO13-1 decreases AICAR intracellular concentration (9). We then isolated mutants that grew when PHO13-1 was expressed (low intracellular AICAR) but were unable to grow in the presence of doxycycline (high intracellular AICAR). One of these AICAR-sensitive mutants (Fig. 2A) was further studied. This mutant showed an atypical metabolite profile characterized by the massive intracellular accumulation of adenine, S-adenosylmethionine (AdoMet) and S-adenosylhomocysteine (SAH) (Fig. 2B). The intracellular adenine accumulation was particularly intriguing because this metabolite is usually hardly detectable, most probably as a result of its active utilization by adenine deaminase (Aah1) and adenine phosphoribosyltransferase (Apt1) (Fig. 1B). Adenine accumulation suggested that APT1 or AAH1 could be affected in this mutant strain. Metabolite profiling of apt1 and aah1 single knock-out mutants in the same conditions revealed that only the aah1 mutant accumulates high levels of adenine, AdoMet, and SAH (Fig. 2C, red lines). Together, these results suggested that AAH1 could be the mutated gene resulting in the synthetic lethality when combined with the ade16 ade17 mutations. Accordingly, sequencing of the AAH1 locus in this mutant revealed a stop codon at position 308, resulting in a protein lacking the 39 C-terminal residues. As expected, the mutant could grow in the absence of the tet-PHO13-1 plasmid on hypoxanthine (Fig. 2D), a purine source that bypasses the requirement for Aah1 (Fig. 1B), and the synthetic lethality of the mutant on adenine medium was complemented by the AAH1 gene carried on a plasmid (Fig. 2E). Finally, a reconstituted ade16 ade17 aah1 triple knock-out mutant was unable to grow on adenine but grew perfectly well on hypoxanthine (Fig. 2F), just as the original mutant from the screen (Fig. 2D). As expected, accumulation of adenine, SAH, and SAM was resumed in the reconstituted mutant (data not shown). In addition, we observed that this mutant had ATP and GTP pools lower by 30 and 60%, respectively (see next section). We conclude that ade16 ade17 and aah1 are synthetically lethal specifically on adenine-containing medium.
FIGURE 2.

AICAR toxicity is strongly increased in an adenine deaminase mutant using adenine as an external purine source. A, the AICAR-sensitive mutant (Y7205) and the cognate reference strain (Y4499), both containing the Tet-OFF PHO13-1 expression plasmid (P4291), were grown overnight, serial diluted, and spotted on SDcasaWA medium with or without doxycycline (Dox, 10 mg/l). B, strains from Fig. 2A (wild type, blue line; mutant, red line) were kept in the exponential phase in SDcasaWA medium for 24 h and were then incubated with doxycycline (10 mg/liter) for 4 h prior to metabolite extraction and separation. Asterisks indicate unidentified peaks found in the control strain that do not correspond to SAH. C, metabolite content was determined from wild type (WT, Y178, blue line), aah1 (Y6750, red line), and apt1 (Y1759, green line) cells grown in SDcasaWAU medium. D, the ade16 ade17 (AAH1, Y4499) and ade16 ade17 aah1-308 (aah1-308, Y9303) strains were grown overnight, serial diluted, and spotted on SDcasaW medium containing either adenine or hypoxanthine as an extracellular purine source. E, plasmids expressing (P2088) or not (control, pCM189) the AAH1 gene were transformed in the ade16 ade17 aah1-308 (aah1-308, Y9303) strain, and transformants were serial diluted and spotted on SDcasaW medium containing either adenine or hypoxanthine. F, the ade16 ade17 aah1 (Y8793) and ade8 aah1 (Y2340) strains were transformed with either the tet-PHO13-1 expression plasmid (P4291) or the pCM189 empty vector (Control). Transformants were grown overnight, serial diluted, and spotted on SDcasaW medium supplemented with either adenine or hypoxanthine, as an external purine source. G, mutant cells (ade16 ade17 ade8 his1 (Y2950); aah1 ade16 ade17 ade8 his1 (Y9798)) were grown overnight, serial diluted, and spotted on SDcasaWAU medium with or without external AICAr (5 mm). In A and D–G, the plates were imaged after 2 days at 30 °C.
Importantly, this synthetic lethality of ade16 ade17 aah1 on adenine was indeed caused by AICAR accumulation because an ade8 aah1 mutant showed no growth impairment, and the overexpression of PHO13-1 restored growth of the triple ade16 ade17 aah1 mutant (Fig. 2F). Finally, an aah1 ade16 ade17 ade8 his1 mutant, unable to produce endogenous AICAR, grew perfectly well on adenine unless AICAr riboside was added to the growth medium (Fig. 2G). Importantly, the ade16 ade17 ade8 his1 strain carrying the wild-type AAH1 allele was able to grow in the presence of AICAr (Fig. 2G). We conclude that AICAR accumulation enables proliferation when Aah1 is not functional.
AICAR Affects ATP and GTP Intracellular Concentration
The low ATP and GTP pools, found in the aah1 ade16 ade17 mutant, suggested that AICAR affected the purine interconversion pathway. Because AICAR can act as an AMP mimetic on AMPK, we reasoned that it could affect enzymes responsible for synthesis (Apt1) or consumption (Adk1, Amd1) of AMP (Fig. 1B). However, in vitro enzymatic assays showed no major effect of AICAR on Apt1, Adk1, and Amd1 specific activities (Fig. 3A). Although Apt1 enzymatic activity was not affected by AICAR in vitro, we found that APT1 overexpression robustly suppressed the aah1 ade16 ade17 lethality on adenine (Fig. 3B). Importantly, none of the other purine salvage pathway genes could allow growth on adenine when overexpressed (Fig. 3B).
FIGURE 3.
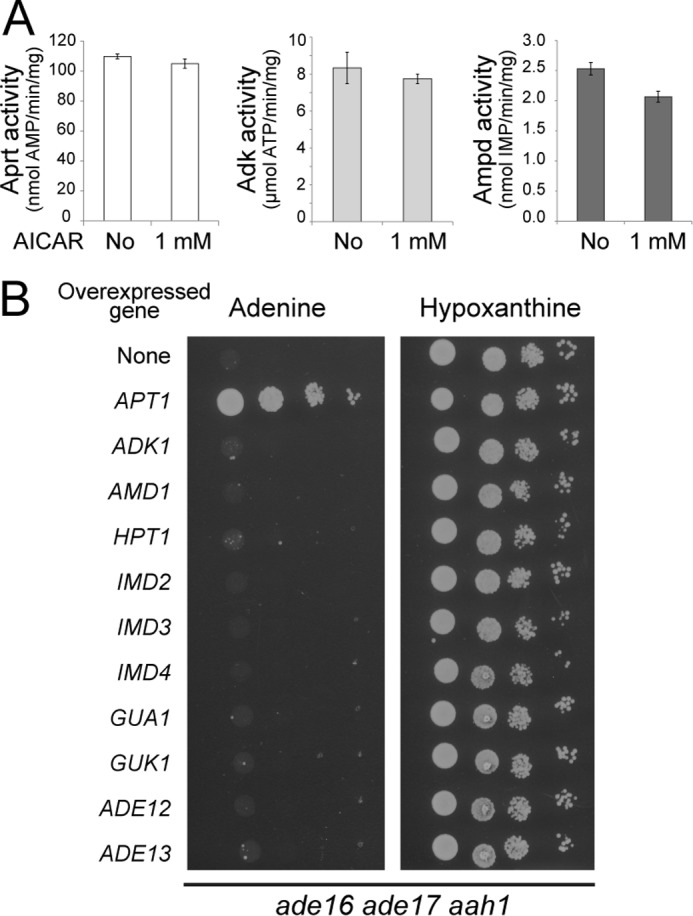
AICAR does not affect enzymatic activities of AMP-metabolizing enzymes A, adenine phosphoribosyl transferase (Aprt), Adk, and AMP deaminase (Ampd) enzymatic activities were determined as described under “Experimental Procedures.” For each enzyme, specific activities were determined in the presence or the absence of AICAR. B, the ade16 ade17 aah1 (Y8793) strain was transformed with plasmids allowing overexpression of the indicated genes or the pCM189 empty vector (none). Transformants were grown overnight, serial diluted, and spotted on SDcasaW medium supplemented with either adenine or hypoxanthine as an external purine source.
We next attempted to understand the basis of AICAR cytotoxicity by analyzing the metabolite profiles of both an ade16 ade17 aah1 triple mutant and an aah1 single mutant grown in various conditions. Among the eight combinations tested, a strong growth defect was only observed in the triple ade16 ade17 aah1 mutant grown on adenine containing medium and not overexpressing APT1 (condition marked by an arrow in Fig. 4A). The metabolite profiling first showed that accumulation of adenine, AdoMet, and SAH is unlikely to cause the growth defect, because levels of these metabolites were similar in the aah1 and aah1 ade16 ade17 strains (Fig. 4, B–D), which are respectively able and unable to grow in the presence of adenine (Fig. 4A). Second, we observed that the ATP and GTP intracellular concentrations were lowered in the ade16 ade17 aah1 strain grown on adenine (Fig. 4, E and F). This result suggested that, in this strain, synthesis of AMP from adenine by Apt1 was not sufficient to satisfy cellular needs for GTP and ATP. Indeed, overexpression of Apt1 restored higher levels of both ATP and GTP (Fig. 4, E and F) and allowed robust growth in the presence of adenine (Fig. 4A). In good agreement with a role of both ATP and GTP pools in AICAR sensitivity, overexpression of Adk1 or Hpt1 that only partially restored the ATP and GTP pools, respectively (Fig. 5, A and B), did not allow growth (Fig. 3B). Consistently, the growth defect of the mutant was much more efficiently rescued by inosine that can refuel the pathway for both ATP and GTP than by guanosine that only contributes to GTP synthesis (Figs. 1B and 5C). This effect on nucleotide triphosphate appears limited to purine nucleotides because UTP levels were not affected by the purine source or by overexpression of Apt1 (Fig. 4G). Finally and most importantly, overexpression of APT1 significantly lowered the intracellular AICAR content (Fig. 4H). Thus, in the AICAR-sensitive condition, AICAR is overaccumulated, whereas purine nucleotide triphosphate levels are lowered.
FIGURE 4.

Impairment of growth and metabolite content of the ade16 ade17 aah1 mutant is suppressed by APT1 overexpression. A, strains (aah1: Y6750; ade16 ade17 aah1: Y8793) were transformed with the APT1 expressing plasmid (P2091) or the pCM189 empty vector (control). Transformants were grown overnight, serial diluted, and spotted on SDcasaW medium supplemented by either adenine or hypoxanthine. B–H, transformants from Fig. 4A were kept in exponential growth for 24 h in SDcasaW medium supplemented with hypoxanthine, harvested by centrifugation, resuspended in SDcasaW medium supplemented with either adenine or hypoxanthine, and grown for 4 h at 30 °C prior to metabolic extraction and separation. Quantifications were determined from at least three independent metabolite extractions for each condition. Error bars indicate variations to the mean. In all panels, the no growth condition is highlighted by the arrowhead. Metabolite content of the ade16 ade17 aah1 mutant strain containing the empty vector and grown in hypoxanthine medium was used as the reference and was set at 1.
FIGURE 5.
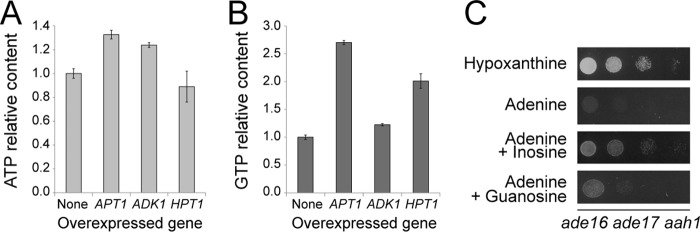
ATP and GTP are both limiting for growth of the ade16 ade17 aah1 mutant on adenine. A and B, the ade16 ade17 aah1 (Y8793) strain was transformed with plasmids allowing or not allowing (None, pCM189) overexpression of the indicated genes. Transformants were kept in exponential phase for 24 h in SDcasaW medium supplemented with hypoxanthine, harvested by centrifugation, resuspended in SDcasaWA medium, and grown for 4 h at 30 °C prior to metabolic extraction and separation. Quantifications were determined from at least three independent metabolite extractions. Error bars indicate variations to the mean. ATP and GTP contents in cells containing the empty vector were used as reference and were set at 1. C, the ade16 ade17 aah1 mutant strain (Y8793) transformed with a plasmid expressing the hCNT3 nucleoside transporter was grown overnight in SDcasaWHypox medium, serial diluted, and spotted on SDcasaW medium supplemented with the indicated nucleobase and/or nucleoside used as purine sources.
Mutations in the HPT1 and ADE13 Genes Are Synthetically Lethal with AICAR Accumulation
We then asked whether this effect was Aah1-specific or whether blocking adenine utilization downstream of Aah1 would also result in AICAR-dependent lethality. We therefore mated a hpt1 mutant that cannot transform hypoxanthine into IMP (Fig. 1B) with an ade16 ade17 double mutant accumulating AICAR. The tetrads obtained from the resulting diploid were dissected on an adenine containing medium. Strikingly, among 14 tetrads, no triple ade16 ade17 hpt1 mutant was recovered (Fig. 6A and not shown). We conclude that hpt1 is synthetically lethal in combination with ade16 ade17. Importantly, the knock-out of hpt1 is not synthetically lethal with other mutations in the de novo pathway such as ade2 (13, 19). To circumvent the ade16 ade17 hpt1 synthetically lethality, we used an ade17 hpt1 double mutant that is viable but showed a growth defect (Fig. 6A, underlined). Of note, Ade17 is the AICAR transformylase IMP-cyclohydrolase isoform that contributes more than 90% of the enzymatic activity (20). This growth impairment was further enhanced when AICAR production was stimulated by growth in the absence of histidine (Fig. 6B) (9). Importantly, overexpression of APT1 suppressed the growth impairment of the hpt1 ade17 mutant in the absence of histidine (Fig. 6C). Metabolite profiling of the ade17 hpt1 mutant revealed that this double mutant, grown in the absence of histidine, accumulated a significant amount of AICAR and that overexpression of APT1 strongly reduced this AICAR accumulation (Fig. 6D). Importantly, in the ade17 hpt1 mutant, overexpression of APT1 also increased ATP and GTP levels, whereas it had little or no effect on UTP (Fig. 6D). Together, our results establish that accumulation of AICAR is highly toxic when yeast cells are unable to synthesize purine de novo and to utilize hypoxanthine.
FIGURE 6.
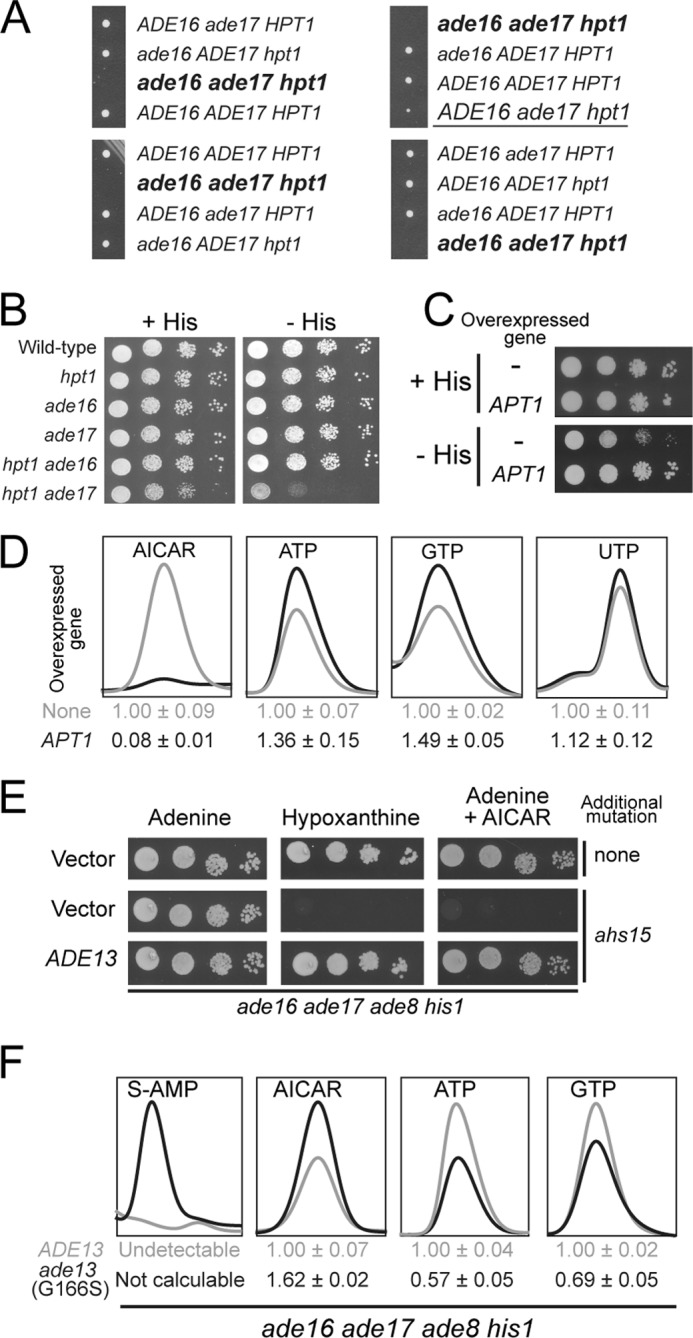
AICAR accumulation is highly toxic for cells unable to use hypoxanthine as an external purine source. A, the diploid strain obtained by crossing ade16 ade17 (Y1095) and hpt1 (Y1127) strains was sporulated, and spores were separated by micromanipulation on YPDAUW medium. The ade17 hpt1 double mutant is underlined. Deduced genotype of lacking spores corresponding to the unviable triple mutant is shown in bold type. B, cells were grown overnight, serial diluted, and spotted on SC medium containing adenine and lacking (− HIS) or not (+ HIS) histidine. Wild type: Y4479; hpt1: Y5864; ade16: Y5870; ade17: Y5876; ade16 ade17: Y5868 and hpt1 ade17: Y5873. C, cells (hpt1 ade17: Y5873) were transformed with the APT1 expressing plasmid (P2091) or the empty vector (pCM189). Transformants were grown overnight, serial diluted, and spotted on SC medium containing adenine and lacking (− HIS) or not (+ HIS) histidine. D, transformants from C were kept in exponential growth for 24 h in SC-U medium supplemented with histidine, harvested by centrifugation, resuspended in SC-U medium lacking histidine, and grown for 4 h at 30 °C prior to metabolic extraction. Quantifications were determined from at least three independent metabolite extractions, and the standard deviations are presented. Metabolite content of cells not overexpressing APT1 were used as reference and set at 1. E, mutant cells (ade16 ade17 ade8 his1 (Y6986); ahs15 ade16 ade17 ade8 his1 (Y7320)) were grown overnight, serial diluted, and spotted on SDCasaWU medium supplemented with either adenine or hypoxanthine and with or without external AICAr (5 mm). The plates were imaged after 2 days at 30 °C. F, cells from Fig. 6E were kept in exponential growth for 24 h in SDCasaWU medium and subjected to metabolic extraction. Quantifications were determined from at least three independent metabolite extractions, and standard deviations are presented. Metabolite content in the ADE13 cells was used as a reference and set at 1.
This prompted us to search for mutants unable to use hypoxanthine as a purine source in our collection of AICAr-sensitive mutants (5). This was indeed the case for the ahs15 mutant (Fig. 6E). Metabolite profiling of ahs15 revealed massive accumulation of succinyl-AMP (Fig. 6F), the substrate of Ade13, the adenylosuccinate lyase (Fig. 1B). Accordingly, both hypoxanthine utilization defect and AICAr sensitivity were fully complemented by the ADE13 gene carried on a plasmid (Fig. 6E). Furthermore, AICAr sensitivity co-segregated with the incapacity to use hypoxanthine in 10 tetrads (not shown). Sequencing of the ADE13 locus in the ahs15 mutant revealed a single mutation resulting in a glycine to serine substitution at position 166 in the protein. This mutation co-segregated with AICAr sensitivity and hypoxanthine auxotrophy in all 10 tetrads (not shown). We conclude that ahs15 is allelic to ADE13. Finally, metabolite profiling of the ahs15 mutant revealed that this mutant had higher levels of AICAR and lower levels of purine nucleotide triphosphate (Fig. 6F). Thus, as for the aah1 mutant, AICAr sensitivity in the hpt1 and ade13 mutants is associated to high AICAR and low ATP and GTP levels.
We propose that ATP and AICAR exert a negative cross-effect on their reciprocal synthesis and that the AICAR/ATP balance is crucial for growth. This was confirmed by analyzing a set of 18 yeast strains (all derived from an ade16 ade17 ade8 his1 background) accumulating various amounts of AICAR (Fig. 7A, gray dots). We indeed found a strong negative correlation between the concentrations of AICAR and ATP. In particular, mutants stimulating AICAR uptake (thi3, thi80, or pdc2 (4)) all strongly increased intracellular AICAR while severely decreasing ATP concentration (Fig. 7A, gray dots) compared with the ade16 ade17 ade8 his1 control strain (Fig. 7A, black dot). Because these uptake mutants are not directly connected to purine metabolism, we conclude that the AICAR-ATP connection is a general phenomenon not restricted to a subset of specific mutants.
FIGURE 7.

AICAR/ATP balance is critical for AICAR toxicity. A, strains were kept in exponential growth for 24 h in SDcasaWAU medium at 30 °C and shifted at 37 °C for 30 min, and AICAr (1.72 mm) was added for 1 h prior to metabolite extraction. The ade16 ade17 ade8 his1 (Y6986) control is shown in black, and the thi3 ade16 ade17 ade8 his1 (Y7321), thi80 ade16 ade17 ade8 his1 (Y8845), and pdc2 ade16 ade17 ade8 his1 (Y7314) strains are indicated. The other mutants shown in gray correspond to unidentified AICAr-sensitive mutants. Quantifications were done on at least three independent metabolite extractions, and error bars indicate variation to the mean. AICAR and ATP contents of the control strain (black dot) were set at 1. B, the ade16 ade17 ade8 his1 mutant strain (Y2950) was kept in exponential phase for 24 h in SDcasaWAU medium containing the indicated concentrations of external AICAr prior to metabolite extraction and separation by liquid chromatography. AICAR (light gray dots) and ATP (dark gray dots) content was set at 1 for cells grown in medium containing either 1 mm or 0 mm of AICAr, respectively. C and D, ATP and AICAR content were determined on cells (ade16 ade17 ade8 his1 (Y2950; light gray bars); kcs1 ade16 ade17 ade8 his1 (Y10399, dark gray bars)) exponentially grown for 24 h in SDcasaWAU medium with (+) or without (−) AICAr (1 mm). ATP (C) and AICAR (D) contents were set at 1 for KCS1 cells grown in the presence of AICAr. E, mutant cells (ade16 ade17 ade8 his1 (Y2950); kcs1 ade16 ade17 ade8 his1 (Y10399)) were grown overnight, serial diluted, and spotted on SDcasaWAU medium with or without external AICAr at the indicated concentration. The plates were imaged after 2 days at 30 °C.
This was further established by studying the ATP/AICAR balance in the ade16 ade17 ade8 his1 strain supplemented with various concentrations of AICAr. Once again, we found a strong inverse connection between the two metabolites (Fig. 7B).
Finally, we took advantage of a kcs1 knock-out mutant previously shown to accumulate higher concentrations of ATP than the wild-type strain (21). The kcs1 knock-out was combined with the ade16 ade17 ade8 his1 mutations. As expected, the kcs1 mutation resulted in increased intracellular ATP concentration (Fig. 7C), whereas AICAR was not significantly affected (Fig. 7D). Of note, in this mutant ATP accumulation does not impact on AICAR concentration because AICAR is provided from the medium in its riboside form and not from ATP-repressible de novo pathway. Most importantly, the kcs1 mutation allowed a robust growth in the presence of AICAR, even though it showed reduced growth in the absence of AICAR (Fig. 7E).
Together, these results establish that AICAR impacts on the ATP concentration and that ATP affects AICAr toxicity. We conclude that disruption of nucleotide homeostasis by AICAR is critical for its effects on yeast cell proliferation.
AICAR Affects Glucose Utilization and Slows Down Growth Recovery after Glucose Replenishment
How does AICAR impact on ATP synthesis? Based on the suppression results, we reasoned that expression of Apt1 could be affected by AICAR. Indeed, the Apt1 steady state level was 4–5-fold lower in an AICAR accumulating strain (Fig. 8A). This could in part be due to the reduced transcription of APT1 (1.7-fold) found in microarray experiments comparing an ade16 ade17 strain accumulating AICAR to an isogenic wild-type strain (4). We conclude that down-expression of Apt1 in the presence of AICAR could limit AMP synthesis and contribute to the low purine nucleotide pools found in the AICAR-accumulating yeast cells. However, we found that overexpression of APT1 could neither fully suppress the AICAR sensitivity of the ade13 mutant nor increase the ability of a wild-type strain to grow in the presence of AICAR (Fig. 8B). We conclude that Apt1 down-expression in the presence of AICAR contributes to AICAR sensitivity but does not account for all of it.
FIGURE 8.
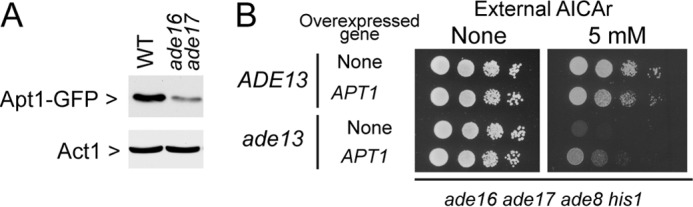
The adenine phosphoribosyl transferase steady state level is affected by AICAR. A, total protein extracts from cells that accumulated AICAR (ade16 ade17 APT1-GFP, Y10351) or that did not accumulate AICAR (APT1-GFP, Y10314) were separated by SDS-PAGE, and Apt1-Gfp (anti-GFP, Roche catalog no. 11814460001 dilution 1/1,000) and Act1 (anti-Act1, generous gift from I. Sagot (Bordeaux, France); 1/100,000) were detected by Western blotting. B, strains (ade16 ade17 ade8 his1 (ADE13; Y6986) and ade16 ade17 ade8 his1 ade13 (G166S) (ade13; Y7320) were transformed with the APT1 expressing plasmid (P2091) or the cognate empty vector (pCM189). Transformants were grown overnight, serial diluted, and spotted on SDcasaWA medium containing AICAr.
Then the question remains as to how AICAR impacts on ATP synthesis aside from its effects on the Apt1 steady state level. One obvious possibility would be that AICAR affects ATP production from ADP, which in yeast mainly takes place via the glycolytic pathway when cells are grown on glucose as a carbon source. Should this be true, such AICAR effects would be lost on carbon sources that do not require the glycolytic pathway. To address this issue, we grew the aah1 ade16 ade17 triple mutant on various carbon sources in the presence of adenine or hypoxanthine as a purine source. Clearly the triple mutant could grow on all media containing hypoxanthine as a purine source (Fig. 9A), although growth was slower on strict respiratory media. When adenine was the purine source, the triple mutant did not grow on glucose or fructose but grew on galactose, lactate, and glycerol/ethanol media (Fig. 9, A and B). This result thus establishes that AICAR cytotoxicity is strongly modulated by the carbon source.
FIGURE 9.

AICAR affects ATP synthesis via alteration of glucose utilization. A, cells (aah1 ade16 ade17; Y8793) were grown overnight, serial diluted, and spotted on SDcasaWU medium containing the indicated carbon source (2% w/v) and either adenine or hypoxanthine. The plates were imaged after 2 days (glucose, fructose, and galactose panels) or 3 days (lactate and glycerol + ethanol) at 30 °C. B, schematic representation of carbon source utilization in yeast. Hxk, hexokinase; Pfk, phosphofructokinase. C, cells (aah1 ade16 ade17; Y8793) were kept in the exponential growth phase for 24 h in ScasaWHypox containing either 2% glucose or 2% galactose as carbon source. The cells were then harvested by centrifugation, resuspended in the same two media but containing adenine instead of hypoxanthine, and grown for 4 h at 30 °C prior to metabolic extraction. Quantifications were determined from at least five independent metabolite extractions, and the standard deviation is indicated. D, cells grown for 48 h in SDcasaWA medium were then diluted in fresh SDcasaWA medium, and cell growth was followed for 6 h at 30 °C using a Multisizer IV (Beckman Coulter). WT: Y4479; ade16 ade17: Y6681 and ade16 ade17 ade8 his1: Y6986. E, AICAR accumulation is required for the lag phase delay of the ade16 ade17 mutant. WT and ade16 ade17 mutant strains were transformed with the plasmid expressing PHO13-1 (P4291) or the empty vector (pCM189).
Importantly, metabolite profiling of the aah1 ade16 ade17 strain revealed that growth in galactose resulted in higher ATP and GTP levels, whereas AICAR levels were concomitantly significantly reduced (Fig. 9C). Thus, the growth phenotype (Fig. 9A) was once again strictly correlated to the AICAR balance with ATP and GTP (Fig. 9C). Of note, the UTP pool was reduced on galactose compared with glucose, possibly because of utilization of UTP for synthesis of UDP derivatives necessary for galactose utilization. These data showing that growth on galactose resulted in a much lower AICAR/ATP and AICAR/GTP ratios and allowed growth further support our assumption that the high AICAR/NTP ratio on glucose is detrimental for growth on this carbon source.
Consistently, we noticed that yeast strains accumulating AICAR, when shifted from post-diauxic growth conditions to fresh glucose medium, required a longer adaptation time before entry into exponential growth. During this adaptation period, known as “lag phase,” cell number remains relatively constant prior to rapid growth. The double ade16 ade17 mutant, unable to metabolize AICAR and thus accumulating it, was compared with a wild-type strain and to a quadruple ade16 ade17 ade8 his1 mutant that cannot synthesize nor metabolize AICAR. Clearly, the ade16 ade17 AICAR-accumulating mutant had a much longer lag phase than the isogenic wild-type strain (Fig. 9D), although its doubling time during exponential growth was not increased (84 min compared with 89 min for the wild-type strain). Importantly, the longer lag-phase was not found in the ade16 ade17 ade8 his1 mutant that does not accumulate AICAR (Fig. 9D), thereby indicating that AICAR and not the ade16 ade17 block per se is responsible for this phenotype. Consistently, the lag-phase of the ade16 ade17 mutant was shortened when these cells carried a plasmid expressing the Pho13-1 phosphatase that leads to decreased intracellular concentration of AICAR (9) (Fig. 9E). These results indicate that AICAR accumulation affects the capacity of yeast cells to adapt to the new glucose rich medium and initiate exponential growth.
All together these results reveal glucose utilization as an important basis of AICAR toxicity in yeast. We then asked whether this could also be the case in human cells.
Growth on Galactose Allows to Bypass AICAR Toxicity in Human Cells
We investigated the effect of AICAr on human HeLa cells propagated in glucose and galactose-containing medium. HeLa cells grown on galactose exhibited a lower mitotic index than in the presence of glucose (doubling times ∼60 h versus 24 h, respectively) (Fig. 10, A and B), as previously reported by others (22). AICAr seriously impaired the ability of HeLa cells to grow in the presence of glucose in a dose-dependent manner (Fig. 10A). In comparison, no significant inhibition of growth was obtained under AICAr treatment when substituting glucose with galactose (Fig. 10B). Indeed, the proliferation of HeLa cells was unaffected at higher doses (1 and 3 mm) of AICAr in the presence of galactose, whereas it was almost completely inhibited when using the glucose medium (Fig. 10, A and B). Comparison of metabolite profiles of HeLa cells grown on glucose and galactose revealed lower AICAR levels in cells grown in galactose compared with glucose (Fig. 10, C and D). In the absence of AICAR, cells grown in galactose accumulated more of all four NTPs (Fig. 10, E–L). AICAR treatment resulted in an increased amount of purine nucleotides and a severe drop of pyrimidine triphosphate pools (Fig. 10, E–L). This effect was strongly reduced in cells grown on galactose compared with glucose, suggesting that it could at least in part account for AICAR toxicity. The differential effects of AICAr on proliferation depending on the carbon source could not be simply attributed to differences in AICAR nor pyrimidine nucleotide contents. Indeed, AICAR accumulation was analogous in glucose + 1 mm AICAr and galactose + 3 mm AICAr (Fig. 10, C and D), whereas pyrimidine nucleotide pools (Fig. 10, I–L) and proliferation (Fig. 10, A and B) were clearly differently affected. In addition, at lower AICAr concentration (0.3 mm) on glucose, pyrimidine nucleotides were severely reduced (Fig. 10, I–K), whereas proliferation was only slightly affected (Fig. 10A). However, by combining both parameters and considering the AICAR/(UTP+CTP) ratio, a strong negative correlation between this ratio and proliferation is found (Fig. 10, A and B, ratios are shown in parentheses).
FIGURE 10.
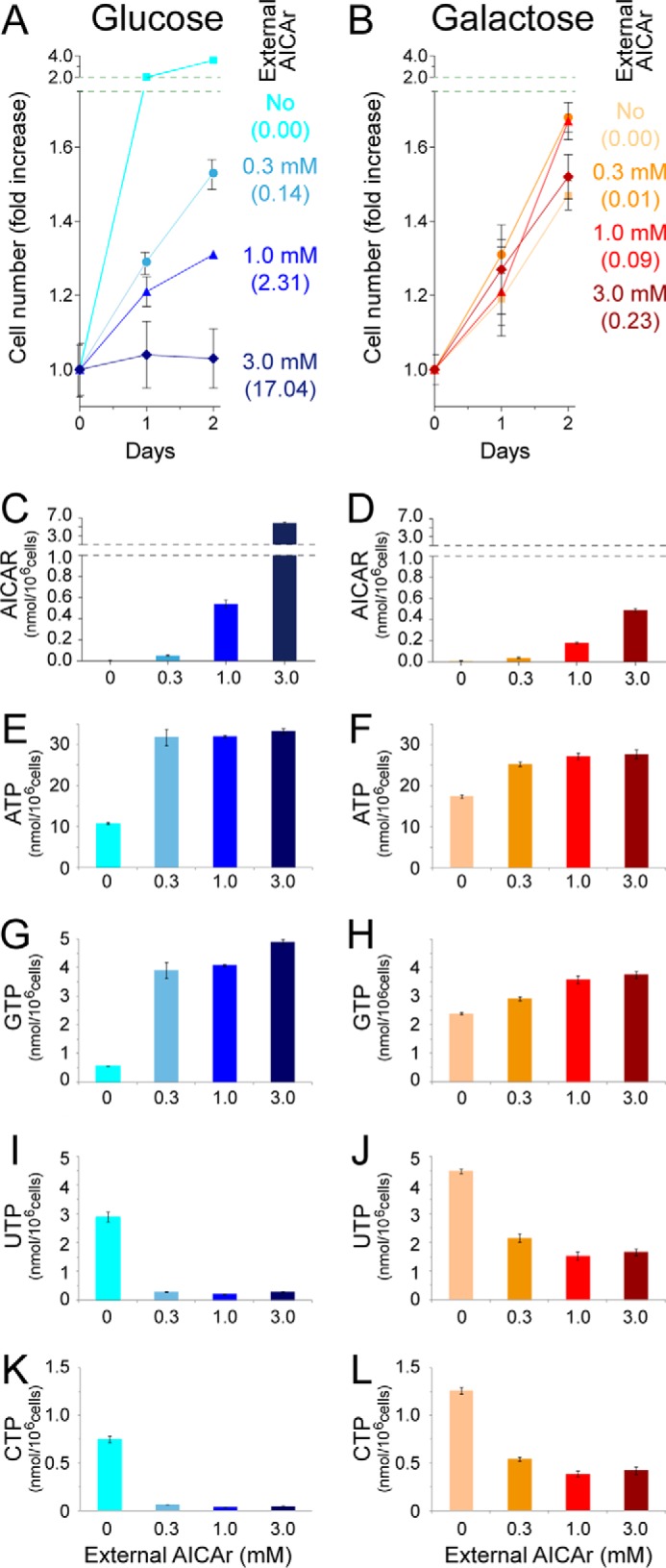
Human cells grown in galactose instead of glucose are significantly more resistant to AICAr. A and B, HeLa cells were seeded in 24-well plates in media containing 5 mm of either glucose (A) or galactose (B) and allowed to settle for 24 h. AICAr was then added (day 0) at the indicated concentrations, and cells were numerated each day. The results are means of triplicate experiments, and the error bars represent standard deviation. The values in parentheses correspond to the intracellular ratios [AICAR]/([CTP]+[UTP]) at day 2. C–L, intracellular contents of AICAR and of NTPs (ATP, GTP, UTP, and CTP) at day 2 as a function of external AICAr concentrations and carbon sources.
Discussion
This work addressed the toxicity of AICAR in eukaryotic cells. AICAR is known to disturb cellular functions through its well established AMP-mimetic properties and in particular through its activating effect on AMPK. However, a growing number of reports have established that the cellular effects of AICAR, initially thought to be mainly due to activation of AMPK, are proving to be much more complicated (1, 2). In particular, the recent demonstration that the antiproliferative effects of AICAR are AMPK-independent (5, 6) has stressed the necessity to identify new functions affected by AICAR.
AICAR Strongly Interferes with NTPs Metabolism
Our search for AICAR-synthetically lethal mutants revealed that adenine deaminase (Aah1) is critical for yeast cells to maintain adequate ATP levels when adenine is provided as a purine source and when AICAR accumulates. Our previous work indicated that the adenine deaminase route is the major adenine utilization route in yeast (23), and accordingly we found that aah1 mutants accumulate much more adenine than the apt1 mutant (Fig. 2C). In the course of this work, we observed that adenine accumulation was associated to accumulation of AdoMet and SAH and that this effect was AICAR-independent (Figs. 2 and 4). We propose that accumulation of AdoMet and SAH could be due to in vivo inhibition of SAH hydrolase by adenine because such an inhibition had been reported to occur in vitro (24). However, our data do not support a role for AdoMet and SAH accumulation in the AICAR synthetically lethal phenotype.
In the aah1 mutant, AICAR accumulation was associated to a lower ATP and GTP concentration. Because ATP exerts an allosteric negative regulation on the purine pathway, lowering ATP results in increased concentration of AICAR as previously described (25). The decreased ATP and GTP levels would in part be due to the aah1 mutation restraining AMP synthesis to the Apt1 route. This phenomenon was found to be amplified by low expression of Apt1 when AICAR accumulates. Consistently, overexpression of Apt1 allowed replenishing the ATP and GTP pools. However, because the NTP pools were not affected in the aah1 single mutant grown with adenine, it is clear that AICAR accumulation, resulting from the ade16 ade17 knock-out, contributes to lowering ATP and GTP levels (see “AICAR Affects Glucose Utilization in Vivo”). Under conditions restoring growth (hypoxanthine or overexpression of APT1), AICAR decreased and ATP + GTP increased, suggesting that the AICAR/(ATP + GTP) balance is crucial for AICAR toxicity.
Importantly, the effect of AICAR on nucleotide homeostasis, initially observed in the aah1 mutant, was not restricted to this particular genetic background because it was also found in other mutants (hpt1 and ade13). In addition, in a nonmutant background, we could establish that AICAR strongly affected ATP concentration (Fig. 7B). Conversely, using the kcs1 mutant, we could show that an increase of the ATP pool was sufficient to alleviate AICAR toxicity (Fig. 7, C–E). Together, our results establish that AICAR balance with nucleotides triphosphate is a critical factor contributing to its toxicity.
Our data establish that AICAR accumulation is synthetically lethal with the hpt1 knock-out abolishing hypoxanthine-guanine phosphoribosyltransferase activity. Strikingly, AICAR is accumulated in body fluids of patients suffering purine metabolism diseases, including hypoxanthine-guanine phosphoribosyltransferase-deficient patients (15, 26). It should be stressed that the levels of AICAR accumulated by these patients (15) are way below those resulting in synthetic lethality in yeast cells (this work). However, should AICAR have similar effects in human cells than the ones observed in yeast, it could contribute to the phenotypic outcome of these complex developmental genetic diseases. Accordingly, we recently found that, in addition to AICAR accumulation, NTP levels were significantly lower in erythrocytes from a large cohort of hypoxanthine-guanine phosphoribosyltransferase-deficient patients (15).
AICAR Affects Glucose Utilization in Vivo
As mentioned previously, our results show that AICAR and ATP negatively impact on their reciprocal synthesis. The control exerted by ATP on AICAR synthesis is likely due to the fact that AICAR synthesis is modulated through feedback inhibition of the first step of the purine de novo pathway by ATP (25). How then does AICAR impact on ATP synthesis?
We found that AICAR quite specifically affects growth on glucose as a carbon source. Growth on fructose is also affected, but utilization of both carbon sources relies on hexokinase activity for the first step of glycolysis. Meanwhile, AICAR is far less toxic on galactose, which allows bypassing the hexokinase step and feeding the glycolytic pathway with glucose-6-phosphate (Fig. 9B). Importantly, this carbon source specific effect was found in both yeast and human cells, suggesting a possibly conserved mechanism of AICAR on glucose utilization, even though in mammalian cells AICAR mostly affected pyrimidine triphosphate nucleotides. The underlying mechanism leading to this difference between yeast and mammalian cells remains to be explored. In vitro inhibitory effects of AICAR on glycolysis have been documented on mammalian enzymes (27, 28), but the in vivo consequences had not been evaluated. Suppression of AICAR toxicity by galactose, but not by fructose, points to glucose-6-phosphate synthesis as a major step affected by AICAR. In hepatocytes, AICAR moderately affects glucokinase enzymatically (28) and inhibits glucokinase translocation (29). In yeast, we could not find any inhibitory effect of AICAR on glucose phosphorylation in vitro.5 In tumor cells, the basis of AICAR effects on glucose utilization is to be established. Whatsoever the exact mechanism(s) involved in vivo, our work established that AICAR inhibition of glucose utilization can significantly contribute to its antiproliferative properties. AICAR effects on glucose utilization are likely to affect more specifically tumor cells that strictly rely on the glycolytic pathway because of the Warburg effect.
Author Contributions
B. D.-F. and B. P. conceived the study, participated in its design and coordination, and drafted the manuscript. J. C., H. C. H, C. S.-M., D. A., T. V., M. M., and B. P. carried out the experiments. All authors read and approved the final manuscript.
Acknowledgments
We thank J. E. Gomes for helpful discussions and J. Tissot-Dupont for technical support.
This work was supported by Association de la Recherche contre le Cancer Grant SFI-2012-120−5915 (to B. D.-F.) and Ligue contre le Cancer Project LARGE (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
J. Ceschin, B. Pinson, and B. Daignan-Fornier, unpublished results.
- AMPK
- AMP-activated protein kinase
- AdoMet
- S-adenosylmethionine
- AICAR
- 5-amino-4-imidazole carboxamide ribonucleotide 5′-phosphate
- AICAr
- 5-amino-4-imidazole carboxamide ribonucleoside
- SAH
- S-adenosylhomocysteine
- Adk
- adenylate kinase.
References
- 1. Daignan-Fornier B., Pinson B. (2012) 5-Aminoimidazole-4-carboxamide-1-beta-d-ribofuranosyl 5′-monophosphate (AICAR), a highly conserved purine intermediate with multiple effects. Metabolites 2, 292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vincent E. E., Coelho P. P., Blagih J., Griss T., Viollet B., Jones R. G. (2014) Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 34, 3627–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mayer F. V., Heath R., Underwood E., Sanders M. J., Carmena D., McCartney R. R., Leiper F. C., Xiao B., Jing C., Walker P. A., Haire L. F., Ogrodowicz R., Martin S. R., Schmidt M. C., Gamblin S. J., Carling D. (2011) ADP regulates SNF1, the Saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metab. 14, 707–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pinson B., Vaur S., Sagot I., Coulpier F., Lemoine S., Daignan-Fornier B. (2009) Metabolic intermediates selectively stimulate transcription factor interaction and modulate phosphate and purine pathways. Genes Dev. 23, 1399–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ceschin J., Saint-Marc C., Laporte J., Labriet A., Philippe C., Moenner M., Daignan-Fornier B., Pinson B. (2014) Identification of yeast and human 5-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside (AICAr) transporters. J. Biol. Chem. 289, 16844–16854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu X., Chhipa R. R., Pooya S., Wortman M., Yachyshin S., Chow L. M., Kumar A., Zhou X., Sun Y., Quinn B., McPherson C., Warnick R. E., Kendler A., Giri S., Poels J., Norga K., Viollet B., Grabowski G. A., Dasgupta B. (2014) Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. U.S.A. 111, E435–E444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang Y. C., Williams B. R., Siegel J. J., Amon A. (2011) Identification of aneuploidy-selective antiproliferation compounds. Cell 144, 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rébora K., Laloo B., Daignan-Fornier B. (2005) Revisiting purine-histidine cross-pathway regulation in Saccharomyces cerevisiae: a central role for a small molecule. Genetics 170, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hürlimann H. C., Laloo B., Simon-Kayser B., Saint-Marc C., Coulpier F., Lemoine S., Daignan-Fornier B., Pinson B. (2011) Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J. Biol. Chem. 286, 30994–31002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sherman F., Fink G. R., Hicks J. B. (1986) Methods in Yeast Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York [Google Scholar]
- 11. Gauthier S., Coulpier F., Jourdren L., Merle M., Beck S., Konrad M., Daignan-Fornier B., Pinson B. (2008) Co-regulation of yeast purine and phosphate pathways in response to adenylic nucleotide variations. Mol Microbiol 68, 1583–1594 [DOI] [PubMed] [Google Scholar]
- 12. Saint-Marc C., Pinson B., Coulpier F., Jourdren L., Lisova O., Daignan-Fornier B. (2009) Phenotypic consequences of purine nucleotide imbalance in Saccharomyces cerevisiae. Genetics 183, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Breton A., Pinson B., Coulpier F., Giraud M. F., Dautant A., Daignan-Fornier B. (2008) Lethal accumulation of guanylic nucleotides in Saccharomyces cerevisiae HPT1-deregulated mutants. Genetics 178, 815–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garí E., Piedrafita L., Aldea M., Herrero E. (1997) A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13, 837–848 [DOI] [PubMed] [Google Scholar]
- 15. Ceballos-Picot I., Le Dantec A., Brassier A., Jaïs J. P., Ledroit M., Cahu J., Ea H. K., Daignan-Fornier B., Pinson B. (2015) New biomarkers for early diagnosis of Lesch-Nyhan disease revealed by metabolic analysis on a large cohort of patients. Orphanet J. Rare Dis. 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merkler D. J., Wali A. S., Taylor J., Schramm V. L. (1989) AMP deaminase from yeast. Role in AMP degradation, large scale purification, and properties of the native and proteolyzed enzyme. J. Biol. Chem. 264, 21422–21430 [PubMed] [Google Scholar]
- 17. Alfonzo J. D., Sahota A., Taylor M. W. (1997) Purification and characterization of adenine phosphoribosyltransferase from Saccharomyces cerevisiae. Biochim. Biophys. Acta 1341, 173–182 [DOI] [PubMed] [Google Scholar]
- 18. Konrad M. (1988) Analysis and in vivo disruption of the gene coding for adenylate kinase (ADK1) in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 263, 19468–19474 [PubMed] [Google Scholar]
- 19. Guetsova M. L., Crother T. R., Taylor M. W., Daignan-Fornier B. (1999) Isolation and characterization of the Saccharomyces cerevisiae XPT1 gene encoding xanthine phosphoribosyl transferase. J. Bacteriol. 181, 2984–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tibbetts A. S., Appling D. R. (1997) Saccharomyces cerevisiae expresses two genes encoding isozymes of 5-aminoimidazole-4-carboxamide ribonucleotide transformylase. Arch. Biochem. Biophys. 340, 195–200 [DOI] [PubMed] [Google Scholar]
- 21. Szijgyarto Z., Garedew A., Azevedo C., Saiardi A. (2011) Influence of inositol pyrophosphates on cellular energy dynamics. Science 334, 802–805 [DOI] [PubMed] [Google Scholar]
- 22. Rossignol R., Gilkerson R., Aggeler R., Yamagata K., Remington S. J., Capaldi R. A. (2004) Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 64, 985–993 [DOI] [PubMed] [Google Scholar]
- 23. Guetsova M. L., Lecoq K., Daignan-Fornier B. (1997) The isolation and characterization of Saccharomyces cerevisiae mutants that constitutively express purine biosynthetic genes. Genetics 147, 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Knudsen R. C., Yall I. (1972) Partial purification and characterization of S-adenosylhomocysteine hydrolase isolated from Saccharomyces cerevisiae. J. Bacteriol. 112, 569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rébora K., Desmoucelles C., Borne F., Pinson B., Daignan-Fornier B. (2001) Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol. Cell. Biol. 21, 7901–7912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sidi Y., Mitchell B. S. (1985) Z-nucleotide accumulation in erythrocytes from Lesch-Nyhan patients. J. Clin. Invest. 76, 2416–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Javaux F., Vincent M. F., Wagner D. R., van den Berghe G. (1995) Cell-type specificity of inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside. Lack of effect in rabbit cardiomyocytes and human erythrocytes, and inhibition in FTO-2B rat hepatoma cells. Biochem. J. 305, 913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vincent M. F., Bontemps F., Van den Berghe G. (1992) Inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside in isolated rat hepatocytes. Biochem. J. 281, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., Hue L. (2006) 5-Aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55, 865–874 [DOI] [PubMed] [Google Scholar]