

Background: Previous human and animal studies suggest lipin1 plays a role in myopathies, but whether lipin1 regulates skeletal muscle regeneration remains unknown.
Results: Lipin1 regulates myoblast differentiation through cytosolic activation of ERK1/2 and a downstream effector cyclin D3-mediated cell cycle withdrawal.
Conclusion: Lipin1 is a key regulator of myoblast differentiation.
Significance: Our study reveals a previously unknown pathway in skeletal muscle regeneration.
Keywords: cell cycle, muscle regeneration, nuclear translocation, protein phosphorylation, skeletal muscle, ERK, lipin1, myoblast differentiation
Abstract
Lipin1, an intracellular protein, plays critical roles in controlling lipid synthesis and energy metabolism through its enzymatic activity and nuclear transcriptional functions. Several mouse models of skeletal muscle wasting are associated with lipin1 mutation or altered expression. Recent human studies have suggested that children with homozygous null mutations in the LPIN1 gene suffer from rhabdomyolysis. However, the underlying pathophysiologic mechanism is still poorly understood. In the present study we examined whether lipin1 contributes to regulating muscle regeneration. We characterized the time course of skeletal muscle regeneration in lipin1-deficient fld mice after injury. We found that fld mice exhibited smaller regenerated muscle fiber cross-sectional areas compared with wild-type mice in response to injury. Our results from a series of in vitro experiments suggest that lipin1 is up-regulated and translocated to the nucleus during myoblast differentiation and plays a key role in myogenesis by regulating the cytosolic activation of ERK1/2 to form a complex and a downstream effector cyclin D3-mediated cell cycle withdrawal. Overall, our study reveals a previously unknown role of lipin1 in skeletal muscle regeneration and expands our understanding of the cellular and molecular mechanisms underlying skeletal muscle regeneration.
Introduction
Lipin1, encoded by the Lpin1 gene, is an intracellular protein and has dual functions as demonstrated by previous studies (1–4). In the cytoplasm, lipin1 acts as a phosphatidic acid phosphohydrolase catalyzing dephosphorylation of phosphatidic acid and is involved in triglyceride and glycerophospholipid synthesis. In the nucleus lipin1 acts as a transcriptional co-activator factor of PPARα in hepatocytes (3). Lipin1 translocation between the cytoplasm and nucleus is influenced by its phosphorylation status (5–7). In response to insulin, lipin1 serine/threonine sites are phosphorylated, which promotes its translocation out of the nucleus into the cytoplasm (7). In contrast, disruption of 17 serine/threonine phosphorylation sites induces exclusive nuclear localization (6).
Recently, the role of lipin1 in skeletal muscle-related diseases/myopathies has gained interest (8–11). Concurrent lipin1 deficiency in the neuron/Glia-related cell adhesion molecule (Nrcam) of mutant mice contributed to skeletal muscle wasting in hindquarters and hind limb paralysis (12). Down-regulated lipin1 mRNA expression levels were observed in dystrophin-deficient myotubes from C57BL10 mice limb muscle (13) and nemaline myopathies (14). Moreover, human studies have identified a population carrying a LPIN1 gene mutation, due to a premature stop codon insertion or as a result of a large intragenic deletion, which is the major cause of rhabdomyolysis (8–10). Despite well known roles of lipin1 in lipid biosynthesis and transcriptional regulation, the pathogenic mechanisms leading to impaired muscle repair are still unknown. It remains to be determined whether lipin1 plays a role in postnatal skeletal muscle regeneration.
Muscle regeneration is a rapid and extensive self-renewal process relying on the presence of satellite cells. These cells are activated and then proliferate to provide a myoblast population that either fuses to each other to create new myofibers or fuses to existing damaged myofibers for repair (15–18). This process of myogenesis is regulated by the myogenic regulatory factors including Myf5, MyoD, and MyoG. Mononucleated myoblast proliferation is driven by Myf5 expression (19). MyoG is a master regulator of myoblast differentiation and fusion into myofibers (17). Mice lacking the MyoG gene die at birth due to severe skeletal muscle deficiency, as myoblasts are unable to fuse into multinucleated myofibers (20). MyoD is not only responsible for the activation of MyoG expression in the regeneration process but also governs cell cycle withdrawal from the G1 to G0 phase partly through its synergistic interactions with retinoblastoma (21–25). In the G1 phase retinoblastoma is hyperphosphorylated by the cyclin D·CDK4/CDK6 complex, causing the release and activation of the E2F·DP transcription factor complex, which in turn activates expression of the genes required for S-phase progression (26).
The aim of this study is to investigate the role of lipin1 in skeletal muscle regeneration. We employed a mouse regeneration model induced by BaCl2 injection and compared the process of regeneration between lipin1-deficient (fatty liver dystrophy (fld)2) mice with their wild-type (WT) littermates. We evaluated the expression of the myogenic transcription factors and proteins involved in cell cycle regulation and identified a lipin1-associated regeneration pathway that was further verified in lipin1-depleted myoblasts and lipin1-deficient primary myoblasts. We suggest that this new observation provides new insights into the role of lipin1 in the skeletal muscle regeneration process.
Experimental Procedures
BaCl2 Induced Muscle Injury Model
Age and gender matched lipin1-deficient fld mice and their littermates (around 4 months of age) were purchased from The Jackson Laboratory. Protocols and procedures conformed to current United States Public Health Service policies and were approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
Induction of BaCl2-induced muscle injury was undertaken by injecting around 50 μl (adjusted by their body weight) of 1.2% BaCl2 solution intramuscularly into the tibialis anterior (TA) muscles of adult mice using a Hamilton syringe and 27-gauge needle. Contralateral TA muscles injected with phosphate-buffered saline (PBS) only served as uninjured controls. Injured and uninjured muscles were then harvested at day 0 and post mortem at 3, 5, 7, or 14 days after injury (n = 4–8 mice) and processed for immunohistochemistry, mRNA, or protein analysis.
Histology and Immunohistochemistry in Muscle Cryosections
TA muscles were embedded in optimal cutting temperature compound, immediately frozen in isopentane cooled in liquid nitrogen, and stored at −80 °C until analysis. Sections of 10 mm were cut and collected from the mid-belly muscles and stained with hematoxylin/eosin (H&E, Sigma) using standard procedures (27). Immunohistochemistry of muscle cryosections was performed with the following antibodies: anti-MyHC (myosin heavy chain, F1.652), anti-mf20 (Developmental Studies Hybridoma Bank), and laminin (Abcam). Labeling of cryosections with mouse monoclonal primary antibodies was performed using the peroxidase or fluorescein M.O.M kit staining (Vector Laboratories) according to the manufacturer's instructions. Double immunostaining was performed by sequential addition of each primary and secondary antibody using appropriate positive and negative controls. Sections were air-dried, fixed on 4% paraformaldehyde, washed with PBS, and incubated with primary antibodies according to the manufacturer's instructions after blocking for 1 h at room temperature with 1% BSA. Subsequently, the slides were washed by PBS and incubated with appropriate secondary antibodies conjugated with Alexa Fluor 488 or 555 (Invitrogen), and nuclei were stained with DAPI (Invitrogen). After washing, tissue sections were mounted.
Microscope, Image Processing, and Morphometric Analysis
Bright-field images were acquired with a microscope (Nikon eclipse 80i) coupled with a camera (Nikon DXM 1200F) using Nikon ACT1 (Version 2.63) or Metamorph software (Molecular Devices). The objectives used were Plan Fluor 20 × 0.50 or Plan Fluor 40 × 1.30 oil (Nikon). Fluorescent micrographs were taken with a confocal microscope (Nikon Eclipse TE2000-E) using Nikon NIS Elements software (Carl Zeiss). Lenses used with the confocal microscope were either CFI Plan Fluor 40× Oil or CFI Plan Fluor DLL 20× (Nikon). All digital microscopic images were acquired at room temperature.
Myofiber cross-sectional areas in injured TA muscle were quantified via manual masking using Nikon NIS Elements software based on laminin staining in 40× fields. A minimum of 400 myofibers and three sections were analyzed per biological replica. To examine the efficiency of myoblast differentiation, a fusion index (FI) was calculated as the number of nuclei present in myotubes over the total number of nuclei present in the observed field. Data were selected from 10 different randomly chosen microscopic fields.
Cell Culture
C2C12 mouse myoblasts were grown to confluency under 5% CO2 at 37 °C in growth medium, DMEM medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco), and penicillin-streptomycin (100 mg/ml, 100 units/ml) (Gibco). To differentiate the myoblasts, they were then switched to differentiation medium containing DMEM supplemented with 2% horse serum (v/v) (Gibco) and penicillin-streptomycin (100 mg/ml, 100 units/ml) for 4 days. Adenoviruses expressing short hairpin RNA to knockdown lipin1 (shLpin1) and control shLacZ were generous gifts from Dr. Thurl Harris (University of Virginia). For knockdown of lipin1 expression, C2C12 myoblasts were infected with shLpin1 and shLacZ control in growth medium containing 8 μg/ml Polybrene for 24 h. The medium was then replaced with DMEM supplemented with 10% FBS. The myoblasts were incubated for an additional 24 h until they were just confluent.
Primary myoblasts were isolated from fld and WT mice (4 weeks of age) according to Yaffe and Saxel (28) with modifications. Briefly, hind limb muscles were dissected and minced into pieces. Cells were dissociated in 20 ml of 0.25% trypsin, 1 mm EDTA at 37 °C for 2 h and filtered through an 85-mm nylon mesh filter (Spectrum, Houston, TX). Dissociated single cells were washed in PBS (Gibco/Invitrogen) and plated in collagen-coated culture plates with F-10-based primary myoblast growth medium (Ham's F-10 nutrient mixture containing 20% fetal calf serum, 2.5 ng/ml basic fibroblast growth factor (Promega Corp.)), streptomycin, and penicillin and incubated at 37 °C in 5% CO2. Differentiation of myoblasts into myotubes was initiated at ∼90% confluence by cultivation in differentiation medium (DMEM, 5% horse serum) for up to 6 days.
When required, ERK phosphorylation inhibitor (100 μm U0126) was added in the differentiation medium. To analyze whether cyclin D acts as a downstream target of ERK, C2C12 myoblasts were incubated with the differentiation medium in the presence of an inhibitor for 4 days. To induce endogenous lipin1 nuclear accumulation, C2C12 myoblasts were treated with 150 nm Torin-1 for 24 h.
Western Blot
Cells were lysed using a protein lysis buffer containing 1% Nonidet P-40, 150 mm NaCl, 20 mm Tris-HCl, and protease inhibitor mixture. The protein expression levels were examined by incubation with Myf5 (1/500; Santa Cruz Biotechnology), MyoG (1/200; BD Biosciences), MyoD (1/300; Santa Cruz), ERK, P-ERK, cyclin D1, cyclin D3, CDK4, CDK6 (1/1000; Cell Signaling Technology), and α-tubulin (1/1000; Sigma) primary antibodies followed by incubation with goat anti-mouse IgG and goat anti-rabbit IgG IR Dye 680 or 800 (LI-COR) secondary antibodies.
Immunoprecipitation
C2C12 myoblasts were co-transfected with HA-lipin1 together with EGFP-ERK2. 24 h after transfection, cells were immunoprecipitated with either mouse anti-GFP or rabbit anti-HA antibody. The interaction between lipin1 and ERK was determined by Western blot using rabbit an-HA or mouse anti-GFP antibody. EGFP-ERK2 plasmid was obtained from Addgene (plasmid #40777).
Statistical Analysis
All data are presented as the means ± S.E. Student's t test (unpaired and two tailed) or one-way analysis of variance was used to determine the statistical significance of individual datasets. Differences were considered significant for p < 0.05 and very significant for p < 0.01.
Results
Lipin1-deficient fld Mice Exhibit Impaired Skeletal Muscle Regeneration Characterized by Smaller Muscle Fiber Cross-sectional Area and Reduced Muscle Mass
We employed a BaCl2 injury model with the lipin1deficient (fld) mice to investigate the role of lipin1 in skeletal muscle regeneration. In both wild-type and fld groups, 50 μl (adjusted by body weight) of 1.2% BaCl2 in PBS was injected into one TA muscle, and PBS only was injected into the contralateral TA muscle as a control (Fig. 1A). H&E staining and immunostaining with antibodies against laminin and embryonic myosin heavy chain were employed to examine the effect of lipin1 deficiency on muscle regeneration in the TA muscle at days 0, 3, 5, 7, and 14 after injury. Before injury, no significant histological defects were observed in the TA muscle of fld mice (Fig. 1B). Extensive myofiber damage in both WT and lipin1-deficient TA muscle was observed at day 3. Five days after injury, regeneration was evident in both WT and lipin1-deficient muscles with the presence of newly formed myotubes containing central nuclei, which is a hallmark of regeneration. By day 14 after injury, the overall tissue architecture was restored in wild-type mice indicating a complete regeneration. In contrast, although the muscle in fld mice did initiate regeneration, the persistence of small myofibers expressing embryonic myosin heavy chain at day 14 after injury suggested that regeneration of new myofibers was still under way (Fig. 1B). Lipin1-deficient fld mice showed significantly reduced mean myofiber cross-sectional area of 1254 μm2 compared with 1790 μm2 in wild-type TA muscles injected with BaCl2. In contrast, PBS injection alone into TA muscles had no effect on myofiber cross-sectional area (Fig. 1C).
FIGURE 1.
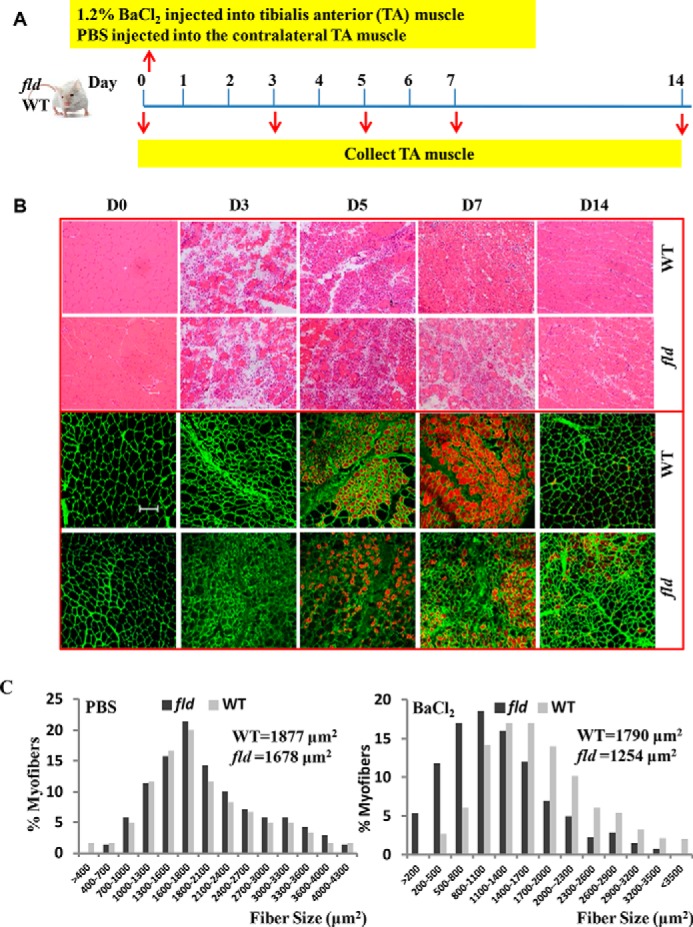
Lipin1-deficient fld mice exhibit impaired skeletal muscle regeneration characterized by smaller muscle fiber cross-sectional area and reduced muscle mass. A, schematic representation of experimental procedures to induce skeletal muscle regeneration. To induce acute skeletal muscle injury, 1.2% BaCl2 solution or PBS was injected into the TA muscle of each mouse. Injured and uninjured muscles were harvested at 0 day (before injury) and post mortem at 3, 5, 7, or 14 days after injury. B, H&E staining and immunostaining with antibodies against embryonic myosin heavy chain and laminin of uninjured TA muscles (day 0) and injured TA muscles harvested at day 3, 5, 7, and 14 after injury. Bars, 100 μm. C, quantification of reduced myofiber area at 14 days after BaCl2 injury are shown; mean cross-sectional area for WT myofibers is 1790 μm2, and fld myofibers is 1254 μm2 (p < 0.001). All values are the means ± S.E. A minimum of 500 myofibers and 3 sections were analyzed for three mice per genotype.
Lipin1 Regulates Skeletal Muscle Regeneration by Mediating ERK Phosphorylation and Cyclin D·CDK6 Complex Expression in Vivo
Myogenic regulatory factors including myoD and myoG are master regulators for the commitment of myogenic precursor cells and the terminal differentiation of myoblasts (17, 29). Western blot analysis showed up-regulated levels of myoD and myoG at day 3 after injury in both WT and lipin1-deficient muscle (Fig. 2). Their expression levels then rapidly decreased and by day 7 had returned back to the baseline. Compared with WT mice, however, myoD and myoG expression levels were suppressed by 60 and 86%, respectively, in the TA muscles of fld mice at day 3.
FIGURE 2.
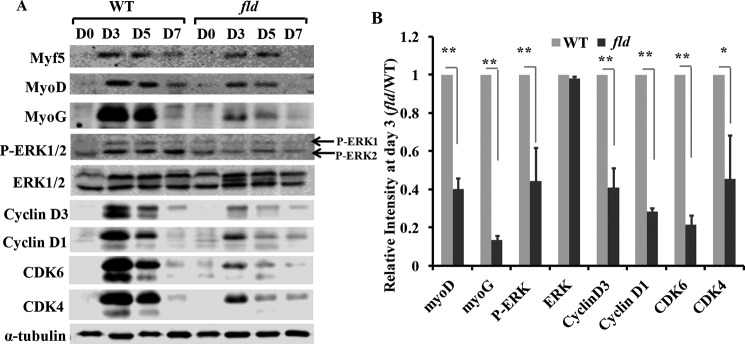
Lipin1 deficiency leads to impaired myogenic differentiation and reduced cyclin D·CDK4/CDK6 complex expression during regenerative myogenesis. A, the expression of myogenic factors and cell cycle regulators was detected by Western blot at days 0, 3, 5, and 7 after injury in TA muscle of fld and WT mice. B, protein expression levels of fld TA muscles at day 3 in response to muscle injury were quantified by densitometry and expressed as -fold change of WT controls. Data are representative of three independent experiments.*, p < 0.05; **, p < 0.01.
To examine the mechanisms underlying defective regeneration in lipin1-deficient skeletal muscle, we also analyzed the protein expression levels of MAPK and cyclin D complexes. These proteins have been shown to be important regulators for myoblast differentiation and myoblast cell cycle withdrawal (30–32). As shown in Fig. 3, although total ERK1/2 expression was not altered between fld and WT mice, a lower phosphorylation of ERK1/2 was detected in the TA muscles of fld mice. Densitometric analysis indicated a respective reduction of 55 and 48% ERK1/2 phosphorylation at days 3 and 5 after injury. As indicated in Fig. 3, cyclin D1, D3, CDK4, and CDK6 expression levels were markedly increased at day 3 after injury and rapidly decreased to almost baseline levels at day 14. However, compared with the control group, cyclin D1, D3, CDK4, and CDK6 levels were reduced by 71, 59, 54, and 78%, respectively, in fld muscle at day 3. These data suggest that lipin1 regulates skeletal muscle regeneration by mediating ERK phosphorylation and cyclin D·CDK complex-mediated cell cycle withdrawal.
FIGURE 3.
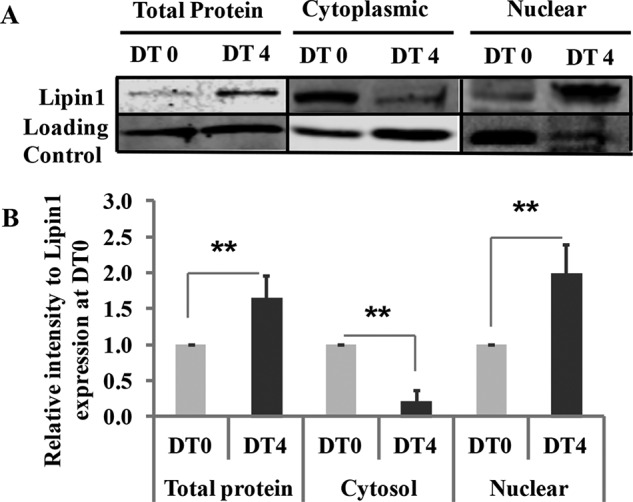
Nuclear translocation of Lipin1 is required for myoblast differentiation. Total cell lysates or cytoplasmic and nuclear protein fractions from differentiated C2C12 were collected at 0 and 4 days after differentiation treatment (DT). A, the expression levels of endogenous lipin1 were measured by Western blot. Loading control for whole cell protein is α-tubulin, for cytoplasmic protein is GAPDH, and for nuclear protein is histone H3. B, densitometry quantification of lipin1 expression at DT4 was quantified and expressed as -fold change over lipin1 expression at DT0. Data are representative of three independent experiments. **, p < 0.01.
Lipin1 Is Required for Myoblast Differentiation and Lipin1-deficient Myoblasts Had Reduced Capacity for Differentiation in Vitro
To examine the role of lipin1 in myoblast differentiation, C2C12 myoblasts were used to measure lipin1 expression. Compared with day 0, lipin1 mRNA (data not shown) and protein expression levels (Fig. 3A) were significantly enhanced at day 4 after differentiation treatment, suggesting that lipin1 is required for myoblast differentiation. This is consistent with a previous study showing increased lipin1 mRNA expression in primary human myoblasts from skeletal muscle biopsies after differentiation treatment (33). Interestingly, we also observed that lipin1 exhibited enhanced nuclear redistribution at day 4 after differentiation treatment (Fig. 3B). These data indicate that lipin1 is required for myoblast differentiation, and lipin1 nuclear function could be critical for myoblast differentiation.
To test whether lipin1 deficiency could affect myogenic differentiation, C2C12 myoblasts were infected with adenovirus for knockdown of lipin1 expression (shLpin1). Differentiation was examined at days 0, 2, and 4 after differentiation treatment, and the myoblasts were shifted from growth medium to differentiation medium when reaching confluence. A severe lack of multinucleated myotube formation and enhanced mononucleated myoblasts were evident in shLpin1-treated myoblasts compared with shLacZ control-treated myoblasts, suggesting that lipin1 plays a critical role in myoblast differentiation (Fig. 4A). A calculation of the FI (i.e. percentage of nuclei found in myosin heavy chain-positive cells) revealed that the lipin1 siRNA-treated myotubes had a significantly reduced FI of 23% compared with 52% of the control siRNA-treated myotubes (Fig. 4B). ERK1/2 phosphorylation and cyclin D3 signaling pathways were suppressed at day 4 after differentiation treatment in lipin1-depleted C2C12 myoblasts (Fig. 4, C and D).
FIGURE 4.

Suppression of lipin1 expression suppresses myoblast differentiation. C2C12 myoblasts were infected with adenovirus to knockdown lipin1 (shLpin1) or control shLacZ before differentiation. A, cells were fixed and stained for mf20+ and DAPI (bars, 50 μm). B, fusion index was determined by counting the percentage of nuclei within mf20+ myotubes over total nuclei within five randomly selected fields per sample. C, immunoblots show lipin1 deficiency induces decreased myogenin and (MyoG) cyclin D, and CDK4/CDK6 expression 4 days after the induction of myoblast differentiation. D, densitometry quantification of protein expression in differentiated cells treated with shLpin1 versus control (shLacZ). E, protein expression levels of myogenic differentiation markers and cell cycle markers in differentiated primary myoblasts isolated from WT or fld mice. *, p < 0.05; **, p < 0.01.
Because the C2C12 cell line may behave differently, we prepared primary myoblasts from the hind limb muscles of fld and WT mice and induced differentiation. The protein expression levels of myogenic factors and cell cycle regulators were examined by Western blot at days 0, 2, 4, and 6 after differentiation treatment. Compared with wild-type controls, ERK1/2 phosphorylation and cyclin D3 expression levels were suppressed at day 6 after differentiation treatment in differentiated fld myotubes, which is consistent with what we observed in C2C12 myotubes (Fig. 4E). Moreover, these results match those found in vivo and confirm that lipin1 is essential for effective differentiation during the early stages of myogenesis. Our data are in agreement with previous studies that ERK2 and cyclin D3 are critical for myoblast terminal differentiation (30, 31).
Cyclin D Is a Downstream Target of the ERK Pathway
In both muscle tissues, differentiated C2C12 myoblasts and primary myoblasts, we observed a reduction in ERK phosphorylation and cyclin D3 expression. We thus tested whether cyclin D3 might function as a downstream target of the ERK pathway in myoblast differentiation. C2C12 myoblasts were induced to differentiate in the presence or absence of the ERK phosphorylation inhibitor U0126. In the absence of U0126, C2C12 myoblasts became elongated by day 4 in differentiation medium (Fig. 5A). In contrast, U0126-treated myoblasts remained mononuclear with very few multinucleated myotubes. As anticipated, Western blot results showed that ERK phosphorylation was inhibited (Fig. 5B). U0126 treatment reduced the expression of myoD and myoG. Consistent with what we observed in the in vitro and in vivo studies, U0126 also decreased cyclin D3, cyclin D1, CDK4, and CDK6 expression, suggesting that cyclin D functions as a downstream target of ERK.
FIGURE 5.

Cyclin D complex is the downstream target of ERK. C2C12 cells were induced to differentiation in the presence or absence of ERK phosphorylation inhibitor 10 μm U0126 for 48 h. A, 4 days after differentiation, cells were fixed and stained with mf20 and DAPI (bars, 50 μm). B, cell lysate was harvested, and protein expression was measured by Western blot. NT, non-transfected cells.
Lipin1 and Lipin1-activated ERK in Cytosol Undergo Enhanced Nuclear Translocation during Myoblast Differentiation
Dynamic subcellular translocation of many regulatory proteins is critical to induce transcription and regulates many intracellular processes including proliferation, differentiation, development, and survival. Upon activation, ERKs translocate from the cytoplasm to the nucleus (34). Lipin1 is continuously trafficked between the nucleus and cytoplasm to perform its enzymatic activity and nuclear functions (1, 4, 6, 7, 35, 36). The translocation of lipin1 within the cells is closely associated with its phosphorylation status. To examine how lipin1 activates ERK, we measured the cytosolic and nuclear distribution of lipin1 and phosphorylated ERK during the differentiation process. We found that ERK phosphorylation increased at day 2 after differentiation, suggesting that ERK was activated at the early stages of myoblast differentiation. From day 2 to day 6 after differentiation, the distribution of both lipin1 and phosphorylated ERK was increased in the nucleus and decreased in the cytosol. If lipin1 activates ERK in the nucleus we would not expect the concurrent decrease in lipin1 and phospho-ERK expression in the cytosol. Therefore, our data suggest that lipin1 activates ERK in the cytosol, and during myoblast differentiation both lipin1 and ERK exhibit enhanced nuclear translocation (Fig. 6A). To further examine whether ERK phosphorylation is regulated by lipin1 dephosphorylation, we transfected C2C12 myoblasts with either lipin1 9A, 17XACA, or lipin1 WT and monitored the ERK phosphorylation by Western blot. Lipin1 9A contains alanine substitutions of all contiguous basic residues within a polybasic motif of lipin1 sequence (4) and is predominantly localized in the cytoplasm, whereas 17XACA, a mutation where the serine and threonine residues are replaced by alanines at 17 of the 19 phosphorylation sites in lipin1, was mainly localized in the nucleus (Fig. 6B). Interestingly, we found that lipin1 mutants localized to the nucleus were more able to induce ERK phosphorylation than in the cytoplasm (Fig. 6C). To further test this finding, C2C12 cells were treated with Torin-1, which is an mTOR inhibitor and has been shown to cause a complete redistribution of lipin1 from the cytoplasm to the nucleus (6). Consistent with Peterson et al. (6), immunostaining for lipin1 revealed that endogenous lipin1 in C2C12 myoblasts was found primarily within the cytosol, whereas treatment with Torin-1 caused a dramatic re-localization to the nucleus (Fig. 6D). Additionally, we observed that Torin-1 treatment enhanced ERK1/2 phosphorylation by 3.6-fold from three independent experiments (Fig. 6E). Cyclin D3 expression also increased.
FIGURE 6.

Lipin1 and lipin1-activated ERK in cytosol undergo enhanced nuclear translocation during myoblast differentiation. A, C2C12 myoblasts were induced to differentiation for 6 days in differentiation medium containing 2% horse serum. Proteins from cytosol and nuclear fractions were separated from myoblasts (day 0), or differentiated myotubes were collected at days 2, 4, and 6 after differentiation treatment. Protein expression levels of lipin1, total ERK, and phosphorylated ERK were examined in the cytosol and nuclear fractions. GAPDH and histone were used as loading controls for cytosol and nuclear proteins, respectively. C2C12 cells were transfected with lipin1 mutants including 9A (cytoplasmic), 17XACA (nucleus), and lipin1 WT (both cytoplasmic and nucleus) with differing nucleo-cytoplasmic distributions. 24 h after transfection, cells were fixed, and subcellular localization of 9A, 17XACA, and lipin1 WT were examined by anti-HA antibody (red) and DAPI (blue) (bars, 20 μm) (B), or cell lysate was harvested, and ERK1/2, phosphorylated ERK1/2, and cyclin D3 expression levels were identified by Western blot (C). Non-transfected cells (NT) were used as a negative control. To examine the effect of the nuclear accumulation of endogenous lipin1 on ERK1/2 phosphorylation, C2C12 cells were treated with 250 nm Torin-1 for 24 h. Cells were fixed, endogenous lipin1 expression was determined by anti-lipin1 antibody (red), and nuclei were identified by DAPI (blue) (bars, 50 μm) (D), or cell lysate was harvested, and ERK1/2, P-ERK1/2, and cyclin D3 expression levels were identified by Western blot (E). Immunoblots are from one of five independent experiments. F, lipin1 may interact with ERK by forming a complex during nucleus translocation. C2C12 myoblasts were transfected with HA-lipin1 and EGFP-ERK2 for 48 h. Subcellular localization of lipin1 and ERK2 was examined by immunostaining with anti-HA and anti-GFP antibodies. IP, immunoprecipitation; WB, Western blot. G, after transfection, the cell lysates were subjected to immunoprecipitation with mouse anti-GFP or rabbit-HA antibody. The immunoprecipitates were then immunoblotted with rabbit anti-HA or mouse anti-GFP antibody. Cell lysates were also immunoblotted with anti-GFP and anti-HA antibodies.
The correlation of lipin1 and ERK in cellular compartments raises a question about whether the two proteins translocate to the nucleus as a complex. To answer that, we co-transfected myoblasts with HA-tagged lipin1 and EGFP-tagged ERK2. Subcellular localization of HA-lipin1 and EGFP-ERK2 was examined by indirect immunofluorescence microscopy with antibodies against anti-HA and anti-GFP (Fig. 6G). Both lipin1 and ERK2 were mainly localized in cytoplasm and partially localized in nucleus. We also measured the interaction of lipin1 and ERK2 by protein immunoprecipitation using either anti-GFP or anti-HA antibody. The interaction between lipin1 and ERK2 was determined by Western blot using anti-HA or anti-GFP antibody. We found that by pulldown of either ERK or lipin1, we detected lipin1 or ERK2, respectively, suggesting that lipin1 and ERK may translocate into the nucleus by forming a complex (Fig. 6F). These data suggest that lipin1 activates ERK in the cytoplasm, and the two are combined to form a nuclear translocation complex that up-regulates cyclin D3 expression.
Discussion
This study was designed to investigate the role of lipin1 in skeletal muscle regeneration. For this purpose we employed an in vivo model of skeletal muscle regeneration and characterized the time course of regeneration in lipin1-deficient fld mice after muscle injury. We evaluated the consequence of lipin1 deficiency in skeletal muscle regeneration and explored the signaling pathway by which lipin1 regulates the regeneration process. We then performed a series of in vitro experiments to mechanistically define the role of lipin1 in myogenesis.
In both animal studies, primary myoblasts and C2C12 myoblasts, we demonstrated that lipin1 plays an important role in skeletal muscle regeneration and implicated the regulation of ERK activity and expression of the cyclin D complex. This is consistent with previous observations that cyclin D3 knock-out mice exhibited decreased myofiber size at 21 days of regeneration and that cyclin D3 deficiency was associated with reduced proliferative capacity and impaired G1/S progression (31, 37, 38). In contrast, cyclin D3 overexpression promotes myogenic differentiation as demonstrated by increased MyoD and MyoG expression (37). Unlike cyclin D3, cyclin D1·CDK4 activity is focused on myoblast proliferation and is reported to suppress skeletal muscle differentiation by blocking the activity of the MEF2 family of transcriptional regulators (39, 40).
Down-regulation of cyclin D3 due to lipin1 deficiency after muscle injury is consistent with impaired myoblast proliferation. However, the mechanism driving these effects on myoblast proliferation and differentiation is unclear but is likely more complex than what we present. Overall, lipin1 deficiency significantly suppressed MyoD and MyoG expression during regeneration, suggesting that lipin1 deficiency affected both the skeletal muscle proliferation and differentiation process.
In skeletal muscle myoblasts, we verified that lipin1 depletion suppressed skeletal muscle regeneration through inhibition of ERK phosphorylation and cyclin D3 expression. Knockdown of lipin1 expression inhibits myoblast differentiation characterized by increased mononuclear myoblast accumulation and decreased myoblast fusion. Furthermore, with the ERK inhibitor, U0126 treatment resulted in impaired myogenesis and down-regulation of cyclin D3, suggesting that cyclin D3 acts as a downstream target of ERK during myogenesis. Consistent with what we observed, ERK1/2 is required for myoblast terminal differentiation and multinucleated muscle fiber formation (30). Activation of ERK signaling occurs at early stages of muscle regeneration (42, 43) and in wound repair (43, 44), suggesting that ERK phosphorylation has some beneficial effects. Phosphatidic acid accumulation in peripheral nerves of lipin1-deficient fld mice increases ERK1/2 activation, mediating Schwann cell dedifferentiation and proliferation (45). However, although lipin1-deficient TA muscles have increased phosphatidic acid levels due to the lack of phosphatidic acid phosphatase activity, the baseline levels of phosphorylated ERK did not increase in fld mice compared with WT mice (Fig. 3). Similarly, Kok et al. (46) did not find significant differences in ERK1/2 phosphorylation in fld hearts, suggesting that the effect of phosphatidic acid on ERK phosphorylation may be tissue-specific. ERK1/2 is required for the transcriptional activation of D-type cyclins and plays a fundamental role in the G0/G1 to the S phase transition. ERK1/2 phosphorylation induces c-Myc, which promotes the transcriptional activation of D-type cyclins (47, 48). Sustained ERK activation results in c-Fos, Fra-1, Fra-2, c-Jun, and JunB accumulation, which stabilizes these transcription factors, enabling the activation of cyclin D expression several hours after stimulation (49, 50). ERK nuclear influx causes the immediate dislodgement of retinoblastoma from its association to lamin A, triggering cell cycle entry (51). All these results unveil that cyclin D3 is the downstream target of ERK1/2.
We demonstrated that lipin1 promotes ERK1/2 phosphorylation, the two form a complex, and the nuclear translocation of this complex is required during skeletal muscle regeneration. This suggests that lipin1 activates ERK in the cytosol, and during myoblast differentiation both lipin1 and ERK translocate to the nucleus. However, we do not exclude the possibility that dephosphorylated lipin1 may also induce ERK phosphorylation, as nuclear localization of the lipin1 mutant (17XACA) promotes ERK1/2 phosphorylation and cyclin D3 expression. Furthermore, we verified the impact of nuclear lipin1 on ERK activation using the mTOR inhibitor, Torin-1, to increase lipin1 nuclear accumulation in C2C12 muscle cells. Lipin1 protein, which predominantly resides in the cytosol, also exhibited nuclear localization. Lipin1 nuclear entry could be regulated by the nuclear localization sequence (4) or dephosphorylation (6, 7). Recent studies suggest that lipin1 is dephosphorylated in response to oleic acid or epinephrine (7), CTDNEP1·NEP1-R1 complex (52), and serine/threonine protein phosphatase-1 catalytic subunit (PP1c) (53) in vitro, suggesting that skeletal muscle regeneration capability could be improved by increasing lipin1nuclear distribution.
Previous studies have demonstrated that ERK1/2 is activated by a diverse array of transmembrane receptors usually via GTP-loading of Ras isoforms (54) or the activation of second messenger kinases such as protein kinase C (55). Activated Raf kinases phosphorylate two serine residues in the activation segment of MEK1/2. Activated MEK1/2, in turn, phosphorylates ERK1/2 on threonine and tyrosine residues (56). In our study we measured MEK and pMEK expression with unexpected results. Moreover, the Raf/MEK1/ERK1/2 pathway has been reported to inhibit myogenesis through cytoplasmic retention of MEF2 (41), whereas lipin1-induced ERK1/2 phosphorylation promotes myogenesis. Collectively, this suggests that lipin1 is a unique and important regulator for ERK1/2 activation during skeletal muscle regeneration. However, further studies are needed to examine whether the effect of lipin1 on ERK activation is through direct interaction with lipin1 or indirectly through adaptor proteins.
In this study we characterized the role of lipin1 in skeletal muscle regeneration in response to acute injury. Although skeletal muscle regeneration mimics a developmental process, the actual role of lipin1 in skeletal muscle development remains to be explored. Cyclin D3 plays a cell-autonomous and non-redundant function in regulating the satellite cell pool involved in proliferation, differentiation, and self-renewal (31). Because lipin1 deficiency suppresses cyclin D3 expression, it would be important to evaluate this effect on satellite cell population and function.
In conclusion, these results reveal a previously unknown role of lipin1 in muscle regeneration in addition to its well known functions as a phosphatidic acid phosphatase enzyme and a transcriptional co-regulator. In response to injury, lipin1 is up-regulated and activates ERK in the cytoplasm to form a complex, which subsequently translocates to the nucleus during myoblast differentiation to regulate the downstream effector cyclin D3-mediated cell cycle withdrawal (Fig. 7). These results significantly expand our understanding of skeletal muscle regeneration and provide insight into the complicated pathophysiology of muscle diseases.
FIGURE 7.
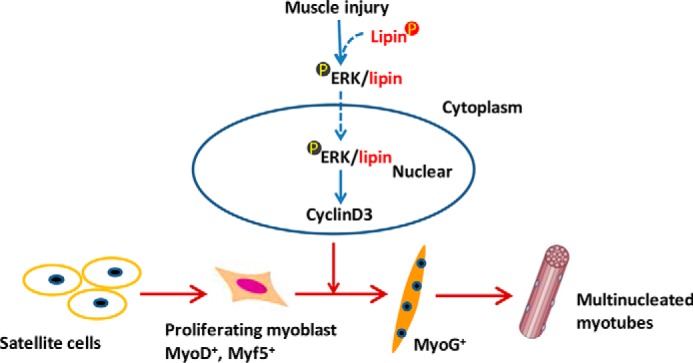
Lipin1 plays an important role in skeletal muscle regeneration. In response to injury, lipin1 promotes ERK1/2 phosphorylation in the cytoplasm, and the two are combined to form a complex. The nuclear translocation of this complex is required to up-regulate cyclin D3 expression during skeletal muscle regeneration.
Author Contributions
H. R. and K. A. E. conceived and designed this work. W. J., J. Z., X. Zhuang, X. Zhang, T. L., and H. R. contributed to the design, performed and analyzed the experiments, and interpreted the data. H. R. wrote the manuscript. H. R. and K. A. E. revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
This work was supported, in part, by resources at the Saha Cardiovascular Center and Gill Heart institute in the Internal Medicine Department, and Center of Biomedical Research Excellence on Obesity and Cardiovascular Diseases at the University of Kentucky, Lexington, KY. We sincerely thank Dr. Thurl Harris (University of Virginia) for the generous gift of lipin1 adenoviruses (shLpin1) and control shLacZ, and Dr. Andrew Morris and Dr. Susan Smyth (University of Kentucky) for their continuous encouragement for this work.
This work was supported, in whole or in part, by National Institutes of Health Grants P20 GM103527-06 (Center of Biomedical Research Excellence on Obesity and Cardiovascular Diseases) and NIH AR061939 (to K. A. E.). This work was also supported by American Heart Association Beginning Grant-in-aid 11BGIA7710059 and National Scientist Development Grant 12SDG12050697 (to H. R.). The authors declare that they have no conflicts of interest with the contents of this article.
- fld
- fatty liver dystrophy
- TA
- tibialis anterior
- FI
- fusion index
- EGFP
- enhanced GFP.
References
- 1. Harris T. E., Finck B. N. (2011) Dual function lipin proteins and glycerolipid metabolism. Trends Endocrinol. Metab. 22, 226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nadra K., Médard J. J., Mul J. D., Han G. S., Grès S., Pende M., Metzger D., Chambon P., Cuppen E., Saulnier-Blache J. S., Carman G. M., Desvergne B., Chrast R. (2012) Cell autonomous lipin 1 function is essential for development and maintenance of white and brown adipose tissue. Mol. Cell. Biol. 32, 4794–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Finck B. N., Gropler M. C., Chen Z., Leone T. C., Croce M. A., Harris T. E., Lawrence J. C. Jr., Kelly D. P. (2006) Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab. 4, 199–210 [DOI] [PubMed] [Google Scholar]
- 4. Ren H., Federico L., Huang H., Sunkara M., Drennan T., Frohman M. A., Smyth S. S., Morris A. J. (2010) A phosphatidic acid binding/nuclear localization motif determines lipin1 function in lipid metabolism and adipogenesis. Mol. Biol. Cell 21, 3171–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eaton J. M., Mullins G. R., Brindley D. N., Harris T. E. (2013) Phosphorylation of lipin 1 and charge on the phosphatidic acid head group control its phosphatidic acid phosphatase activity and membrane association. J. Biol. Chem. 288, 9933–9945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peterson T. R., Sengupta S. S., Harris T. E., Carmack A. E., Kang S. A., Balderas E., Guertin D. A., Madden K. L., Carpenter A. E., Finck B. N., Sabatini D. M. (2011) mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harris T. E., Huffman T. A., Chi A., Shabanowitz J., Hunt D. F., Kumar A., Lawrence J. C. Jr. (2007) Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J. Biol. Chem. 282, 277–286 [DOI] [PubMed] [Google Scholar]
- 8. Michot C., Hubert L., Romero N. B., Gouda A., Mamoune A., Mathew S., Kirk E., Viollet L., Rahman S., Bekri S., Peters H., McGill J., Glamuzina E., Farrar M., von der Hagen M., Alexander I. E., Kirmse B., Barth M., Laforet P., Benlian P., Munnich A., JeanPierre M., Elpeleg O., Pines O., Delahodde A., de Keyzer Y., de Lonlay P. (2012) Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise-induced myalgia. J. Inherit Metab. Dis. 35, 1119–1128 [DOI] [PubMed] [Google Scholar]
- 9. Michot C., Hubert L., Brivet M., De Meirleir L., Valayannopoulos V., Müller-Felber W., Venkateswaran R., Ogier H., Desguerre I., Altuzarra C., Thompson E., Smitka M., Huebner A., Husson M., Horvath R., Chinnery P., Vaz F. M., Munnich A., Elpeleg O., Delahodde A., de Keyzer Y., de Lonlay P (2010) LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum. Mutat. 31, E1564–E1573 [DOI] [PubMed] [Google Scholar]
- 10. Zeharia A., Shaag A., Houtkooper R. H., Hindi T., de Lonlay P., Erez G., Hubert L., Saada A., de Keyzer Y., Eshel G., Vaz F. M., Pines O., Elpeleg O. (2008) Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am. J. Hum. Genet. 83, 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tseng B. S., Zhao P., Pattison J. S., Gordon S. E., Granchelli J. A., Madsen R. W., Folk L. C., Hoffman E. P., Booth F. W. (2002) Regenerated mdx mouse skeletal muscle shows differential mRNA expression. J. Appl. Physiol. 93, 537–545 [DOI] [PubMed] [Google Scholar]
- 12. Douglas D. S., Moran J. L., Bermingham J. R. Jr., Chen X. J., Brindley D. N., Soliven B., Beier D. R., Popko B. (2009) Concurrent Lpin1 and Nrcam mouse mutations result in severe peripheral neuropathy with transitory hindlimb paralysis. J. Neurosci. 29, 12089–12100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghahramani Seno M. M., Trollet C., Athanasopoulos T., Graham I. R., Hu P., Dickson G. (2010) Transcriptomic analysis of dystrophin RNAi knockdown reveals a central role for dystrophin in muscle differentiation and contractile apparatus organization. BMC Genomics 11, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanoudou D., Haslett J. N., Kho A. T., Guo S., Gazda H. T., Greenberg S. A., Lidov H. G., Kohane I. S., Kunkel L. M., Beggs A. H. (2003) Expression profiling reveals altered satellite cell numbers and glycolytic enzyme transcription in nemaline myopathy muscle. Proc. Natl. Acad. Sci. U.S.A. 100, 4666–4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ciciliot S., Schiaffino S. (2010) Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 16, 906–914 [DOI] [PubMed] [Google Scholar]
- 16. Carlson B. M., Faulkner J. A. (1983) The regeneration of skeletal muscle fibers following injury: a review. Med. Sci. Sports Exerc. 15, 187–198 [PubMed] [Google Scholar]
- 17. Bentzinger C. F., Wang Y. X., Rudnicki M. A. (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 4, a008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chargé S. B., Rudnicki M. A. (2004) Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238 [DOI] [PubMed] [Google Scholar]
- 19. Ustanina S., Carvajal J., Rigby P., Braun T. (2007) The myogenic factor Myf5 supports efficient skeletal muscle regeneration by enabling transient myoblast amplification. Stem Cells 25, 2006–2016 [DOI] [PubMed] [Google Scholar]
- 20. Hasty P., Bradley A., Morris J. H., Edmondson D. G., Venuti J. M., Olson E. N., Klein W. H. (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501–506 [DOI] [PubMed] [Google Scholar]
- 21. Parker M. H., von Maltzahn J., Bakkar N., Al-Joubori B., Ishibashi J., Guttridge D., Rudnicki M. A. (2012) MyoD-dependent regulation of NF-κB activity couples cell-cycle withdrawal to myogenic differentiation. Skelet Muscle 2, 6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Micheli L., Leonardi L., Conti F., Maresca G., Colazingari S., Mattei E., Lira S. A., Farioli-Vecchioli S., Caruso M., Tirone F. (2011) PC4/Tis7/IFRD1 stimulates skeletal muscle regeneration and is involved in myoblast differentiation as a regulator of MyoD and NF-κB. J. Biol. Chem. 286, 5691–5707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kiess M., Gill R. M., Hamel P. A. (1995) Expression of the positive regulator of cell cycle progression, cyclin D3, is induced during differentiation of myoblasts into quiescent myotubes. Oncogene 10, 159–166 [PubMed] [Google Scholar]
- 24. Cenciarelli C., De Santa F., Puri P. L., Mattei E., Ricci L., Bucci F., Felsani A., Caruso M. (1999) Critical role played by cyclin D3 in the MyoD-mediated arrest of cell cycle during myoblast differentiation. Mol. Cell. Biol. 19, 5203–5217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang K., Sha J., Harter M. L. (2010) Activation of Cdc6 by MyoD is associated with the expansion of quiescent myogenic satellite cells. J. Cell Biol. 188, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ezhevsky S. A., Nagahara H., Vocero-Akbani A. M., Gius D. R., Wei M. C., Dowdy S. F. (1997) Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc. Natl. Acad. Sci. U.S.A. 94, 10699–10704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Richter T., Nährig J., Komminoth P., Kowolik J., Werner M. (1999) Protocol for ultrarapid immunostaining of frozen sections. J. Clin. Pathol. 52, 461–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yaffe D., Saxel O. (1977) Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725–727 [DOI] [PubMed] [Google Scholar]
- 29. Shi X., Garry D. J. (2006) Muscle stem cells in development, regeneration, and disease. Genes Dev. 20, 1692–1708 [DOI] [PubMed] [Google Scholar]
- 30. Li J., Johnson S. E. (2006) ERK2 is required for efficient terminal differentiation of skeletal myoblasts. Biochem. Biophys. Res. Commun. 345, 1425–1433 [DOI] [PubMed] [Google Scholar]
- 31. De Luca G., Ferretti R., Bruschi M., Mezzaroma E., Caruso M. (2013) cyclin D3 critically regulates the balance between self-renewal and differentiation in skeletal muscle stem cells. Stem Cells 31, 2478–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Knight J. D., Kothary R. (2011) The myogenic kinome: protein kinases critical to mammalian skeletal myogenesis. Skelet Muscle 1, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sterrenburg E., Turk R., 't Hoen P. A., van Deutekom J. C., Boer J. M., van Ommen G. J., den Dunnen J. T. (2004) Large-scale gene expression analysis of human skeletal myoblast differentiation. Neuromuscul. Disord. 14, 507–518 [DOI] [PubMed] [Google Scholar]
- 34. Plotnikov A., Chuderland D., Karamansha Y., Livnah O., Seger R. (2011) Nuclear extracellular signal-regulated kinase 1 and 2 translocation is mediated by casein kinase 2 and accelerated by autophosphorylation. Mol. Cell. Biol. 31, 3515–3530 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Liu G. H., Gerace L. (2009) Sumoylation regulates nuclear localization of lipin-1α in neuronal cells. PLoS ONE 4, e7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Péterfy M., Harris T. E., Fujita N., Reue K. (2010) Insulin-stimulated interaction with 14–3-3 promotes cytoplasmic localization of lipin-1 in adipocytes. J. Biol. Chem. 285, 3857–3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gurung R., Parnaik V. K. (2012) cyclin D3 promotes myogenic differentiation and Pax7 transcription. J. Cell Biochem. 113, 209–219 [DOI] [PubMed] [Google Scholar]
- 38. Mariappan I., Gurung R., Thanumalayan S., Parnaik V. K. (2007) Identification of cyclin D3 as a new interaction partner of lamin A/C. Biochem. Biophys. Res. Commun. 355, 981–985 [DOI] [PubMed] [Google Scholar]
- 39. Shiomi K., Kiyono T., Okamura K., Uezumi M., Goto Y., Yasumoto S., Shimizu S., Hashimoto N. (2011) CDK4 and cyclin D1 allow human myogenic cells to recapture growth property without compromising differentiation potential. Gene Ther. 18, 857–866 [DOI] [PubMed] [Google Scholar]
- 40. Lazaro J. B., Bailey P. J., Lassar A. B. (2002) cyclin D-cdk4 activity modulates the subnuclear localization and interaction of MEF2 with SRC-family coactivators during skeletal muscle differentiation. Genes Dev. 16, 1792–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Winter B., Arnold H. H. (2000) Activated raf kinase inhibits muscle cell differentiation through a MEF2-dependent mechanism. J. Cell Sci. 113, 4211–4220 [DOI] [PubMed] [Google Scholar]
- 42. Stratos I., Richter N., Rotter R., Li Z., Zechner D., Mittlmeier T., Vollmar B. (2012) Melatonin restores muscle regeneration and enhances muscle function after crush injury in rats. J. Pineal. Res. 52, 62–70 [DOI] [PubMed] [Google Scholar]
- 43. Yeow K., Cabane C., Turchi L., Ponzio G., Dérijard B. (2002) Increased MAPK signaling during in vitro muscle wounding. Biochem. Biophys. Res. Commun. 293, 112–119 [DOI] [PubMed] [Google Scholar]
- 44. Huh J. E., Nam D. W., Baek Y. H., Kang J. W., Park D. S., Choi D. Y., Lee J. D. (2011) Formononetin accelerates wound repair by the regulation of early growth response factor-1 transcription factor through the phosphorylation of the ERK and p38 MAPK pathways. Int. Immunopharmacol. 11, 46–54 [DOI] [PubMed] [Google Scholar]
- 45. Nadra K., de Preux Charles A. S., Médard J. J., Hendriks W. T., Han G. S., Grès S., Carman G. M., Saulnier-Blache J. S., Verheijen M. H., Chrast R. (2008) Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev. 22, 1647–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kok B. P., Kienesberger P. C., Dyck J. R., Brindley D. N. (2012) Relationship of glucose and oleate metabolism to cardiac function in lipin-1 deficient (fld) mice. J. Lipid Res. 53, 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seth A., Alvarez E., Gupta S., Davis R. J. (1991) A phosphorylation site located in the NH2-terminal domain of c-Myc increases transactivation of gene expression. J. Biol. Chem. 266, 23521–23524 [PubMed] [Google Scholar]
- 48. Daksis J. I., Lu R. Y., Facchini L. M., Marhin W. W., Penn L. J. (1994) Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene 9, 3635–3645 [PubMed] [Google Scholar]
- 49. Weber J. D., Raben D. M., Phillips P. J., Baldassare J. J. (1997) Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem. J. 326, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Balmanno K., Cook S. J. (1999) Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP-1 proteins in CCL39 cells. Oncogene 18, 3085–3097 [DOI] [PubMed] [Google Scholar]
- 51. Rodríguez J., Calvo F., González J. M., Casar B., Andrés V., Crespo P. (2010) ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J. Cell Biol. 191, 967–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han S., Bahmanyar S., Zhang P., Grishin N., Oegema K., Crooke R., Graham M., Reue K., Dixon J. E., Goodman J. M. (2012) Nuclear envelope phosphatase 1-regulatory subunit 1 (formerly TMEM188) is the metazoan Spo7p ortholog and functions in the lipin activation pathway. J. Biol. Chem. 287, 3123–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kok B. P., Skene-Arnold T. D., Ling J., Benesch M. G., Dewald J., Harris T. E., Holmes C. F., Brindley D. N. (2014) Conserved residues in the N terminus of lipin-1 are required for binding to protein phosphatase-1c, nuclear translocation, and phosphatidate phosphatase activity. J. Biol. Chem. 289, 10876–10886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raman M., Chen W., Cobb M. H. (2007) Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112 [DOI] [PubMed] [Google Scholar]
- 55. Zhao Y., Zhang L., Longo L. D. (2005) PKC-induced ERK1/2 interactions and downstream effectors in ovine cerebral arteries. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R164–R171 [DOI] [PubMed] [Google Scholar]
- 56. Roskoski R., Jr. (2012) MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem. Biophys. Res. Commun. 417, 5–10 [DOI] [PubMed] [Google Scholar]