

Background: Patients with pemphigus vulgaris develop antibodies to both cell adhesion molecules and intracellular proteins.
Results: On the keratinocyte cell membrane, pemphigus autoantibodies form complexes with FcRn internalizing and trafficking them to mitochondria.
Conclusion: The pathogenic role of antimitochondrial autoantibodies is complementary to that of anti-desmoglein autoantibodies, because both are required to disrupt the epidermal barrier.
Significance: FcRn represents a common acceptor protein for internalization of autoantibodies to intracellular proteins.
Keywords: antibody, antigen, autoimmunity, keratinocyte, mitochondria, mitochondrial apoptosis, pemphigus, skin
Abstract
Pemphigus vulgaris (PV) is a life-long, potentially fatal IgG autoantibody-mediated blistering disease targeting mucocutaneous keratinocytes (KCs). PV patients develop pathogenic anti-desmoglein (Dsg) 3 ± 1 and antimitochondrial antibodies (AMA), but it remained unknown whether and how AMA enter KCs and why other cell types are not affected in PV. Therefore, we sought to elucidate mechanisms of cell entry, trafficking, and pathogenic action of AMA in PV. We found that PVIgGs associated with neonatal Fc receptor (FcRn) on the cell membrane, and the PVIgG-FcRn complexes entered KCs and reached mitochondria where they dissociated. The liberated AMA altered mitochondrial membrane potential, respiration, and ATP production and induced cytochrome c release, although the lack or inactivation of FcRn abolished the ability of PVIgG to reach and damage mitochondria and to cause detachment of KCs. The assays of mitochondrial functions and keratinocyte adhesion demonstrated that although the pathobiological effects of AMA on KCs are reversible, they become irreversible, leading to epidermal blistering (acantholysis), when AMA synergize with anti-Dsg antibodies. Thus, it appears that AMA enter a keratinocyte in a complex with FcRn, become liberated from the endosome in the cytosol, and are trafficked to the mitochondria, wherein they trigger pro-apoptotic events leading to shrinkage of basal KCs uniquely expressing FcRn in epidermis. During recovery, KCs extend their cytoplasmic aprons toward neighboring cells, but anti-Dsg antibodies prevent assembly of nascent desmosomes due to steric hindrance, thus rendering acantholysis irreversible. In conclusion, FcRn is a common acceptor protein for internalization of AMA and, perhaps, for PV autoantibodies to other intracellular antigens, and PV is a novel disease paradigm for investigating and elucidating the role of FcRn in this autoimmune disease and possibly other autoimmune diseases.
Introduction
Pemphigus vulgaris (PV)4 is a life-long, potentially fatal IgG autoantibody-mediated blistering disease affecting oral and/or esophageal surfaces and, sometimes, also the skin. The ultimate goal of pemphigus research is to develop an effective treatment modality that would allow patients to achieve and maintain clinical remission without the need for systemic corticosteroids. PV patients develop pathogenic antibodies against desmogleins (Dsgs) 3 ± 1 and other self-antigens expressed in keratinocytes (KCs) (1, 2). Since the first demonstration of autoimmunity to Dsg molecules in the 1990s, they have been studied in great detail. However, understanding PV pathophysiology is still incomplete, because the ancillary pathways triggered by pathogenic antibodies of other specificities remain largely unknown. Modern research has provided overwhelming evidence for the “multipathogenic,” rather than “monopathogenic,” nature of PV autoimmunity, wherein a constellation of autoantibodies of different antigenic specificities, rather than a single type of autoantibody, such as anti-Dsg 3 ± 1, are responsible for blistering (3–6). Various PVIgG species may concur to cause blistering by acting synergistically with anti-Dsg antibodies, as predicted by the “multiple hit” hypothesis (7). In addition to blocking function of adhesion molecules on keratinocyte cell membrane, PVIgGs elicit pro-apoptotic signaling events causing cell shrinkage, detachment from neighboring KCs, and rounding up, the unique process of keratinocyte detachment and death termed apoptolysis (8).
Antimitochondrial antibodies (AMA) are a critical link of the PV pathophysiology because of the following: 1) AMA can launch the intrinsic apoptotic program, which has been documented in KCs exposed to PVIgGs (9–11); and 2) adsorption of AMA abolishes the ability of PVIgGs to cause acantholysis (i.e. keratinocyte detachment) both in vitro and in vivo (9). AMA produced by PV patients can disrupt the electron transfer chain, resulting in a loss of electrochemical gradient across the inner membrane, increase production of reactive oxygen species, and reduce the ability of KCs to respond to stress (12). In turn, the pharmacological agents known to alleviate disease severity in PV patients protect KCs from the mitochondrial damage by AMA and abolish acantholysis in the passive antibody transfer neonatal mouse model of PV (12). Among mitochondrial proteins targeted by AMA are the mitochondrial nicotinic acetylcholine receptors coupled to inhibition of intrinsic apoptosis by preventing formation of mitochondrial permeability transition pore (13). Binding of PVIgGs to these receptors compromises their anti-apoptotic activities, as evidenced by cytochrome c (CytC) release. The mitochondrial permeability transition pore opening causes massive swelling of mitochondria, rupture of outer membrane, and release of intermembrane components, such as CytC, that induce formation of the apoptosome, a multimeric complex formed by Apaf-1 and procaspase-9 providing for activation of caspase-9, which is known to be activated by PVIgGs in KCs (10). It remained unknown, however, whether and how AMA enter KCs and why the cell types other than KCs that possess the same mitochondrial self-antigens are not affected by AMA produced by PV patients who do not develop pathological changes outside of the stratified squamous epithelium lining the mucocutaneous surfaces.
Although it is well established that FcRn (“neonatal” Fc receptor for IgG) contributes to IgG-mediated autoimmune diseases by extending the life span of autoantibodies (14, 15), recent research has demonstrated a possibility of IgG internalization through interactions with FcRn (16–20). FcRn is a heterodimer of a β2-microglobulin light chain and a major histocompatibility complex class I-like heavy chain. At neutral pH, IgG uptake into cells occurs via fluid phase pinocytosis with subsequent binding to FcRn in the acidified environment of the endosomal compartment. The FcRn-IgG complex is then trafficked through cellular conduits to bypass lysosomal degradation and is finally released from the cell. However, FcRn can also bind IgG on the cell membrane, followed by internalization of the FcRn-IgG complex (16–20).
The critical role of FcRn in pemphigus pathophysiology was illustrated by the inability to induce acantholysis in the neonatal knock-out mouse lacking FcRn by passive transfer of anti-Dsg antibodies (21). Furthermore, an excess of normal IgG (NIgG), which can saturate FcRn, protects cultured KCs from PVIgG-induced apoptolysis (10), and high doses of intravenous IgG are therapeutic in PV (22). The role of FcRn in pemphigus pathophysiology, however, has always been interpreted as protection of pathogenic PVIgGs from degradation. Indeed, KCs express FcRn (23) and PVIgGs internalized by KCs are seen within endosomes (24). However, an intracellular release of IgG from its endosomal complex with FcRn and subsequent binding to and inactivation of its intracellular targets have been demonstrated in epithelial cells (25–27), suggesting that in addition to protecting PVIgGs from degradation the FcRn may be directly involved in delivery of AMA to their intracellular target antigens.
In this study, we sought to elucidate the mechanisms of cell entry, trafficking, and pathogenic action of AMA produced by patients with PV. We found that PVIgGs form complexes with FcRn on the cell membrane of KCs, which are then internalized and trafficked to the mitochondria. Inactivation of FcRn abolishes the ability of PVIgG to reach and damage mitochondria, and cause shrinkage of KCs. We also demonstrated that AMA synergize with anti-Dsg antibodies to induce epidermal acantholysis. These results shed new light on the mechanism of cell entry of pathogenic PVIgGs and emphasized the importance of pharmacological protection of mitochondria in nonsteroidal treatment of PV patients.
Experimental Procedures
Test Sera and Reagents
We used five serum specimens from patients with acute mucocutaneous PV containing AMA (1) and normal human sera purchased from Bioreclamation, Inc. (Westbury, NY). This research has been approved by Institutional Review Boards at University of California at Irvine. The diagnosis of PV was made based on the results of comprehensive clinical and histological examinations and immunological studies that included both direct immunofluorescence of skin biopsies and indirect immunofluorescence of the patients' sera on various epithelial substrates. The titer of “intercellular” antibodies determined on monkey esophagus ranged from 1/320 to 1/2560. The presence of anti-Dsg1 and Dsg3 antibodies in each PV serum was established using the MESACUP Dsg1 and Dsg3 ELISA test system (MBL, Nagoya, Japan). All test sera were treated for 30 min at 56 °C to inactivate complement. The IgG fractions of PV sera (i.e. PVIgGs) and NIgG were isolated by FPLC protein G affinity chromatography using the FPLC system purchased from Amersham Biosciences and following the manufacturer's protocol, as detailed by us elsewhere (10). Mouse anti-FcRn (SC-271745) and anti-voltage-dependent anion channel 1 (VDAC1; SC-390996) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Rabbit anti-E1 (GTX104015) and anti-apoptosis-inducing factor (ab32516) antibodies were purchased from Gentex (Irvine, CA) and Abcam (Cambridge, MA), respectively. Mouse anti-β-tubulin antibody (T4026) was from Sigma. MitoTracker Green (M7514), Alexa Fluor 350 anti-mouse IgG (A11068), Alexa Fluor 594 anti-human IgG (A11014), and Alexa Fluor 488 anti-human IgG (A11013) antibodies were purchased from Life Technologies, Inc. JC-1 dye was purchased from Biotium, Inc. (Hayward, CA; catalog no. 70014). Polyclonal rabbit anti-Dsg1 antibody was purchased from OriGene (Rockville, MD), and goat anti-Dsg3 was from R&D Systems Inc. (Minneapolis, MN). Both recognized mouse Dsg molecules. The human CytC immunoassay kit was purchased from R&D Systems, Inc., and the assay was performed following a protocol provided by the manufacturer. The Px4-3 clone of the single-chain variable fragment (scFv) binding mouse Dsg1 was provided by Dr. John Stanley (University of Pennsylvania).
Cell Culture Experiments
The HET-1A cell line, an established clonal population of SV40-immortalized human esophageal squamous epithelial cells (i.e. KCs) widely used for the studies of apoptosis (28), was purchased from American Type Culture Collection (ATCC; Manassas, VA; catalog no. CRL-2692) and propagated in the Clonetics brand bronchial cell medium without retinoic acid (Cambrex Bio Sciences, Walkersville, MD), as detailed by us elsewhere (29). The HET-1A line was used instead of primary human KCs to avoid donor- and passage-related variations and for consistency with our previous studies of AMA in PV (12). The human embryonic kidney epithelial cell line HEK293, human bladder carcinoma line T24, human normal breast epithelial cell line HBL100, and human breast carcinoma cell line MB231 were purchased from ATCC and propagated in the recommended media. Mitochondria were isolated using the mitochondrial/cytosol fractionation kit from BioVision Research Products (Mountain View, CA) following the manufacturer's instruction. Briefly, the cells were detached by a brief trypsinization, isolated by centrifugation, washed in PBS, resuspended in the Cytosol Extraction Buffer containing a mix of DTT and protease inhibitors, homogenized in an ice-cold tissue homogenizer, and centrifuged at 700 × g for 10 min at 4 °C. The supernatant was recentrifuged at 10,000 × g for 30 min at 4 °C, and the pelleted mitochondrial fraction was resuspended in 100 μl of the Mitochondrial Extraction Buffer. The protein concentration was determined by a Bradford protein assay kit (Bio-Rad). To obtain AMA and non-AMA, the PVIgGs were preabsorbed with either nonmitochondrial or mitochondrial proteins, respectively. As determined by the MESACUP Dsg1 and Dsg3 ELISA test system, the AMA fraction did not contain anti-Dsg1 and Dsg3 antibodies. In co-precipitation experiments, the cells were fractionated by sequential differential centrifugation. After exposure to test antibodies (see under “Results”), experimental and control cells were lysed with a lysis buffer (50 mm Tris-HCl, pH 7.6; 250 mm NaCl; 5 mm EDTA; 0.1% Nonidet P-40; and 50 μm NaF); the salt concentration was adjusted to 125 mm, and the IgGs that penetrated the cells were bound to protein A-Sepharose during an overnight incubation at 4 °C. The resin-bound protein complexes were washed 10 times with PBS supplemented with 0.1% Nonidet P-40 and then were subjected to standard Western blot (WB) analysis.
Assay of Cell-Cell Adhesion
The effects of AMA and non-AMA on keratinocyte cell-cell adhesion were measured using the monolayer permeability assay detailed by us elsewhere (30, 31). Briefly, a confluent monolayer of KCs was formed in the Costar® Transwell® cell culture chambers inserted into the 24-well culture plates 2–3 days after KCs were seeded at a cell density of 1 × 104/100 μl of culture medium into the chambers and cultured at 37 °C in a humid atmosphere with 5% CO2. The monolayers were incubated with test antibodies for different periods of time (see under “Results”) and washed, and the permeability of the monolayer was measured by adding 100 μl of PBS containing [3H]dT (1 μCi/insert; 6.7 Ci/mmol; PerkinElmer Life Sciences) to each culture. Five minutes later, 100-μl aliquots of solution containing [3H]dT were taken in triplicate from each lower chamber. The more the KCs were separated from each other, the more tracer penetrated to the lower chamber through the porous membrane of the upper chamber, and the higher the permeability coefficient (PC) values were obtained as shown in Equation 1,
 |
Analysis of Mitochondrial O2 Respiration by Extracellular Flux Measurement
To measure mitochondrial function in test cell types, we employed a Seahorse Bioscience XF24 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA) and followed the manufacturer's protocol, as detailed by us elsewhere (12). Briefly, the cells were plated in a 0.2% gelatin-coated 24-well Seahorse XF-24 assay plate at 3 × 104 cells/well and were grown for 16 h before being treated for another 24 h with a quintuplicate of test sera at a final concentration of 4% of the total volume of culture medium. On the day of metabolic flux analysis, cells were washed once with freshly prepared Krebs-Henseleit buffer (KHB: 111 mm NaCl, 4.7 mm KCl, 2 mm MgSO4, 1.2 mm Na2HPO4, 2.5 mm glucose and 0.5 mm carnitine; pH 7.4) and incubated in KHB at 37 °C in a non-CO2 incubator for 1 h. Three baseline measurements of oxygen consumption rate (OCR) were taken before sequential injection of the following mitochondrial inhibitors and final concentrations as follows: oligomycin (1 μg/ml), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (3 μm), and rotenone (0.1 μm). Measurements were taken after addition of each of the three inhibitors. The OCR values were automatically calculated and recorded by the Seahorse XF-24 software. The basal respiration was calculated by averaging the three measurements of OCR before injection of inhibitors.
Measurement of Mitochondrial Membrane Potential (ΔΨm)
The changes in ΔΨm induced by test sera were measured using a standard protocol, as detailed by us elsewhere (12). Briefly, KCs were plated in a 6-well plate at a density of 3 × 104 per well and incubated for 16 h to allow cells to adhere to the dish surface, after which the cells were either left untreated (negative control) or exposed to mouse anti-FcRn antibody (2 μg/ml) for 10 min before adding test sera at a final concentration of 4% of the total volume per well. After 24 h of incubation, experimental and control cells were exposed to the fluorescent cationic dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) for 25 min and observed at green and red emission wavelengths using a Zeiss Axioplan II fluorescence microscope (Carl Zeiss, Thornwood, NY). Intact mitochondrial membrane retained the JC-1 dye and forms J-aggregate (orange-red fluorescence), whereas depolarization of membrane decreased the ability to retain the dye that remained as a monomer (green fluorescence). Increased green fluorescence and decreased orange-red fluorescence thus indicated a loss of ΔΨm. In the absence of apoptosis, the lipophilic JC-1 dye bearing a delocalized positive charge accumulates in the mitochondrial matrix and stains the mitochondria bright red, whereas in apoptotic mitochondria ΔΨm dissipates, and the cells display green fluorescence.
ATP Level by Mass Spectrometry Analysis
The keratinocyte monolayers were incubated with PV sera with or without pretreatment with anti-FcRn antibody for 24 h, after which the metabolites were extracted by treatment with 80% HPLC grade methanol for 24 h at room temperature. After centrifugation at low speed, supernatant was separated and dried in an Eppendorf Vacufuge. The dried metabolites were resuspended in 30% acetonitrile with 5% TFA and spotted onto an assay plate. The metabolites were co-crystallized with matrix (α-cyano-4-hydroxycinnamic acid) before being analyzed with an AB Sciex MALDI 5800 TOF/TOF mass spectrophotometer (Foster City, CA). The relative level of ATP was calculated as ratio of 506 m/z (ATP) over 187 m/z (internal control).
Statistical Analysis
The data were analyzed using analysis of variance against an α-level of 0.05 and presented as mean ± S.D. The graphs were created using GraphPad Prism 5.
Results
Blockade of FcRn Abolishes the Ability of PVIgGs to Reach Mitochondria
To investigate the possible contribution of FcRn on the ability of AMA to reach mitochondrial targets, the effects of anti-FcRn antibody on PVIgG activities were measured in monolayers of KCs (i.e. HET-1A cells). In keeping with our previous observations (9), PVIgGs entered KCs and reached mitochondria (Fig. 1A). Preincubation of KCs with anti-FcRn antibody almost completely abolished staining for PVIgGs, indicating that AMAs could not reach mitochondria when the FcRn pathway was blocked (Fig. 1A). The specificity of the anti-FcRn role in the delivery of AMA to the mitochondria was validated by dose-dependent inhibition of PVIgG entry of KCs by anti-FcRn antibody (Fig. 1B). The specificity of anti-FcRn activity to the observed phenomenon was validated by the dose dependence of competitive blockade of AMA entry of KCs (Fig. 1B).
FIGURE 1.

Indispensable role of FcRn in the AMA-mediated damage of KCs. A, WB study of the ability of PVIgGs to reach mitochondrial targets in KCs. The monolayers of KCs grown to ∼80% confluence at 37 °C and 5% CO2 were preincubated for 15 min with 2 μg/ml mouse monoclonal antibody against the extracellular domain of human FcRn or an equal amount of nonimmune mouse IgG (control) and then were exposed for 16 h to 1 mg/ml PVIgG from four patients or NIgG. The mitochondrial protein fraction of exposed cells was isolated as detailed under “Experimental Procedures,” separated by SDS-PAGE, and probed with HRP-conjugated anti-human IgG antibody that visualized PVIgG's heavy and light chains. Pretreatment of keratinocyte monolayer with anti-FcRn antibody (+), but not with control mouse IgG (−), abolished detection of PVIgGs in the subcellular mitochondrial fraction. N = NIgG. B, dose-dependent inhibition of AMA entry of KCs by anti-FcRn antibody. Following the protocol described in the legend to A, the monolayers of KCs were exposed to 1 mg/ml pooled PVIgGs (PV1 + PV3 + PV4) in the presence of 0 (lane 1), 1 (lane 2), or 2 (lane 3) μg/ml anti-FcRn antibody, after which each mitochondrial fraction was isolated and stained for human IgG. The molecular mass (lane MW) markers are shown on the left lane. C and D, control WB experiments showing purity of the nonmitochondrial (NM) and mitochondrial (M) fractions of KCs. The NM fraction, which was composed of the cell membrane and cytosol proteins, and the M fraction were isolated as described under “Experimental Procedures.” The staining for the cell membrane protein Dsg3 is present in the nonmitochondrial but not in the mitochondrial fraction (C), whereas that for the mitochondrial protein apoptosis-inducing factor (AIF) in the mitochondrial but not the nonmitochondrial fraction (D). E, time course study of CytC release in human KCs treated with AMA with or without the anti-FcRn antibody or non-AMA. To obtain AMA, the IgGs isolated from sera four PV patients were pooled and preabsorbed with a mixture of both the cytosolic and the cell membrane protein fractions of KCs, whereas non-AMA were obtained by preabsorption of PVIgGs with the mitochondrial protein fraction (both protein fractions were tested for purity in C and D). Both the AMA and non-AMA were used in the CytC release as detailed under “Experimental Procedures.” KCs were incubated at 37 °C and 5% CO2 for the indicated periods of time in culture medium containing 1.6 mm Ca2+ in the presence of test PVIgGs. Before exposure to AMA, some monolayers were preincubated for 15 min with 2 μg/ml anti-FcRn antibody. The data were obtained in three independent experiments. ▴, AMA; ●, AMA + anti-FcRn antibody; ○, non-AMA; □, NIgG. F, time course study of permeability of keratinocyte monolayers treated with AMA with or without anti-FcRn antibody or non-AMA. KCs grown to confluence on the membrane-bottomed tissue culture insets were exposed to test antibodies, and permeability of the monolayer was determined by measuring the amount of the radioactive tracer moving between cells across the monolayer into the lower chamber as described under “Experimental Procedures.” The data were obtained in three independent experiments. The designations are the same as in E.
Blockade of FcRn Abolishes the Ability of AMA to Trigger CytC Release
The effects of AMA and non-AMA on CytC release were measured in monolayers of KCs after incubation with each of these PVIgG fractions versus NIgG. The purity of the protein fraction used to prepare AMA and non-AMA fractions was confirmed by WB with the cell membrane and mitochondrial marker proteins (Fig. 1, C and D). After 1 h of incubation, CytC release significantly (p < 0.05) increased (Fig. 1E). After 2 h, the AMA-dependent elevation of CytC significantly (p < 0.05) exceeded that of non-AMAs, indicating that binding of AMA to their mitochondrial target antigens increases permeability to CytC, which might trigger the intrinsic apoptotic pathway in KCs. Preincubation of KCs with anti-FcRn antibody prevented the AMA-dependent release of CytC (Fig. 1E), thus providing evidence for the requirement of FcRn for the pathobiological action of AMA. Notably, an increased level of CytC in KCs treated with AMA spontaneously declined by the 4th h of incubation (Fig. 1E), at which point cell viability measured by the trypan blue dye-exclusion test still exceeded 80% (data not shown). This unexpected observation suggested that damage of KCs by AMA is reversible, perhaps due to a self-repair. The minor CytC release induced by non-AMA (Fig. 1E) indicated that other pathways might also be involved, in keeping with a synergistic “cross-talk” of the apoptosis and oncosis pathways in KCs exposed to PVIgGs (10).
Blockade of FcRn Abolishes the Ability of AMA to Cause Keratinocyte Shrinkage
The effect of anti-FcRn antibody on the ability of AMA to increase permeability of the keratinocyte monolayer, resulting from cell shrinkage and detachment from each other (31), was studied using the monolayer permeability assay. AMA increased by severalfold permeability of the monolayer (Fig. 1F). The shrinkage of KCs reached its maximum at the same time as CytC release (Fig. 1E), i.e. 2 h after exposure to PVIgGs, and then spontaneously reversed. Pretreatment with anti-FcRn antibody significantly (p < 0.05) decreased the permeability coefficient (Fig. 1F), indicating that blockade of FcRn protected KCs from the morphological changes resulting from the pathobiological action of AMA.
Functional Inactivation of FcRn Protects Cells from AMA
To confirm that FcRn renders cells accessible to pathogenic PVIgGs, we evaluated the role of FcRn in the ability of AMA to enter KCs using immunofluorescence, and we assessed their ability to alter mitochondrial function of KC by XF24 extracellular flux analysis. As expected, anti-FcRn antibody blocked both internalization of PVIgG (Fig. 2A) and its deleterious effect on mitochondrial respiration (Fig. 2B). Anti-FcRn antibodies alone had no effect on basal respiration.
FIGURE 2.

FcRn is indispensable for cell entry and mitochondrial damage by AMA. A, effect of anti-FcRn antibody on internalization of PVIgGs. The monolayers of KCs were exposed to two different PV sera at a final concentration of 4% with or without pretreatment by anti-FcRn antibody (2 μg/ml), incubated for 1 h, fixed, and immunostained by Alexa Fluor 488 anti-human IgG antibody. B, effects of anti-FcRn antibody on the PVIgG-dependent alterations of mitochondrial respiration. The keratinocyte monolayers were exposed for 16 h to two PV sera after a 10-min pretreatment with anti-FcRn antibody (2 μg/ml) and then subjected to the Seahorse XF bioenergetics assay measuring basal mitochondrial respiration, as described under “Experimental Procedures.” All experiments were performed in triplicate. *, p < 0.0001; compared with mock treatment. C, analysis of FcRn expression in different epithelial cell lines. The WB analysis of FcRn expression was performed in five epithelial cell lines as follows: HET-1A (i.e. KCs), HEK293, HBL-100, MB-231, and T24 as detailed under “Experimental Procedures.” The specific staining with anti-FcRn antibody was seen in HET-1A and HEK293 cells but not in HBL-100, MB-231, and T24 cells. D, analysis of PVIgG effects on basal mitochondrial respiration in the cell lines expressing and not expressing FcRn. Measurements of OCR in HET-1A, HEK293, HBL-100, MB-231, and T24 cells plated at a density of 4 × 104 per well and exposed to PV versus normal donor (control) sera at a final concentration of 4% were performed using the Seahorse XF bioenergetics assays detailed under “Experimental Procedures.” The data represent the means ± S.D. of quintuplicate measurements of the effects of each individual serum in three independent experiments. *, p < 0.05, compared with control.
The contribution of FcRn to the pathogenic action of AMA was also evaluated by comparing the effects of PVIgGs on mitochondrial respiration in the human cell types found by WB to either express FcRn or not. The former included human immortalized KCs (HET-1A) and embryonic kidney cells (HEK293), and the latter included breast epithelial cells (HBL-100) as well as breast (MBL231) and urinary bladder (T24) carcinoma cells. By WB, FcRn was only detected in HET-1A and HEK293 cells (Fig. 2C). Each cell line was incubated with sera from two PV patients for 16 h, after which mitochondrial respiration was measured by the XF24 extracellular flux analysis. Although the control levels of basal mitochondrial respiration were different in different cell types, PVIgGs reproducibly induced abnormalities only in the FcRn-positive cells and had no effect on those lacking FcRn (Fig. 2D).
Taken together, these results demonstrated the pivotal role of FcRn in cell entry of pathogenic AMA, and they also indicated that in addition to KCs, AMA can enter other cell types expressing FcRn.
Association of PVIgG with FcRn
To examine the presumed direct interaction of PVIgG with FcRn, we performed a co-precipitation assay with KCs treated by sera from five PV patients versus NIgG. The IgGs were pulled down with protein A-Sepharose beads, and the PVIgG-FcRn complexes were detected by WB using relevant antibodies. PVIgGs from all patients, but not NIgG, were found in cell lysates, wherein they formed complexes with FcRn (Fig. 3A).
FIGURE 3.

Interactions between PVIgG and FcRn. A, co-precipitation of PVIgG and FcRn. KCs were treated with PV sera from five patients for 16 h, washed, and lysed, and IgGs were pulled down with protein A-Sepharose, as detailed under “Experimental Procedures.” The resin-bound proteins were resolved by SDS-PAGE and probed with the HRP-conjugated anti-human IgG and FcRn antibodies. B, time course of PVIgG-FcRn complexes in subcellular fractions of KCs. Monolayers of KCs treated with PV sera for indicated periods of time were washed, pelleted, and fractionated as described under “Experimental Procedures.” The purity of the cytosol and membrane fractions is illustrated by opposite reactivities for the respective markers β-tubulin and VDAC1. The cytosol, mitochondrial, and cell membrane fractions were incubated with protein A-Sepharose, and resin-bound PVIgG-FcRn complexes were visualized by WB as described in A. The results were reproduced using sera from three different PV patients (data not shown). C, localization of PVIgG-FcRn complexes at mitochondria. KCs were plated onto 12-mm circular cover glasses in 24-well plates at the density of 1 × 105 per well, incubated for 16 h to allow cells to settle, exposed to PV serum for 1 or 10 min, washed three times with PBS, fixed, and immunostained with anti-human IgG (red) or anti-FcRn (blue) antibodies or MitoTracker (green). Thin arrows show co-localization of PVIgG, FcRn, and mitochondria. Thick arrowheads show co-localization of PVIgG and FcRn outside of mitochondria.
Next, we investigated interactions of PVIgG and FcRn in a time course study. KCs were incubated with PV sera for different time points and subjected to subcellular fractionation, and the presence of PVIgG-FcRn complexes was examined in each cell fraction. PVIgGs were detected in the cytosol, mitochondria, and cell membrane fractions 2 min after exposure of KCs to PV serum (Fig. 3B). At this early time point, the relative amount of PVIgG-FcRn complexes was maximal in the cytosol and minimal in the mitochondrial fraction. The presence of PVIgG-FcRn complexes in the mitochondrial fraction increased with time and reciprocally decreased in the cell membrane fraction (Fig. 3B).
The fate of internalized PVIgG-FcRn complexes was studied in KCs by indirect immunofluorescence, which revealed co-localization of PVIgG, FcRn, and the mitochondrial marker MitoTracker 1 min after exposure of KCs to PV serum (Fig. 3C, thin arrows). At this early, but not the late, time point, we also observed co-localization of PVIgG with FcRn without MitoTracker (Fig. 3C, thick arrowheads), which indicated that the PVIgG/FcRn interaction had occurred before the PVIgG-FcRn complexes reached mitochondria. Taken together, these results demonstrated that FcRn can mediate both internalization and intracellular trafficking of PVIgG to mitochondria.
Molecular Mechanisms of the Pro-apoptotic Action of AMA
Our proteomic studies have demonstrated that AMA produced by PV patients bind to pyruvate dehydrogenase complex (PDC) proteins (1, 12). Therefore, we sought to confirm association of PVIgG with the E1 subunit of PDC and to determine functional consequences of such interaction. The co-precipitation experiments revealed PVIgG-E1 complexes in KCs treated with PV sera from five patients (Fig. 4A). Because PDC plays a key role in the production of acetyl-CoA and is involved in the regulation of oxidative phosphorylation leading to ATP synthesis, and because the PVIgG-mediated increase of basal respiration (Fig. 2) was consistent with an uncoupling effect, targeting of E1 by AMA might modulate ATP production. Experimental testing of this hypothesis in KCs exposed to PV sera demonstrated that AMA indeed up-regulated generation of ATP, which could be abolished when KCs were pretreated with anti-FcRn antibody (Fig. 4B).
FIGURE 4.

Interactions PVIgG with mitochondrial proteins regulating PDC activity and ΔΨm. A, co-precipitation of PVIgG and the E1 protein of PDC. KCs were treated with PV sera from five patients for 5 min, washed, and lysed, and the IgGs were pulled down with protein A-Sepharose. The resin-bound proteins were resolved by SDS-PAGE and probed with the HRP-conjugated anti-human IgG and E1 antibodies. B, measurement of ATP production. KCs treated with PV sera with or without pretreatment with anti-FcRn antibody for 24 h were subjected to the ATP assay using TOF mass spectrometry, as detailed under “Experimental Procedures.” The data are mean ± S.D. of six independent experiments. Ab = pretreatment with 2 μg/ml of anti-FcRn antibody. *, p < 0.001, compared with untreated control (C). C, co-precipitation of PVIgG and VDAC1. KCs were treated with PV sera from five different patients for 5 min, washed, and lysed, and the IgGs were pulled down with protein A-Sepharose. The resin-bound proteins were resolved by SDS-PAGE and probed with the HRP-conjugated anti-human IgG and VDAC1 antibodies. D and E, analysis of the effects of anti-FcRn antibody on the PVIgG-dependent loss of ΔΨm. Percent of cells (D) and representative images (E) of KCs treated with PV sera with or without pretreatment with anti-FcRn antibody and subjected to ΔΨm measurement using the JC-1 dye, as described under “Experimental Procedures.” The loss of red fluorescence indicating a loss ΔΨm due to AMA action could be abolished by pretreatment with anti-FcRn antibody.
Our previous functional studies demonstrated that AMA produced by PV patients cause dramatic changes in ΔΨm (12). One possible mechanism involves phosphorylation of VDAC1, because changes of VDAC1 phosphorylation status cause a similar effect (32, 33). Furthermore, AMA binding to mitochondrial nicotinic acetylcholine receptors, which both associate with VDAC and activate protein kinases (34, 35), can cause mitochondrial permeability transition pore opening (13). Therefore, we hypothesized that VDAC1 might be associated with AMA in multiprotein complexes. To test this hypothesis, we performed co-precipitation experiments with KCs treated by five PV sera. We found that VDAC1 was present in the protein complexes precipitated by serum from each of these patients, but not normal control serum (Fig. 4C). As expected, prevention of AMA entry of KCs due to pretreatment with anti-FcRn antibody protected them from PVIgG-dependent loss of ΔΨm (Fig. 4, D and E).
The PVIgG complexes with PDC or VDAC1 proteins were also detected in the co-precipitation experiments employing mitochondria isolated from KCs treated with PV sera (data not shown). Taken together, these results provided a mechanistic insight into the molecular mechanism of AMA-dependent triggering of mitochondrial pro-apoptotic events in KCs targeted by PVIgGs.
Synergy of AMA and Anti-Dsg Antibodies in Epidermal Acantholysis
To determine whether AMA or non-AMA produced by PV patients can induce acantholysis on their own, we treated organ cultures of neonatal mouse skin with AMA or non-AMA for 24 h, followed by histological examination of the hematoxylin and eosin-stained sections. When given alone, neither AMA or non-AMA could induce acantholysis (Fig. 5). This was consistent with the reversible nature of the pathobiological effects of AMA, as shown for CytC release and cell shrinkage (Fig. 1, E and F). Therefore, we hypothesized that AMA synergize with autoantibodies targeting keratinocyte adhesion molecules such as Dsg3 and Dsg1. To test this hypothesis, neonatal mouse skin explants were incubated with commercial anti-Dsg3 and/or anti-Dsg1 antibodies that did not induce acantholysis on their own (Fig. 5). The acantholysis, developed only when AMA, but not non-AMA, were combined with anti-Dsg antibodies. The AMA/anti-Dsg3 combination induced epidermal splitting suprabasally and the AMA/anti-Dsg1 combination induced epidermal splitting subcorneally (Fig. 5), which is consistent with the respective predominant localization of each Dsg molecule in the epidermis (36). When AMA were combined with both anti-Dsg3 and Dsg1 antibodies, acantholysis involved the entire epidermis (Fig. 5).
FIGURE 5.

Synergistic acantholytic activities of AMA and anti-Dsg antibodies in the neonatal mouse skin explant model. Representative images of epidermis in ∼3 × 3-mm pieces of truncal skin of neonatal BALB/c mice incubated at 37 °C and 5% CO2 for 24 h in PBS containing AMA (i.e. PVIgGs preabsorbed with a mixture of the cytosolic and cell membrane proteins of KCs), polyclonal rabbit anti-Dsg1, and/or goat anti-Dsg3 antibodies known to react with the respective mouse Dsg molecules or high (650 ng/μl; scFvH) versus low (163 ng/μl; scFvL) concentrations of scFv. Each experiment was repeated at least three times. Magnification = ×40.
Furthermore, as an alternative approach to test the possible synergy between AMA and anti-Dsg antibodies, we treated mouse skin cultures with decreasing concentrations of the scFv-binding mouse Dsg1 molecule (37) with or without a constant dose of AMA. When given alone, the higher concentrations of scFv did but the lower did not induce visible changes in the epidermis (Fig. 5). The intraepidermal (subcorneal) split, however, was induced when the lower dose of scFv was combined with AMA (Fig. 5).
Taken together, these results clearly demonstrated a synergy of the pathogenic actions of AMA and anti-Dsg antibodies. The indispensable role of FcRn in such synergy has been indicated by the results of passive transfer experiments in FcRn-deficient mice that did not develop acantholysis when treated with pathogenic PVIgGs (21), which provides a control in the in vivo model.
Discussion
The results of this study shed new light on the mechanisms of entry of KCs and mitochondrial damage by AMA in PV. The fact that adsorption of AMA from PVIgGs abolished the ability of PVIgGs to induce acantholysis (9) provided strong evidence of the indispensable role of mitochondrial damage in the disease mechanism. Conversely, the fact that functional inactivation of FcRn in knock-out mice protects them from PVIgG-induced blistering had been attributed solely to rapid lysosomal degradation of PVIgGs (21). In this study, we demonstrated for the first time that FcRn plays a pivotal role in AMA-dependent abnormalities of KCs targeted by PVIgGs, which, in addition to protecting PVIgG, is mediated by trafficking of AMA to the mitochondria. Co-localization experiments demonstrated that the PVIgG-FcRn complexes formed on the cell membrane of KCs become internalized and reach mitochondria, where they dissociate. Blockade of FcRn or its functional inactivation in both cases protected KCs from both PVIgG internalization and mitochondrial damage. Thus, although the composition of AMA's pool produced by individual PV patients may vary (9, 12, 13), FcRn appears to represent a common acceptor protein for internalization of these and, perhaps, other PVIgG species that may target other intracellular self-antigens besides mitochondrial proteins (1). Therefore, PV may be a novel disease paradigm providing for elucidation of the role of FcRn in basic mechanisms of autoimmunity to intracellular self-antigens.
In this study, the pathogenic role of the FcRn pathway was demonstrated by abolishing AMA's effects by the inhibition of the FcRn expression or function. The contribution of the pathogenic factors of PV other than autoantibodies, such as complement (38) and Fas receptor/ligand pathway (39), has been eliminated by heat inactivation of the patients' sera and preabsorption of PVIgG with the keratinocyte cell membrane proteins containing Fas receptor, respectively. Our results extend the function of FcRn beyond simple endocytosis of IgG. FcRn not only bound and internalized PVIgG, the protein complex also transported PVIgG to mitochondria and allowed PVIgG to reach its mitochondrial targets, such as the PDC complex. Although exactly how PVIgG-FcRn complex reached mitochondria and how PVIgG might enter mitochondria remains unknown, our findings suggested a new role for FcRn in mediating the delivery of PVIgG to its intracellular targets. Further studies will be needed to determine whether the mitochondrial transmembrane protein import machinery such as the Tim/Tom system is involved in this process (40). The major unresolved issue at the current level of understanding of antimitochondrial autoimmunity in PV is the heterogeneity of AMA. It remains to be determined whether and how distinct AMA species produce their pathobiological effects and contribute to PV severity. The results of this study lay the ground work and provide the rationale for future studies toward the generation of monoclonal AMA from patients with PV. By analogy to prior studies of human monoclonal anti-Dsg antibodies (37, 41), such an approach should help identify the exact mechanism of pathogenic action of specific AMA species.
Results of this study helped to resolve a seeming controversy about the apparent ability of AMA to enter various cell types expressing FcRn and the lack of measurable abnormalities in cell types other than KCs in PV patients. The following two sets of data suggest an explanation. First, measurements of CytC release and monolayer permeability (Fig. 1, E and F) demonstrated that the damage of KCs by AMA is reversible, perhaps due to self-repair. Second, analysis of mitochondrial respiration revealed PVIgG-induced abnormities only in the epithelial cell types expressing FcRn (Fig. 2, C and D). The reversibility of the cellular apoptotic process (42) may therefore explain why the cells hit by AMA alone do not undergo apoptolysis. In PV, the pathobiological action of AMA synergizes with that of autoantibodies to Dsg molecules uniquely expressed by KCs, so the irreversible damage becomes specific only to this cell type, as in PV. The fact that FcRn is predominantly expressed within the basal epidermal layer (23) may render basal KCs a preferred “functional” target for PVIgGs, explaining why they shrink more than suprabasal KCs (43), despite the fact that both basal and suprabasal KCs are targeted by PVIgGs (44). Our organ culture experiments demonstrated that a combination of AMA and anti-Dsg3 or Dsg1 antibodies induced epidermal splitting at the levels of predominant localization of corresponding Dsg molecules in the epidermis (36). However, a mixture of AMA with both anti-Dsg3 and Dsg1 antibodies, which is not infrequently found in PV patients (45), induced acantholysis throughout the entire epidermis (Fig. 5). Such a pattern of acantholysis is not seen in PV, indicating that the immunopathology of this disease cannot be adequately explained merely by an interplay between AMA and anti-Dsg antibodies. Other factors are apparently also involved.
The advantage of using scFv blocking Dsg1 (37), compared with commercial anti-Dsg1 antibody, is the ability to achieve a sufficiently high supraphysiological concentration of antibody at which keratinocytes start to separate from each other, perhaps due to steric hindrance. We titrated down scFv to identify the dose at which it did not cause any visible changes in the epidermis. Addition of AMA to that dose of scFv, however, disrupted the epidermal integrity. These new results vividly demonstrated the synergy between AMA and anti-Dsg antibodies, and they further substantiated an indispensable role of AMA in the multifactorial pathophysiology of PV. Thus, it has become evident that PV is a multifactorial disease, wherein KCs lose their ability to maintain epidermal cohesion due to a multiple hit sustained by autoantibodies to both adhesion and nonadhesion molecules, as was predicted by the multiple hit hypothesis (7). Very recently, the multiple hit hypothesis of PV also has been validated by results of the study showing that simply interfering with desmosomal adhesion is not sufficient to induce PV pathology and that there is another “hit” triggering pro-apoptotic events (46).
We believe that in PV both AMA and anti-Dsg antibodies are pathogenic in a sense that they are indispensable elements of the multifactorial pathophysiological mechanism of PV, which may also include autoantibodies of other specificities. The synergistic pathobiological effects of AMA and anti-Dsg3 antibody on KCs might occur at the molecular and/or cellular levels. In the past, we have evaluated dependence on Dsg3 of the signaling events elicited by AMA (9). We compared the AMA-dependent CytC release and activities of signaling kinases in Dsg3+/+ versus Dsg3−/− KCs treated with PVIgGs. Lack of Dsg3 did not affect these parameters, indicating that Dsg3 neither acts as a surrogate for the nominal mitochondrial antigen allowing AMA to enter KCs nor do the AMA-dependent molecular events require binding of anti-Dsg3 antibody to KCs. The synergy of AMAs with anti-Dsg antibody most likely occur at the cellular level, as depicted in Fig. 6. The basal KCs with damaged mitochondria may shrink because they run out of energy and because caspases activated due to CytC release cleave their structural and adhesion molecules resulting in the cytoskeleton collapse (8). During recovery, when KCs extend their cytoplasmic aprons toward neighboring cells, the antibodies can prevent assembly of nascent desmosomes due to steric hindrance, thus rendering acantholysis irreversible.
FIGURE 6.
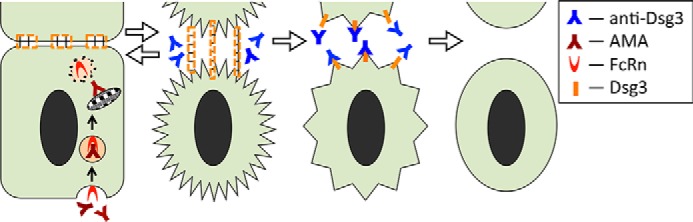
Hypothetical scheme of synergistic acantholytic activities of AMA and anti-Dsg3 antibodies on KCs. AMA enter a keratinocyte in a complex with FcRn, become liberated from the endosome compartment in the cytosol, and are trafficked to mitochondria. The mitochondrial damage triggers pro-apoptotic events leading to cell shrinkage. Stretching of the cell membrane areas anchored to neighboring cells by desmosomes tears them off. Keratinocyte shrinkage, however, is reversible (Fig. 1F). During recovery, KCs extend their cytoplasmic aprons toward neighboring cells. This is when anti-Dsg3 antibodies prevent assembly of nascent desmosomes due to steric hindrance, thus rendering acantholysis irreversible.
According to our explanation of PV pathophysiology, although a single species of PV autoantibodies can on its own alter keratinocyte function, the changes are insufficient to produce disease phenotype. This postulate is supported by the following clinical and experimental data. 1) Although PVIgGs bind to urinary bladder epithelial cells (47), this is not associated with damage of urinary bladder in PV, perhaps because the synergy of AMA and anti-Dsg3 antibody does not occur, as urinary bladder epithelial cells do not express Dsg3 (48) nor do they seem to express FcRn (e.g. T24 cells in Fig. 2C). 2) Cutaneous blisters in PV were associated with the anti-Dsg3 antibody that did not induce keratinocyte separation in vitro (49), suggesting that non-Dsg antibodies, such as AMA, were required to overcome the epidermal integrity. 3) Anti-Dsg antibodies produced by PV patients during remission, people with some other diseases, and some healthy individuals (6, 50) do not produce PV-like lesions, perhaps because they lack partnering autoantibodies. 4) The intraepidermal split in mouse skin was induced when AMA was added to the low dose of scFv that did not produce acantholysis on its own (Fig. 5). Thus, although autoimmunity in pemphigus is directed against multiple organ-specific and non-organ-specific proteins, the constellation of pathogenic autoantibodies capable of disrupting the integrity of the mucocutaneous barrier in PV patients contains antibodies to a combination of self-antigens unique to KCs.
Author Contributions
S. A. G. and P. H. W. designed experiments, obtained funding, and drafted parts of the manuscript. Y. C. and A. C. performed all assays. R. J. W. provided advice on data interpretations and provided important intellectual input to the final version of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We are grateful to Dr. John Stanley (University of Pennsylvania) for providing the Px4-3 clone of scFv.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL096987. This work was also supported by a grant from the Oxnard Foundation (to P. H. W.) and internal funds from the Department of Dermatology, University of California at Irvine. The authors declare that they have no conflicts of interest with the contents of this article.
- PV
- pemphigus vulgaris
- KC
- keratinocyte
- AMA
- antimitochondrial antibody
- Dsg
- desmoglein
- WB
- Western blot
- CytC
- cytochrome c
- FcRn
- neonatal Fc receptor
- PDC
- pyruvate dehydrogenase complex
- scFv
- single-chain variable fragment
- NIgG
- normal IgG
- OCR
- oxygen consumption rate.
References
- 1. Kalantari-Dehaghi M., Anhalt G. J., Camilleri M. J., Chernyavsky A. I., Chun S., Felgner P. L., Jasinskas A., Leiferman K. M., Liang L., Marchenko S., Nakajima-Sasaki R., Pittelkow M. R., Zone J. J., Grando S. A. (2013) Pemphigus vulgaris autoantibody profiling by proteomic technique. PLoS ONE 8, e57587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sinha A. A. (2012) Constructing immunoprofiles to deconstruct disease complexity in pemphigus. Autoimmunity 45, 36–43 [DOI] [PubMed] [Google Scholar]
- 3. Kurzen H., Brenner S. (2006) Significance of autoimmunity to non-desmoglein targets in pemphigus. Autoimmunity 39, 549–556 [DOI] [PubMed] [Google Scholar]
- 4. Lanza A., Cirillo N., Femiano F., Gombos F. (2006) How does acantholysis occur in pemphigus vulgaris: a critical review. J. Cutan. Pathol. 33, 401–412 [DOI] [PubMed] [Google Scholar]
- 5. Cirillo N., Cozzani E., Carrozzo M., Grando S. A. (2012) Urban legends: pemphigus vulgaris. Oral Dis. 18, 442–458 [DOI] [PubMed] [Google Scholar]
- 6. Grando S. A. (2012) Pemphigus autoimmunity: hypotheses and realities. Autoimmunity 45, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grando S. A. (2000) Autoimmunity to keratinocyte acetylcholine receptors in pemphigus. Dermatology 201, 290–295 [DOI] [PubMed] [Google Scholar]
- 8. Grando S. A., Bystryn J. C., Chernyavsky A. I., Frusić-Zlotkin M., Gniadecki R., Lotti R., Milner Y., Pittelkow M. R., Pincelli C. (2009) Apoptolysis: a novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp. Dermatol. 18, 764–770 [DOI] [PubMed] [Google Scholar]
- 9. Marchenko S., Chernyavsky A. I., Arredondo J., Gindi V., Grando S. A. (2010) Antimitochondrial autoantibodies in pemphigus vulgaris: a missing link in disease pathophysiology. J. Biol. Chem. 285, 3695–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arredondo J., Chernyavsky A. I., Karaouni A., Grando S. A. (2005) Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in pemphigus. Am. J. Pathol. 167, 1531–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gil M. P., Modol T., España A., López-Zabalza M. J. (2012) Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp. Dermatol. 21, 254–259 [DOI] [PubMed] [Google Scholar]
- 12. Kalantari-Dehaghi M., Chen Y., Deng W., Chernyavsky A., Marchenko S., Wang P. H., Grando S. A. (2013) Mechanisms of mitochondrial damage in keratinocytes by pemphigus vulgaris antibodies. J. Biol. Chem. 288, 16916–16925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chernyavsky A., Chen Y., Wang P. H., Grando S. A. (2015) Pemphigus vulgaris antibodies target the mitochondrial nicotinic acetylcholine receptors that protect keratinocytes from apoptolysis. Int. Immunopharmacol. 10.1016/j.intimp.2015.04.046 [DOI] [PubMed] [Google Scholar]
- 14. Ward E. S., Velmurugan R., Ober R. J. (2014) Targeting FcRn for therapy: from live cell imaging to in vivo studies in mice. Immunol. Lett. 160, 158–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sesarman A., Vidarsson G., Sitaru C. (2010) The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell. Mol. Life Sci. 67, 2533–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goebl N. A., Babbey C. M., Datta-Mannan A., Witcher D. R., Wroblewski V. J., Dunn K. W. (2008) Neonatal Fc receptor mediates internalization of Fc in transfected human endothelial cells. Mol. Biol. Cell 19, 5490–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kobayashi N., Suzuki Y., Tsuge T., Okumura K., Ra C., Tomino Y. (2002) FcRn-mediated transcytosis of immunoglobulin G in human renal proximal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 282, F358–F365 [DOI] [PubMed] [Google Scholar]
- 18. Antohe F., Rădulescu L., Gafencu A., Gheție V., Simionescu M. (2001) Expression of functionally active FcRn and the differentiated bidirectional transport of IgG in human placental endothelial cells. Hum. Immunol. 62, 93–105 [DOI] [PubMed] [Google Scholar]
- 19. Nagai J., Sato K., Yumoto R., Takano M. (2011) Megalin/cubilin-mediated uptake of FITC-labeled IgG by OK kidney epithelial cells. Drug Metab. Pharmacokinet. 26, 474–485 [DOI] [PubMed] [Google Scholar]
- 20. Gurbaxani B., Dela Cruz L. L., Chintalacharuvu K., Morrison S. L. (2006) Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol. Immunol. 43, 1462–1473 [DOI] [PubMed] [Google Scholar]
- 21. Li N., Zhao M., Hilario-Vargas J., Prisayanh P., Warren S., Diaz L. A., Roopenian D. C., Liu Z. (2005) Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J. Clin. Invest. 115, 3440–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aoyama Y. (2010) What's new in i.v. immunoglobulin therapy and pemphigus: high-dose i.v. immunoglobulin therapy and its mode of action for treatment of pemphigus. J. Dermatol. 37, 239–245 [DOI] [PubMed] [Google Scholar]
- 23. Cauza K., Hinterhuber G., Dingelmaier-Hovorka R., Brugger K., Klosner G., Horvat R., Wolff K., Foedinger D. (2005) Expression of FcRn, the MHC class I-related receptor for IgG, in human keratinocytes. J. Invest. Dermatol. 124, 132–139 [DOI] [PubMed] [Google Scholar]
- 24. Milner Y., Sagi E., Timberg R., Michel B., Me'te'zeau P., Goldberg M. (1989) Binding modes of IgG from pemphigus autoimmune sera onto guinea pig keratinocytes and the fate of bound IgGs. J. Cell Physiol. 139, 441–454 [DOI] [PubMed] [Google Scholar]
- 25. Bai Y., Ye L., Tesar D. B., Song H., Zhao D., Björkman P. J., Roopenian D. C., Zhu X. (2011) Intracellular neutralization of viral infection in polarized epithelial cells by neonatal Fc receptor (FcRn)-mediated IgG transport. Proc. Natl. Acad. Sci. U.S.A. 108, 18406–18411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Armitage C. W., O'Meara C. P., Harvie M. C., Timms P., Blumberg R. S., Beagley K. W. (2014) Divergent outcomes following transcytosis of IgG targeting intracellular and extracellular chlamydial antigens. Immunol. Cell Biol. 92, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schilling R., Ijaz S., Davidoff M., Lee J. Y., Locarnini S., Williams R., Naoumov N. V. (2003) Endocytosis of hepatitis B immune globulin into hepatocytes inhibits the secretion of hepatitis B virus surface antigen and virions. J. Virol. 77, 8882–8892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wan X., Duncan M. D., Nass P., Harmon J. W. (2001) Synthetic retinoid CD437 induces apoptosis of esophageal squamous HET-1A cells through the caspase-3-dependent pathway. Anticancer Res. 21, 2657–2663 [PubMed] [Google Scholar]
- 29. Arredondo J., Chernyavsky A. I., Grando S. A. (2006) Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol. Ther. 5, 511–517 [DOI] [PubMed] [Google Scholar]
- 30. Grando S. A., Dahl M. V. (1993) Activation of keratinocyte muscarinic acetylcholine receptors reverses pemphigus acantholysis. J. Eur. Acad. Dermatol. Venereol. 2, 72–86 [Google Scholar]
- 31. Nguyen V. T., Chernyavsky A. I., Arredondo J., Bercovich D., Orr-Urtreger A., Vetter D. E., Wess J., Beaudet A. L., Kitajima Y., Grando S. A. (2004) Synergistic control of keratinocyte adhesion through muscarinic and nicotinic acetylcholine receptor subtypes. Exp. Cell Res. 294, 534–549 [DOI] [PubMed] [Google Scholar]
- 32. Chen Y., Craigen W. J., Riley D. J. (2009) Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle 8, 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y., Gaczynska M., Osmulski P., Polci R., Riley D. J. (2010) Phosphorylation by Nek1 regulates opening and closing of voltage dependent anion channel 1. Biochem. Biophys. Res. Commun. 394, 798–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lykhmus O., Gergalova G., Koval L., Zhmak M., Komisarenko S., Skok M. (2014) Mitochondria express several nicotinic acetylcholine receptor subtypes to control various pathways of apoptosis induction. Int. J. Biochem. Cell Biol. 53, 246–252 [DOI] [PubMed] [Google Scholar]
- 35. Gergalova G., Lykhmus O., Komisarenko S., Skok M. (2014) α7 nicotinic acetylcholine receptors control cytochrome c release from isolated mitochondria through kinase-mediated pathways. Int. J. Biochem. Cell Biol. 49, 26–31 [DOI] [PubMed] [Google Scholar]
- 36. Mahoney M. G., Wang Z., Rothenberger K., Koch P. J., Amagai M., Stanley J. R. (1999) Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J. Clin. Invest. 103, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamagami J., Payne A. S., Kacir S., Ishii K., Siegel D. L., Stanley J. R. (2010) Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J. Clin. Invest. 120, 4111–4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jordon R. E., Schroeter A. L., Rogers R. S. 3rd, Perry H. O. (1974) Classical and alternate pathway activation of complement in pemphigus vulgaris lesions. J. Invest. Dermatol. 63, 256–259 [DOI] [PubMed] [Google Scholar]
- 39. Puviani M., Marconi A., Cozzani E., Pincelli C. (2003) Fas ligand in pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. J. Invest. Dermatol. 120, 164–167 [DOI] [PubMed] [Google Scholar]
- 40. Harbauer A. B., Zahedi R. P., Sickmann A., Pfanner N., Meisinger C. (2014) The protein import machinery of mitochondria–a regulatory hub in metabolism, stress, and disease. Cell Metab. 19, 357–372 [DOI] [PubMed] [Google Scholar]
- 41. Payne A. S., Ishii K., Kacir S., Lin C., Li H., Hanakawa Y., Tsunoda K., Amagai M., Stanley J. R., Siegel D. L. (2005) Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J. Clin. Invest. 115, 888–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang H. L., Yuen K. L., Tang H. M., Fung M. C. (2009) Reversibility of apoptosis in cancer cells. Br. J. Cancer 100, 118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bystryn J.-C., Grando S. A. (2006) A novel explanation for acantholysis in pemphigus vulgaris–the “Basal Cell Shrinkage” hypothesis. J. Am. Acad. Dermatol. 54, 513–516 [DOI] [PubMed] [Google Scholar]
- 44. Bhogal B., Wojnarowska F., Black M. M., Xu W., Levene G. M. (1986) The distribution of immunoglobulins and the C3 component of complement in multiple biopsies from the uninvolved and perilesional skin in pemphigus. Clin. Exp. Dermatol. 11, 49–53 [DOI] [PubMed] [Google Scholar]
- 45. Amagai M., Tsunoda K., Zillikens D., Nagai T., Nishikawa T. (1999) The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J. Am. Acad. Dermatol. 40, 167–170 [DOI] [PubMed] [Google Scholar]
- 46. Seiffert-Sinha K., Yang R., Fung C. K., Lai K. W., Patterson K. C., Payne A. S., Xi N., Sinha A. A. (2014) Nanorobotic investigation identifies novel visual, structural and functional correlates of autoimmune pathology in a blistering skin disease model. PLoS ONE 9, e106895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ortolan D. G., Souza D. P., Aoki V., Santi C. G., Gabbi T. V., Ichimura L. M., Maruta C. W. (2011) Analysis of the reactivity of indirect immunofluorescence in patients with pemphigus foliaceus and pemphigus vulgaris using rat bladder epithelium as a substrate. Clinics 66, 2019–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang W., Williamson S. R., Rao Q., Lopez-Beltran A., Montironi R., Eble J. N., Grignon D. J., Idrees M. T., Emerson R. E., Zhou X. J., Zhang S., Baldridge L. A., Hahn N. M., Wang M., Koch M. O., Cheng L. (2013) Novel markers of squamous differentiation in the urinary bladder. Hum. Pathol. 44, 1989–1997 [DOI] [PubMed] [Google Scholar]
- 49. Saleh M. A., Hashimoto R., Kase Y., Amagai M., Yamagami J. (2015) Low pathogenicity of anti-desmoglein 3 immunoglobulin G autoantibodies contributes to the atypical clinical phenotypes in pemphigus. J. Dermatol. 42, 685–689 [DOI] [PubMed] [Google Scholar]
- 50. Amagai M., Ahmed A. R., Kitajima Y., Bystryn J. C., Milner Y., Gniadecki R., Hertl M., Pincelli C., Kurzen H., Fridkis-Hareli M., Aoyama Y., Frusić-Zlotkin M., Müller E., David M., Mimouni D., et al. (2006) Are desmoglein autoantibodies essential for the immunopathogenesis of pemphigus vulgaris, or just ‘witnesses of disease’? Exp. Dermatol. 15, 815–831 [DOI] [PubMed] [Google Scholar]