

Background: VDAC1 functions in both cellular metabolism and mitochondria-mediated apoptosis.
Results: New compounds were identified that induce apoptosis by promoting VDAC1 oligomerization and apoptosis in a Bak- and Bak-independent manner.
Conclusion: Bax and Bak are dispensable for VDAC1-mediated apoptosis, revealing a novel mechanism of apoptosis involving VDAC1 oligomerization.
Significance: In cancers with Bax/Bak down-regulated, VDAC1-induced apoptosis offers a novel approach for tumor therapies.
Keywords: B-cell lymphoma 2 (Bcl-2), Bax, mitochondrial apoptosis, protein cross-linking, voltage-dependent anion channel (VDAC), Bcl2, cyathane diterpenoid, oligomerization
Abstract
The pro-apoptotic Bax and Bak proteins are considered central to apoptosis, yet apoptosis occurs in their absence. Here, we asked whether the mitochondrial protein VDAC1 mediates apoptosis independently of Bax/Bak. Upon screening a fungal secondary metabolite library for compounds inducing apoptosis in Bax/Bak-deficient mouse embryonic fibroblasts, we identified cyathin-R, a new cyathane diterpenoid compound able to activate apoptosis in the absence of Bax/Bak via promotion of the VDAC1 oligomerization that mediates cytochrome c release. Diphenylamine-2-carboxilic acid, an inhibitor of VDAC1 conductance and oligomerization, inhibited cyathin-R-induced VDAC1 oligomerization and apoptosis. Similarly, Bcl-2 overexpression conferred resistance to cyathin-R-induced apoptosis and VDAC1 oligomerization. Silencing of VDAC1 expression prevented cyathin-R-induced apoptosis. Finally, cyathin-R effectively attenuated tumor growth and induced apoptosis in Bax/Bak-deficient cells implanted into a xenograft mouse model. Hence, this study identified a new compound promoting VDAC1-dependent apoptosis as a potential therapeutic option for cancerous cells lacking or presenting inactivated Bax/Bak.
Introduction
One of the enigmas in mitochondria-mediated apoptosis concerns the release pathway(s) used by apoptogenic factors residing in the mitochondrial intermembranal space to cross the outer mitochondrial membrane (OMM)5 and reach the cytosol. Indeed, mitochondrial outer membrane permeabilization (MOMP) is a critical event resulting in the release of cytochrome c into the cytosol, where it binds to Apaf1 to promote formation of the apoptosome (1). The apoptosome recruits and cleaves pro-caspase-9 to initiate a caspase cascade, ultimately leading to apoptosis (2). To date, several models of MOMP leading to cytochrome c release have been proposed (3), such as the Bax/Bak pore formation (4) and mitochondrial permeability transition pore (mPTP) models (5).
Bcl-2 family proteins Bax and Bak are believed to serve as central regulators of MOMP and thus of mitochondria-mediated apoptosis (6). In response to apoptotic stimuli, BH3-only proteins like Bim or Bid are activated via transcriptional up-regulation or post-translational modification. These subsequently bind to either Bcl-2 or Bax and Bak, leading to the translocation of Bax to the OMM, where the protein changes conformation. This structural alteration encourages the formation of Bax homo-oligomers or hetero-oligomers with OMM-anchored Bak, yielding porelike structures that mediate MOMP and apoptosis (7–9). Although the importance of Bax and Bak in MOMP and apoptosis is widely recognized, Bax/Bak-independent apoptotic pathways also exist (10–12). Indeed, Bax/Bak down-regulation or inactivation has been shown to be the mechanism for the development of resistance to apoptosis in some cancers (13–15). Therefore, identifying molecules that mediate apoptosis in tumor cells independently of Bax and Bak offers an opportunity for the development of novel tumor therapies.
The voltage-dependent anion channel 1 (VDAC1) is an OMM protein that serves as a mitochondrial gatekeeper, controlling metabolic and energy cross-talk between mitochondria and the rest of the cell (3, 16, 17). The involvement of VDAC1 in mitochondria-mediated apoptosis has been proposed based on several lines of experimental evidence. VDAC1 is involved in cytochrome c release and is associated with pro- and anti-apoptotic members of the Bcl-2 protein family (3, 17–21). siRNA-mediated down-expression of VDAC1 prevents cell death and activation of Bax as induced by cisplatin and strongly reduced cisplatin-induced release of cytochrome c and apoptosis-inducing factor (AIF), as well as the maturation of caspase-3 (22). Similarly, reducing VDAC1 expression by siRNA attenuated endostatin-induced apoptosis (23), whereas knockdown of VDAC1 in non-small cell lung cancer cells inhibited TNF-related apoptosis-inducing ligand (TRAIL)-induced activation of caspase-8 and subsequent apoptosis (24). In addition, anti-VDAC1 antibodies specifically and effectively prevent As2O3-induced cytochrome c release from isolated mitochondria (25) and, when microinjected into cells, prevented Bax-induced cytochrome c release and subsequent apoptosis as well as etoposide-, paclitaxel-, and staurosporine-induced apoptosis (26). Anti-VDAC1 antibodies also inhibited the interaction of Bax with VDAC and the triggering of cell death (25–27). Still, others have questioned VDAC function in apoptosis (28).
Recent studies have indicated that in response to numerous apoptogens acting via different initiating cascades, VDAC1 can mediate MOMP and apoptosis via its oligomerization, forming a protein-conducting channel within a VDAC1 homo-oligomer that mediates cytochrome c release (17, 29–37). It was also proposed that p53 modulates VDAC1 oligomerization toward the formation of high molecular mass complexes (38, 39).
Interestingly, various studies have demonstrated an increase in VDAC1 levels following apoptosis induction (40–42) and the causal relationship between VDAC1 levels and drug sensitivity (43). Accordingly, a new concept for apoptosis induction has been postulated in which agents and conditions that induce apoptosis up-regulate VDAC1 expression in a Ca2+-dependent manner, in turn leading to the formation of VDAC1 oligomers that mediate cytochrome c release and subsequent cell death (36). Nevertheless, in various studies and proposed models, the apoptotic role suggested for VDAC1 is that of an auxiliary component that merely assists more main players, mostly Bax and/or Bak, in mediating MOMP and apoptosis. Thus, it remains a matter of debate whether VDAC1 provides an apoptotic function in the absence of Bax and Bak.
In previous studies, we tested the capability of several compounds to induce apoptosis induction in cells depleted of Bax and Bak. These include gossypol, a compound that induces a conformational change in Bcl-2, converting it into a pro-apoptotic protein (44). Other compounds, such as Spiraea diterpenoid derivative, 15-oxospiramilactone, named S-3, and PAO, induce significant up-regulation of Bim, which interacts with Bcl-2 to form a hetero-oligomeric complex that is sufficient to mediate MOMP and cytochrome c release (45, 46). Here, we screened a library of fungi-derived compounds to identify new molecules acting via VDAC1. We thus identified cyathin-R and confirmed its ability to mediate apoptosis in Bax- and Bak-deficient cells. We further report that cyathin-R induces an increase in VDAC1 levels, promoting VDAC1 oligomerization that subsequently leads to apoptosis in the absence of Bax and Bak in a manner that can be inhibited by the VDAC1-interacting molecules, diphenylamine-2-carboxilic acid (DpC), 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS), and Bcl-2.
Experimental Procedures
Cell Culture and Reagents
E1A- and K-Ras-transformed Bax−/−/Bak−/− MEF, Bax−/− HCT116, and HCT116 colon cancer cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum and antibiotics. Antibodies to β-actin and Bad, Puma, and FLAG were from Sigma; antibodies to Bcl-2, Tim23, cytochrome c, and Bcl-xL were from BD Biosciences; antibody to VDAC1 was from Abcam (ab14734) (Cambridge, UK); and antibody to cyclophilin D was from Calbiochem. FITC-conjugated goat anti-mouse and anti-rabbit antibodies were from Santa Cruz Biotechnology, Inc., and HRP-labeled goat anti-mouse and anti-rabbit IgG antibodies were from Kirkegaard & Perry Laboratories (Gaithersburg, MD). ECL reagents and ethylene glycolbis(succinimidyl succinate) (EGS) cross-linker were from Thermo Scientific (Waltham, MA). MitoTracker Red CMXRos, MitoSOX Red mitochondrial superoxide indicator, and carboxy-H2DCFDA were from Invitrogen, whereas Z-VAD-fmk was from Enzo Life Sciences (Lausen, Switzerland). BAPTA-AM was obtained from Tocris Bioscience (Bristol, UK). Other chemicals were purchased from Sigma, unless otherwise specified.
Extraction and Isolation of Cyathin-R and X-ray Crystallographic Analysis
As reported in our earlier publication (47), Cyathus africanus was incubated on rice at 25 °C for 38 days to afford a solid culture. The culture was extracted repeatedly with ethyl acetate by exhaustive maceration (3 × 4 liters). The organic solvent was evaporated to dryness under vacuum to afford the crude extract (56.5 g). The residue was dissolved in H2O (400 ml) and partitioned with CHCl3 (400 ml). The CHCl3-soluble fraction (36 g) was subjected to silica gel column chromatography and eluted with a gradient of n-hexane-ethyl acetate (100:0, 100:1, 100:2, 100:5, 100:10, 100:15, 100:25, and 100:35 (v/v)) and dichloromethane-acetone (100:2, 100:4, 100:8, 100:15, 100:25, 100:35, 100:100, and 0:100 (v/v)) to give 11 fractions (CA-1 to CA-11). Fraction CA-6 (4.1 g), eluted with n-hexane-ethyl acetate (100:25 (v/v)), was further separated by ODS column chromatography using a gradient of increasing methanol (30–100%) in water to afford 10 subfractions (CA-6-1 to CA-6-10). Cyathin-R (1.1 g) was obtained from CA-6-6 by recrystallization in methanol.
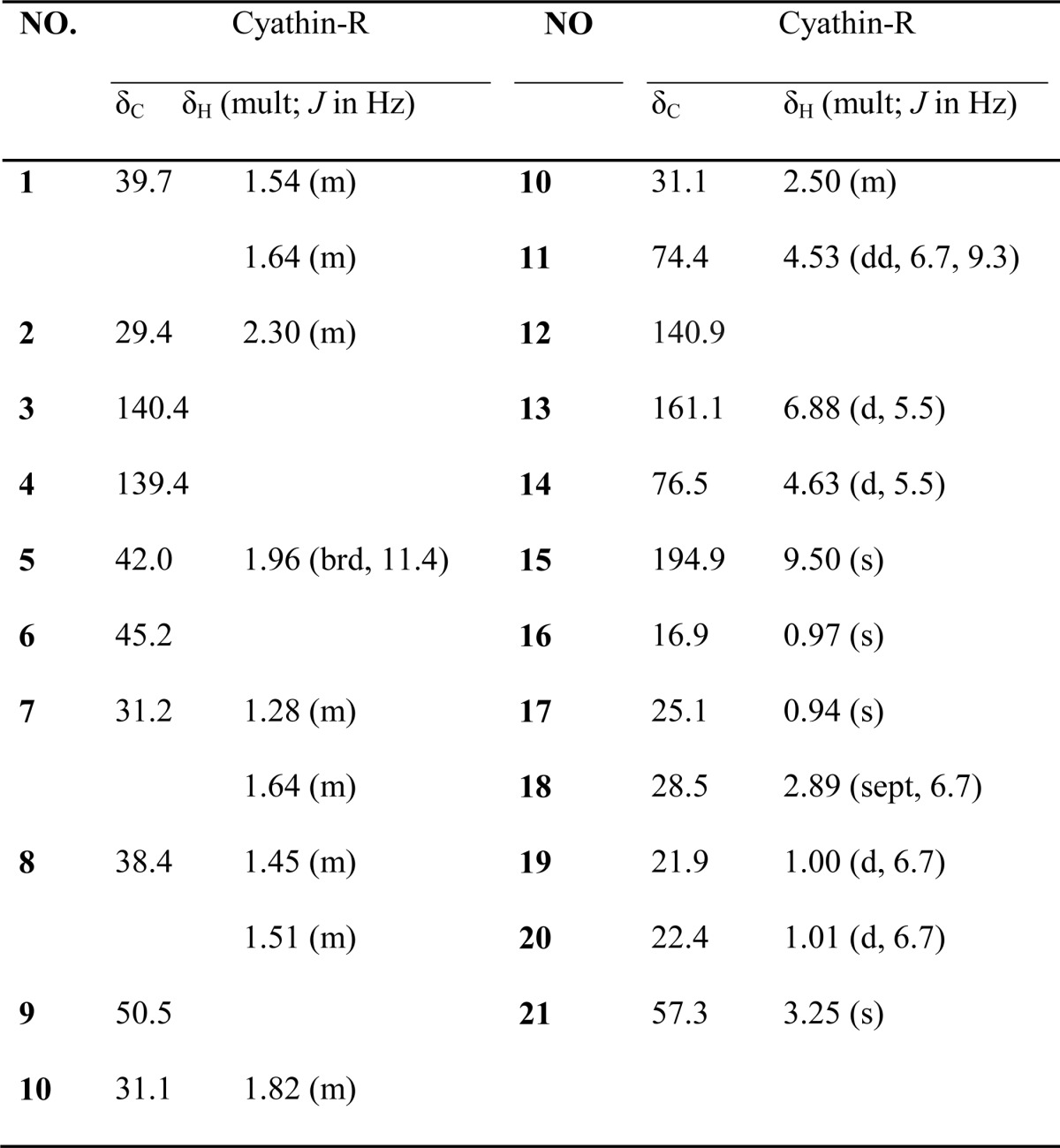
Cyathin-R was isolated as white crystalline. [α]D25 −18.46 (c 0.22, MeOH); CD (c 0.4 × 10-3 m, MeOH); λmax (Δϵ): 209 (−4.11); 241 (1.32); 284 (0.31); 325 (−0.03); UV (MeOH) λmax (log ϵ) 203 (2.89) nm, 235 (3.62) nm; IR (νmax): 3423, 2934, 2869, 1686, 1455, 1379, 1141 cm−1. For 1H NMR and 13C NMR, see Table 1; positive high resolution electrospray ionization mass spectrometry m/z 355.2240 [M + Na]+ (calculated for C21H32O3Na, 355.2244).
TABLE 1.
NMR data of cythin-R
1H NMR at 600 Hz, 13NMR at 150Hz in CD3 OD.
X-ray Crystallographic Analysis of Cyathin-R and Structure Determination
The molecular formula of cyathin-R was established to be C21H32O3 based on high resolution electrospray ionization mass spectrometry and NMR data (Table 1), indicating six degrees of unsaturation. The [1H]NMR spectrum indicated the presence of four methyl groups [δH 0.94 (3H, s, H-17); 0.97 (3H, s, H-16); 1.00 (3H, d, J = 6.7 Hz, H-19) and 1.01 (3H, d, J = 6.7 Hz, H-20)], a methoxyl group at δH 3.25 (3H, s, H-21), an olefinic proton at δH 6.88 (1H, d, J = 5.5 Hz, H-13), and an oxymethine at δH 4.63 (1H, d, J = 5.5 Hz, H-14). The [13C]NMR spectrum of cyathin-R (Table 1) revealed 21 carbons ascribable to four methyl, six methylene, and four methane groups, five quaternary carbons, one methoxyl carbon, and one carbonyl group.
Upon crystallization from MeOH using the vapor diffusion method, colorless needles of cyathin-R were obtained. Data collection was performed on an Agilent Gemini A Ultra diffractometer using graphite-monochromated copper Kα radiation, λ = 1.54184 Å at 100.2 K. Crystal data: C21H32O3, M = 332.47, space group orthorhombic, P212121; unit cell dimensions were determined to be a = 8.2443(2) Å, b = 11.0732(4) Å, c = 20.5762(6) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 1878.42(10)Å3, Z = 4, Dx = 1.176 mg/m3, F (000) = 728, μ (copper Kα) = 0.601 mm−1. 18,044 unique reflections were collected to θmax = 71.90°, in which 3,586 reflections were observed (F2 > 4σ (F2)). The structure was solved by direct methods using the SHELXS-97 program and refined using SHELXL-97 and full-matrix least-squares calculations. In the structure refinements, non-hydrogen atoms were placed on the geometrically ideal positions by the “ride on” method. Hydrogen atoms bonded to oxygen were located via structure factors with isotropic temperature factors. The final refinement gave r = 0.0359, RW = 0.0945, S = 1.049, and Flack = 0.05(16). The absolute configuration of cyathin-R was confirmed as 5R, 6R, 9R, 11R, and 14S by x-ray crystallographic analysis on the basis of the value of the Flack absolute structure parameter 0.05(16) obtained by graphite-monochromated copper Kα radiation.
Detection of Apoptosis
Apoptotic cell death was analyzed by annexin V-FITC and PI staining and by an apoptosis detection kit (PharMingen) according to the recommended protocol, followed by flow cytometry. Caspase activity in cells was quantified using a CaspACE-FITC-VAD-fmk kit. Flow cytometry data were obtained with FACScan and analyzed with CellQuest software (BD Biosciences).
Knockdown of VDAC1 Expression
DNA sequences for mouse VDAC1-shRNA1 (GCTACGGCTTTGGCTTAATAA), shRNA2 (GTTGGCTATAAGACGGATGAA), shRNA3 (ACCAGGTATCAAACTGACGTT), and scrambled shRNA were inserted into the pSilencer 2.1-CMV-Puro vector. Human VDAC1-shRNA (AGTGACGGGCAGTCTGGAA) and its scrambled shRNA were inserted into the pSUPER-retro-puro vector. The resulting plasmids (1.5 μg) were respectively transfected into Bax−/−/Bak−/− MEF or Bax−/− HCT116 cells using MegaTran version 1.0 (OriGene, Rockville, MD). Clones of Bax−/− HCT116 colon cancer cells were selected in culture medium containing 1 μg/ml puromycin.
ROS and Mitochondrial Superoxide Detection
To detect ROS, growing (5 × 105 cells/ml) Bax−/−/Bak−/− MEF or Bax−/− HCT116 cells were incubated with cyathin-R for the indicated times, harvested, washed with phosphate-buffered saline (PBS), and incubated with carboxy-H2DCFDA (5 μm, 37 °C, 10 min). Fluorescence intensity in the cells was examined by FACS, with excitation set at 488 nm. For mitochondrial superoxide detection, cells were incubated with MitoSOX Red (5 μm, 37 °C, 30 min), and fluorescence was measured with excitation at 510 nm. The data were analyzed with Cell Quest software (BD Biosciences).
PTP Opening
Rat liver mitochondria were isolated as described previously (37). The isolated mitochondria were kept on ice and used for PTP opening assays within 2–4 h of preparation. PTP opening was assayed by monitoring Ca2+-induced large amplitude mitochondrial swelling, as described previously (37). Briefly, freshly prepared mitochondria (0.5 mg/ml) were added to a solution containing 225 mm mannitol, 75 mm sucrose, 5 mm Hepes/KOH, 5 mm succinate, and 0.1 mm Pi, pH 7.0, with mitochondrial swelling being initiated by the addition of Ca2+ (50 μm).
Immunofluorescence Assay
Cells grown on coverslips were treated with cyathin-R and stained with MitoTracker Red CMXRos (50 nm, 15 min, 37 °C), fixed with 3.7% formaldehyde, and permeabilized with 0.2% Triton X-100 on ice. Samples were incubated with anti-cytochrome c antibodies (1 h, 25 °C) and then FITC-conjugated goat anti-mouse secondary antibodies (1 h, room temperature). Fluorescence images were visualized using a Zeiss LSM 510 META confocal microscope.
Real-time PCR
mRNA was prepared from Bax−/−/Bak−/− MEFs using TRIzol reagent (Invitrogen). cDNA was synthesized using the SuperScript VILO cDNA synthesis kit (Invitrogen). Real-time PCR was carried out using a Quant One Step quantitative RT-PCR (probe) kit (Tiangen) and a CFX96 real-time PCR detection system (Applied Biosystems).
The resulting cDNAs for Bcl-2 were PCR-amplified using 5′-GTGGCCTTCTTTGAGTTCG-3′ and 5′-CCTACCCAGCCTCCGTTAT-3′ (Bcl-2) as the sense and antisense primers, respectively. Glyceraldehyde 3-phosphate dehydrogenase(GAPDH) cDNA that served as a control was amplified using 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ as the sense and antisense primers, respectively.
Reconstitution of Purified Intact VDAC into a Planar Lipid Bilayer, Single Channel Current Recording, and Data Analysis
VDAC1 purified from rat liver mitochondria was solubilized with LDAO and purified using hydroxyapetite resin, as described previously (48). Purified VDAC was used for channel reconstitution into a planar lipid bilayer prepared from soybean asolectin dissolved in n-decane (50 mg/ml) as described previously (48). Purified VDAC was added to the cis chamber containing 1 m NaCl and 10 mm Hepes, pH, 7.4. After one or more channels were inserted into the planar lipid bilayer, currents were recorded by voltage clamping using a Bilayer ClampBC-525B amplifier (Warner Instruments, Hamden, CT). Current was measured with respect to the trans side of the membrane (ground). The current was digitized on-line using a Digi data 1200 interface board and pCLAMP6 software (Axon Instruments, Union City, CA).
Cellular [Ca2+] Analysis
Fluo-4-AM was used to monitor changes in cytosolic Ca2+ levels. Bax−/−/Bak−/− MEF cells (1 × 106 cells/ml) were harvested after the appropriate treatment, collected (1,500 × g for 10 min), washed with HBSS buffer (5.33 mm KCl, 0.44 mm KH2PO4, 138 Mm NaCl, 4 mm NaHCO3, 0.3 mm Na2HPO4, 5.6 mm glucose, 0.03 mm phenol red), and incubated with 2 mm Fluo-4 in 200 ml of HBSS(+) buffer (HBSS buffer supplemented with 1.8 mm CaCl2) in the dark for 30 min at 37 °C. After washing the excess dye, the cells were incubated with 200 ml of HBSS(+) buffer, and changes in cellular free Ca2+ concentration were measured immediately via FACS analysis. At least 10,000 events were recorded on the FL1 detector, represented as a histogram, and analyzed by FACSCalibur flow cytometer software (BD Biosciences). Positive cells showed a shift to an enhanced level of green fluorescence (FL1).
Gel Electrophoresis and Immunoblotting
Cells were lysed in lysis buffer (150 mm NaCl, 25 mm HEPES, pH 7.4, 1 mm EGTA, 1 mm DTT, 1% Nonidet P-40, 0.25% sodium deoxycholate, 50 μg/ml trypsin inhibitor, and 10 μg/ml each of aprotinin, leupeptin, and pepstatin), diluted with sample buffer, subjected to SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% nonfat milk, and then incubated with specific antibodies overnight at 4 °C. Bands were detected with HRP-conjugated secondary antibodies and an enhanced chemiluminescence system. Quantitative analysis of bands was performed using Image Gauge software (Fujifilm).
Cross-linking of VDAC1
After treatment with compounds, cells were harvested and treated with DMSO as a vehicle control (2%, as used in compound-containing samples) or cross-linked with 0.5 mm EGS for 10 min at 30 °C. The reaction was quenched by the addition of 20 mm Tris-HCl (pH 7.4). Samples were analyzed by SDS-PAGE followed by immunoblotting.
BRET Assay
In BRET2 experiments, Bax−/−/Bak−/− MEF cells were transfected to express the proteins below using JetPRIME transfection agent (Illkirch, France) according to the manufacturer's instructions. Transfections were carried out with siRNA specific to human VDAC1 (50 nm), 0.2 μg of a plasmid coding for rat VDAC1 (rVDAC1)-Rluc (Renilla luciferase fused to the VDAC1 C terminus) and with 0.8 μg of a plasmid coding for rVDAC1-GFP2 (GFP2 fused to the VDAC1 C terminus) (34). Cells were subjected to the BRET2 assay 48–72 h post-transfection. As background, cells were transfected with plasmids encoding rVDAC1-Rluc (0.2 μg) and with plasmid pcDNA4/TO (0.8 μg). Cells transiently expressing rVDAC1-Rluc and rVDAC1-GFP2 were incubated with the apoptosis inducer, harvested, washed twice with PBS, resuspended in 200 μl of PBS, and divided between two wells of a white 96-well clear bottom plate (Grenier). Luciferase activity was assayed using the membrane-permeant substrate DeepBlue C in PBS supplemented with MgCl2 (1 g/liter) and glucose (1 g/liter), with DeepBlue C added to a final concentration of 5 μm just before luminescence measurements. The BRET2 signal represents the ratio of the GFP2 fluorescence, measured at its emission wavelength (510 nm), to the light intensity (luminescence) emitted at 395 nm. All measurements were performed using the Infinite 200 ELISA reader (Tecan). BRET2 signals were defined as the GFP2/Renilla luciferase intensity ratio and calculated as described previously (34).
Xenograft Experiments Using Nude Mice
Bax−/−/Bak−/− MEF and Bax−/− HCT116 and HCT116 colon cancer cells (5 × 105) were subcutaneously injected into the back of the upper limbs of 4–5-week-old nude BALB/c mice. The mice were separated into three groups (6 mice/group). One week postinjection, two groups were treated with 5 or 10 mg/kg cyathin-R, whereas the vehicle control third group was treated with propylene glycol (50% (v/v) in 1.8% NaCl every other day for 2 weeks. The three groups of mice were sacrificed, and tumors were excised. Tumor length (a) and width (b) were measured using a slide gauge to determine tumor volumes (V) according to the formula V = a × b2/2. Apoptotic cells in the tumors were visualized by fluorescence microscopy after TUNEL using an in situ cell death detection kit (Roche Applied Science). Mice were maintained in specific pathogen-free conditions, and all research work was approved by the Animal Care and Use Committee of the Institute of Zoology, Chinese Academy of Sciences.
Statistical Analysis
Data are expressed as means ± S.E. Statistical evaluation was carried out using Student's t test (two-tailed) to test for differences between control and experimental results. The level of significance of differences between control and treated samples was determined using the nonparametric Mann-Whitney U test. A difference was considered statistically significant when the p value was deemed <0.05 (*), <0.01 (**), or <0.001 (***). Tumor volume data were analyzed using one-way repeated measures analysis of variance.
Results
Cyathin-R Is Able to Induce Bax/Bak-independent Apoptosis
To identify new compounds able to induce apoptosis in a Bak- and Bak-independent manner, Bax−/−/Bak−/− MEF cells were employed in a two-round screening strategy (Fig. 1A). We first screened 149 fungus-derived natural compounds for cytotoxicity using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell survival assay. In this manner, we obtained seven compounds that strongly reduced cell viability. As the next step to select for VDAC1-dependent molecules, DpC, identified as an inhibitor of both VDAC1 channel conductance and selenite-induced oligomerization (Fig. 1, B–D), was recruited (Fig. 1E). Purified mitochondrial VDAC1 were reconstituted into a planar lipid bilayer, and channel conductance was monitored under voltage clamp conditions. DpC reduced channel conductance of reconstituted VDAC1 assayed at −10 or 10 mV (Fig. 1B). VDAC1 channel conductance is voltage-dependent, showing decreased conductance at both high negative or positive potentials. DpC decreased the conductance regardless of the voltage gradient applied (Fig. 1C).
FIGURE 1.

Screening for VDAC1-associated apoptosis inducers and structural analysis of cyathin-R. A, diagram of compound-screening strategy. B, DpC reduces VDAC1 channel conductance. VDAC1 was reconstituted into a planar lipid bilayer as described under “Experimental Procedures.” Currents through VDAC1 in response to a voltage step from 0 to 10 mV were recorded before and 20 min after the addition of DpC (100 μm). C, VDAC1 conductance as a function of voltage, from 60 to −60 mV, was recorded before (●) and after (○) the addition of DpC. The average steady-state conductance at a given voltage (G) was normalized to the maximal conductance at 10 mV (G0). D, DpC inhibits VDAC1 oligomerization induced with selenite (30 μm, 3 h) in HeLa cells preincubated with DpC (1 h). VDAC1 oligomerization was revealed using EGS-based cross-linking and immunoblotting using anti-VDAC1 antibodies. E, Bax−/−/Bak−/− MEFs were pretreated with 0.5 mm DpC for 1 h and incubated for 24 h with the indicated compounds (10 μm X15-1, S3, 5 μm H8-5, and 1 μm PAO). Apoptotic cell death was analyzed by annexin-V/PI staining and flow cytometry. Means ± S.E. (error bars) (n = 3) are shown. H8-5, cyathin-R. F and G, Bax−/−/Bak−/− MEFs were treated with cyathin-R for the indicated times, stained with annexin-V/PI, and analyzed by flow cytometry. Data from one of the three independent experiments are shown on the left. Means ± S.E. (n = 3) are shown. H–J, chemical structure of cyathin-R (H), key heteronuclear multiple-bond correlations (HMBC) of cyathin-R (I), and the conformation of cyathin-R with minimized energy (J) are shown. The arrows show the NOE correlations. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. *, p < 0.05; ***, p < 0.001.
DpC inhibited VDAC1 oligomerization as analyzed following chemical cross-linking with cell-permeable EGS and Western blot analysis using anti-VDAC1 antibodies. Protein bands corresponding to VDAC1 dimers (72 kDa) and multimers were obtained upon exposure to selenite (Fig. 1D).
To identify molecules that induce VDAC1-dependent apoptosis, cells were pretreated with DpC before exposing them to the seven fungus-derived hit compounds, and then apoptosis was analyzed by annexin-V/propidium iodide staining and flow cytometry (Fig. 1E). This approach led to the finding that of the seven molecules inducing apoptosis in Bax−/−/Bak−/− MEFs, only H8-5, termed cyathin-R, was able to induce apoptosis that was inhibited by DpC (Fig. 1E) and in a time-dependent manner (Fig. 1, F and G). DpC alone had no effect on cell viability.
Cyathin-R, a cyathane-type diterpenoid from the medicinal fungus C. africanus, was isolated from solid culture by column chromatography on silica gel and C-18 reverse-phase silica, followed by recrystallization in methanol. The molecular formula of cyathin-R was established to be C21H32O3 based on high resolution electrospray ionization mass spectrometry, 1H NMR, and 13C NMR (Table 1). The structure of cyathin-R (Fig. 1H) was unambiguously defined by heteronuclear multiple-bond correlation and NOESY spectral analysis and x-ray crystallographic analysis (Fig. 1, H–J).
Whereas annexin-V/PI staining revealed cyathin-R-induced apoptosis in Bax−/−/Bak−/− MEF cells, other markers of apoptosis were also considered. Cyathin-R induced hallmarks of apoptosis, such as cytochrome c release from mitochondria (Fig. 2, A and B), poly(ADP-ribose) polymerase cleavage (Fig. 2C), and caspase activation (Fig. 2D), as well as nuclear fragmentation and condensation (Fig. 2F), all in the absence of Bax and Bak. Furthermore, the pan-caspase inhibitor Z-VAD prevented cyathin-R-induced apoptosis (Fig. 2E). Similar results were also obtained with Bax−/− HCT116 colon cancer cells (Fig. 3). We thus conclude that cyathin-R induced MOMP and apoptosis in a Bax- and Bak-independent fashion.
FIGURE 2.

Cyathin-R induces apoptosis in Bax−/−/Bak−/− MEFs. A, cytochrome c release from mitochondria; Bax−/−/Bak−/− MEFs treated with 3 μm cyathin-R for the indicated times, as analyzed by immunohistochemistry using anti-cytochrome c antibodies, with nuclei stained with DAPI and mitochondria with MitoTracker. Cells were visualized by confocal microscopy. The enlarged images on the right are from the boxed areas. Scale bar, 20 μm. B, cytochrome c (Cyto c) release as assayed by immunoblotting of cytosolic and mitochondrial fractions. C, poly(ADP-ribose) polymerase (PARP) cleavage; Bax−/−/Bak−/− cells were treated with 3 μm cyathin-R for the indicated times. Poly(ADP-ribose) polymerase cleavage was analyzed by immunoblotting. D, caspase activation; Bax−/−/Bak−/− MEFs were treated as described in the legend to Fig. 1G and then stained with a CaspACE-FITC-conjugated caspase marker. Means ± S.E. (error bars) (n = 3) are shown. E, apoptosis inhibition with casapse inhibitor; Bax−/−/Bak−/− MEFs were pretreated with Z-VAD-fmk casapse inhibitor (1 h) and then incubated with cyathin-R (5 μm, 24 h). Cell death was analyzed by annexin-V/PI staining and flow cytometry. Means ± S.E. (n = 3) are presented. F, cell nuclear fragmentation; Bax−/−/Bak−/− cells were treated with 3 μm cyathin-R, stained with Hoechst 33342, and imaged by confocal microscopy. Scale bar, 20 μm. **, p < 0.01; ***, p < 0.001.
FIGURE 3.

Cyathin-R induces apoptosis in Bax−/− HCT116 cells. A and B, Bax−/− HCT116 cells were treated with the indicated concentrations of cyathin-R for 24 h, harvested, and divided into two parts. One part was stained with annexin-V/PI and the other with a CaspACE-FITC-conjugated caspase marker. Both populations were analyzed by flow cytometry. Data from one of the three independent experiments are shown on the left. Means ± S.E. (error bars) (n = 3) are presented. C, Bax−/− HCT116 cells were pretreated with Z-VAD-fmk caspase inhibitor for 1 h at the indicated concentrations, followed by a 24-h incubation with cyathin-R. Cells were stained with annexin-V-FITC and PI and analyzed by flow cytometry. Means ± S.E. (n = 3) are reported. D, effect of cyathin-R on mitochondria membrane potential. Bax−/− HCT116 cells were treated with the indicated concentrations of cyathin-R (6 h). After staining with TMRM for 10 min at 37 °C, cells were analyzed by flow cytometry. Means ± S.E. (n = 3) are reported. FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone. **, p < 0.01; ***, p < 0.001.
Cyathin-R Induces Increased VDAC1 Protein Levels and Oligomerization That Mediate Apoptosis in Bax- and Bak-deficient Cells
Cyathin-R induced apoptosis in Bax−/−/Bak−/− MEFs and Bax−/− HCT116 colon cancer cells that was inhibited by DpC and by other anion transport inhibitors, DIDS, DNDS, or SITS (Fig. 4, A and B). DIDS was shown to inhibit VDAC1 channel conductance (49) and oligomerization (34). Interestingly, the cyclophilin D-interacting molecule cyclosporin A, a known inhibitor of mPTP opening, failed to inhibit cyathin-R-triggered apoptosis but rather promoted it (Fig. 4, C and D). The direct interaction of cyathin-R with VDAC1 is reflected in the decreased channel conductance of bilayer-reconstituted VDAC1 (Fig. 4, E and F).
FIGURE 4.

VDAC1-interacting molecules but not cyclosporine A inhibit cyathin-R-induced apoptosis. Where indicated, apoptosis was analyzed by annexin-V/PI staining and flow cytometry. A and B, Bax−/−/Bak−/− MEFs (A) and Bax−/− HCT116 colon cancer cells (B) were pretreated with 0.5 mm DpC, DIDS, DNDS, or SITS (1 h) at the indicated concentrations, incubated with cyathin-R (5 μm, 24 h), and analyzed for apoptosis. Means ± S.E. (error bars) (n = 3) are presented. C and D, Bax−/−/Bak−/− MEFs (C) or Bax−/− HCT116 colon cancer cells (D) were pretreated with cyclosporin A (CsA) at the indicated concentrations, incubated with 5 μm cyathin-R, and analyzed for apoptosis. Means ± S.E. (n = 3) are presented. E, cyathin-R reduces VDAC1 channel conductance. VDAC1 was reconstituted into a planar lipid bilayer as described under “Experimental Procedures.” Currents through VDAC1 in response to a voltage step from 0 to 10 mV were recorded before and 30 min after the addition of cyathin-R (5 μm). F, VDAC1 conductance as a function of voltage, from 60 to −60 mV, was recorded before (●) and after (○) the addition of cyathin-R. The average steady-state conductance at a given voltage (G) was normalized to the maximal conductance at 10 mV (G0). G and H, effect of cyathin-R on the mPTP. PTP opening was assayed as described under “Experimental Procedures.” The effect of cyathin-R (5 μm) on mPTP opening in the absence (G) or the presence of Ca2+ (50 μm) (H) was monitored by following changes in absorbance at 520 nm, monitored every 15–20 s with an Ultraspec 2100 spectrophotometer. **, p < 0.01; ***, p < 0.001.
Moreover, cyathin-R did not activate PTP opening in the absence of Ca2+ and had no effect on Ca2+-activated PTP opening (Fig. 4, G and H). These findings imply that VDAC1 and not mPTP opening mediates cyathin-R-induced apoptosis in the absence of Bax and Bak.
We next asked whether oligomerization of VDAC1 is required for cyathin-R-induced cytochrome c release and apoptosis. In both Bax−/−/Bak−/− MEF and HCT116 colon cancer cells, following chemical cross-linking with EGS and immunoblotting, VDAC1 dimers and multimers were obtained upon exposure to cyathin-R in an incubation time- and concentration-dependent manner, concomitant with the disappearance of monomeric VDAC1 (Fig. 5, A–D). The inhibitors DpC, SITS, DIDS, and DNDS completely prevented cyathin-R-induced VDAC1 oligomerization (Fig. 5, C and D).
FIGURE 5.

Cyathin-R elevates VDAC1 levels and oligomerization coupled with apoptosis induction. A–D, Bax−/−/Bak−/− MEFs (A) and Bax−/− HCT116 (B) were treated with cyathin-R at the indicated times, or Bax−/−/Bak−/− MEFs were pretreated for 1 h with 0.5 mm DpC (C), SITS, DIDS, or DNDS (D), incubated with cyathin-R, and then cross-linked with EGS. VDAC1 levels and oligomerization were analyzed by immunoblotting, with the arrow in A pointing to the increase in VDAC1 expression level. E, cyathin-R-induced VDAC1 oligomerization detection by BRET2. Bax−/−/Bak−/− MEFs were co-transfected with siRNA specific to human VDAC1-encoding (50 nm), rVDAC1-Rluc-encoding (0.2 μg), and rVDAC1-GFP2-encoding (0.8 μg) plasmids. As a control, cells were co-transfected with the VDAC1-Rluc-encoding plasmid and siRNA for hVDAC1. Twenty-four h later, the cells were treated with cyathin-R (3 or 5 μm, 24 h) or with the relevant amount of DMSO. Luciferase and GFP signals were measured following 48 h of transfection. The ratios of the bioluminescence resonance energy transfer (BRET) signals were calculated as described. Luciferase activity was measured following the addition of the substrate, DeepBlue C, as luminescent, whereas GFP fluorescence was measured at 510 nm. All readings were performed with an Infinite 200 ELISA reader. F and G, Bax−/−/Bak−/− MEFs were treated with 5 μm cyathin-R and cyclophilin D (Cyp D), and VDAC1 levels were analyzed by Western blotting. Quantitative analysis of VDAC1 levels are presented as -fold increase. H, Bax−/− HCT116 cells stably expressing human VDAC1-shRNA to down-regulate VDAC1 levels or scrambled shRNA (Western blot at the top) were incubated with cyathin-R (5 μm, 24 h) and analyzed for apoptosis. Means ± S.E. (error bars) (n = 3) are presented. I and J, stable human VDAC1 knockdown Bax−/− HCT116 cells were transfected to express native or L277R mutant rat VDAC1. Twenty-four h later, the cells were treated with 5 μm cyathin-R. Aliquots were analyzed for VDAC1 levels and oligomerization by Western blotting (I). A second set of cell aliquots was analyzed by annexin-V/PI staining (J). Means ± S.E. (n = 3) are reported. *, p < 0.05.
Cyathin-R-induced VDAC1 oligomerization was also monitored in living cells by BRET. Energy transfer between VDAC1-luciferase (light-producing enzyme) as the donor and VDAC1-GFP2 (fluorophore) as the acceptor was obtained when the two VDAC1 molecules interacted physically (Fig. 5E). The BRET signal was increased about 7-fold upon apoptosis induced by cyathin-R.
These data are consistent with the ability of DpC, DIDS, DNDS, and SITS to inhibit cyathin-R-induced apoptosis (Figs. 1E and 4 (A and B)) and suggest that VDAC1 oligomerization induced by cyathin-R can mediate MOMP and apoptosis.
Cyathin-R also induced an increase in VDAC1 levels (Fig. 5, A (arrow), F, and G), as recently demonstrated for various other apoptosis stimuli (36). VDAC1 levels were increased severalfold upon cell treatment with cyathin-R, as compared with other mitochondrial proteins, such as cyclophilin D, which maintained a constant level (Fig. 5, F and G).
Stable and specific knockdown of human VDAC1 expression afforded these cells almost complete resistance to cyathin-R-induced apoptosis (Fig. 5H). More importantly, rescue experiments showed that wild type rat VDAC1 could restore cyathin-R-induced apoptosis in such cells, whereas the VDAC1 L277R mutant, which shows weakened oligomerizing ability (33) (Fig. 5I), was significantly less able to restore cyathin-R pro-apoptotic potential (Fig. 5J).
Because different apoptosis inducers elevate cytosolic Ca2+ and induce VDAC1 overexpression and oligomerization (36), we examined the effect of cyathin-R on cytosolic Ca2+ ([Ca2+]i) levels using the Ca2+ indicator Fluo-4 (Fig. 6A). Cyathin-R elevated [Ca2+]i concomitant with increasing apoptosis (Fig. 6B), whereas decreasing [Ca2+]i using the cell-permeable Ca2+-chelating reagent BAPTA-AM inhibited cyathin-R-induced increases in VDAC1 expression level (Fig. 6C), oligomerization (Fig. 6D), and apoptosis (Fig. 6E).
FIGURE 6.

Cyathin-R-induced VDAC1 oligomerization and apoptosis are accompanied by an elevation of intercellular Ca2+ and are inhibited by BAPTA-AM. A, representative flow cytomery analysis of [Ca2+]i levels, as monitored using Fluo-4. Bax−/−/Bak−/− MEF cells were incubated for 16 h with cyathin-R (8 μm) and harvested, and intracellular Ca2+ levels were assessed. The values shown indicate the increase in [Ca2+]i. B, quantitative analysis of [Ca2+]i levels (black circles) and apoptosis (white circles) assayed using annexin-V-FITC and PI staining, as analyzed by flow cytometry. C–E, Bax−/−/Bak−/− MEF cells were incubated with BAPTA-AM (5 μm, 1.5 h) and then with or without cyathin-R (5 or 8 μm, 16 h), and VDAC1 overexpression (C), VDAC1 oligomerization (D), and apoptosis (E) were assayed. Means ± S.E. (error bars) (n = 3) are presented.
Cyathin-R-mediated Changes in the Cellular Redox State and Mitochondrial Membrane Potential
Previous studies revealed that oxidative stress, especially due to the mitochondrial superoxide, could trigger VDAC1-dependent apoptosis (3, 17, 20, 21). Here, we found that cyathin-R induced reactive oxidative species (ROS) production, as measured by MitoSOX Red, a mitochondrial superoxide indicator (Fig. 7A). Cyathin-R elevated overall cellular ROS levels (H2DCFDA fluorescence) in Bax−/−/Bak−/− MEF cells (Fig. 7B), with cyathin-R-induced apoptosis being inhibited by the ROS scavenger NAC (Fig. 7C) or by DTT (Fig. 8A). Moreover, NAC completely inhibited cyathin-R-induced VDAC1 oligomerization (Fig. 7D). Because depletion of GSH, a vital cellular antioxidant, could lead to oxidative stress, we examined the cellular GSH content and found it to be significantly decreased upon cyathin-R treatment (Fig. 7E). Pretreatment with cell-permeable glutathione ethyl ester prevented cyathin-R-induced apoptosis (Fig. 7F).
FIGURE 7.

Cyathin-R induces apoptosis via oxidative stress. A–E, Bax−/−/Bak−/− MEFs were pretreated (1 h) with NAC followed by incubation with cyathin-R (5 μm, 16 h) (A). The cells were stained with MitoSOX Red ROS indicator and analyzed by flow cytometry. Shown are means ± S.E. (error bars) (n = 3). B, cells were incubated with cyathin-R (6 h), stained with H2DCFDA (DCF), and analyzed by flow cytometry. Shown are means ± S.E. (n = 3). C, cells were incubated with cyathin-R (5 μm, 36 h) and analyzed for apoptosis by annexin-V/PI staining. Means ± S.E. (n = 3) are presented. D, cells were incubated with cyathin-R (1 h) and cross-linked by EGS, and VDAC1 oligomerization was analyzed by immunoblotting. E, cells were incubated with cyathin-R (16 h), and GSH content was assayed with a GSH and GSSG assay kit. Means ± S.E. (n = 3) are presented. F, Bax−/−/Bak−/− MEFs were pretreated with glutathione ethyl ester (GSHee) for 1 h and then with cyathin-R (16 h), and apoptosis was analyzed. Means ± S.E. (n = 3) are presented. G, analysis of mitochondrial membrane potential (ψm). Bax−/−/Bak−/− MEF cells were treated with cyathin-R (5 μm, 6 h) or carbonyl cyanide p-trifluoromethoxyphenylhydrazone (10 μm, 6 h) as a positive control and incubated with TMRM (10 min, 37 °C), and fluorescence was analyzed by flow cytometry. The results shown are those obtained after subtracting the value measured with the carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) control. Means ± S.E. (n = 3) are presented. **, p < 0.01; ***, p < 0.001.
FIGURE 8.

DTT prevents apoptosis induced by cyathin-R and activation of NF-κB. A, Bax−/−/Bak−/− MEFs were pretreated with the indicated concentrations of DTT for 1 h and then incubated with cyathin-R (3 μm). Cell death was analyzed by annexin-V/PI staining and flow cytometry. Means ± S.E. (error bars) (n = 3) are reported. B and C, Bax−/−/Bak−/− MEF cells were treated with cyathrin-R (3 μm) for the indicated times in the absence and the presence of NAC (10 mm). Immunofluorescence analysis revealed p65 to be mainly found in the cytosol, although cyathrin-R induced translocation of p65 to the nucleus (B, arrows). Analysis of the nuclear fraction further indicates p65 translocation to the nucleus (C). Such translocation was prevented when NAC was present (B and C).
Because cyathin-R induces ROS production, with ROS known to activate NF-κB, we analyzed the location of the NF-κB component p65 and found it to have translocated to the nucleus, as analyzed by immunocytochemistry and immunoblotting of the nuclear fraction. Such translocation was prevented by NAC (Fig. 8, B and C).
Finally, because loss of mitochondrial membrane potential (Δψm) is often considered to be indicative of MOMP, the effect of cyathin-R on Δψm was monitored using TMRM. Cyathin-R induced a complete loss of Δψm in a dose-dependent manner (Fig. 7G).
Cyathin-R Attenuates the Growth of Bax−/−/Bak−/− MEFs, Bax−/− HCT116, and HCT116 Colon Cancer Cells in Vivo
Because Bax or Bak mutation or deficiency was correlated with the occurrence of cancer and drug resistance in cancer cells (13–15), we examined the effect of cyathin-R on MEFs and cancer cells growth in a xenograft mouse model. Immortalized Bax−/−/Bak−/− MEFs or wild type or Bax−/− HCT116 colon cancer cells were implanted into nude mice, and tumor growth was monitored. One week later, mice implanted with a specific cell type were divided into three groups (6 mice/group) and underwent intraperitoneal treatment every 2 days with the vehicle control (10% DMSO) or cyathin-R (5 or 10 mg/kg) for 2 weeks. Cyathin-R effectively inhibited the growth of Bax−/−/Bak−/− MEFs (Fig. 9, A and B) and of wild type and Bax−/− HCT116 colon cancer cells in the mice in a concentration-dependent manner (Fig. 9, E, F, H, and I). Comparing tumor sizes at the end point revealed that the average tumor volume in the 10 mg/kg cyathin-R-treated tumors was decreased by about 75% of that of control, DMSO-injected tumors (Fig. 9A). At the dosage used, cyathin-R was tolerable, because no obvious weight loss was observed (Fig. 9, D, G, and J). In situ TUNEL staining showed that cyathin-R also effectively induced apoptosis of the neoplasm in vivo (Fig. 9C). These data indicate that as with cells in culture, cyathin-R induced cell death in the xenograft mouse model.
FIGURE 9.

Cyathin-R attenuates tumor growth by inducing apoptosis of immortalized Bax−/−/Bak−/− cells and Bax−/− HCT116 or HCT116 colon cancer cells in a xenograft mouse model. Immunodeficient nude mice were implanted with Bax−/−/Bak−/− MEFs (A–D), Bax−/− HCT116 (E–G), or HCT116 (I–J) colon cancer cells. One week later, the mice were divided into three groups (6 mice/group) and underwent intraperitoneal treatment with the vehicle control (DMSO, 10%) or cyathin-R (5 or 10 mg/kg/2 days) for 2 weeks, during which time tumor size was measured. Two weeks post-cyathin-R treatment, the mice were euthanized, and the xenografts were excised for further studies. Tumor volume curves (A, E, and H) and representative images of isolated tumors (B, F, and I) and average body weights of nude mice after 2 weeks of treatment (D, G, and J) are shown. C, fluorescence images show staining of apoptotic Bax−/−/Bak−/− cells by the in situ TUNEL assay. Scale bar, 100 μm. Error bars, S.E.
Bcl-2 Protects against Cyathin-R-induced Apoptosis
Bcl-2 and Bcl-xL are important antagonists of MOMP and apoptosis and were shown to directly interact with VDAC1 (25, 48, 50, 51). We observed that the overall levels and mitochondrial localization of Bcl-2 (and, to a lesser extent, of Bcl-xL) were dramatically decreased upon cyathin-R treatment of Bax−/−/Bak−/− MEFs (Fig. 10, A and B). Quantitative real-time PCR analysis showed that cyathin-R treatment decreased Bcl-2 transcript levels (Fig. 10C). As expected, cells stably expressing Bcl-2 showed resistance to cyathin-R-induced apoptosis and VDAC1 oligomerization (Fig. 10D). Interestingly, the Bcl-2/Bcl-xL inhibitor ABT737 failed to induce apoptosis in Bax-/Bak-deficient cells and had no effect on cyathin-R-induced apoptosis. Moreover, ABT737 did not abrogate Bcl-2-mediated protection against cyathin-R-induced apoptosis (data not shown).
FIGURE 10.

Bcl-2 regulates cyathin-R-induced VDAC1 oligomerization and apoptosis. A–C, Bax−/−/Bak−/− MEFs were treated with 3 μm cyathin-R for the indicated times, and the indicated proteins were analyzed by immunoblotting. A, cells were fractionated into mitochondrial and cytosol fractions, and Bcl-2 was analyzed by immunoblotting, with actin and Tim23 serving as input controls (B). Bcl-2 mRNA levels were monitored by quantitative real-time PCR (C). Means ± S.E. (error bars) (n = 3) are presented. D, a FLAG-tagged Bcl-2 vector was transfected into Bax−/− HCT116 cells, and stable Bcl-2-overexpressing cells were acquired using puromycin. Stable Bcl-2-overexpressing Bax−/− HCT116 and control cells were treated with cyathin-R (5 μm, 24 h) and analyzed for apoptosis. Means ± S.E. (n = 3) are presented. E, proposed model of cyathin-R-induced apoptosis. Cyathin-R, through a triggering of oxidative stress, causes augmentation of VDAC1 levels that in turn shifts the equilibrium toward VDAC1 oligomerization, allowing cytochrome c release, which leads to apoptosis. The decrease of Bcl-2 facilitates oligomerization of VDAC1, leading to apoptosis. *, p < 0.05.
Discussion
Investigations into the mechanisms of MOMP and cytochrome c release from mitochondria have centered mainly on Bax and Bak as major players in these critical processes that ultimately set off the apoptotic cascade. However, mitochondria-mediated apoptosis can be activated in the absence of Bax/Bak (10–12). This is significant because in several tumors, resistance to chemotherapy is due to down-regulation of Bax and Bak (13–15, 52), thus reflecting a substantial clinical challenge. As such, it is important to identify novel apoptosis inducers that bypass Bax/Bak. The results presented here showing that a new compound, cyathin-R, is able to induce apoptosis in a Bax/Bak-independent manner by activating a novel mechanism involving VDAC1 oligomerization are, therefore, of importance.
In recent years, accumulated evidence has pointed to VDAC1 involvement in apoptosis (3, 16–21, 29, 30, 32–37, 53, 54), although VDAC function in apoptosis has also been also questioned (28). Upon apoptosis induction, VDAC1 oligomerizes to form a large pore that allows the passage of a folded protein like cytochrome c, leading to apoptosis (31, 34–39). Although evidence for coupling between VDAC1 oligomerization and apoptosis abounds, it remains unclear whether Bax and Bak are dispensable for VDAC1-mediated cytochrome c release. With this in mind, we undertook a chemical biology approach to identify compounds that induce apoptosis in Bax-/Bak-deficient cells in a manner that can be inhibited by the VDAC1 interaction and conductance inhibitors DpC, DIDS, or Bcl-2. Accordingly, we have identified a cyathane diterpenoid compound, cyathin-R, that enhanced VDAC1 protein levels and promoted VDAC1 oligomerization and apoptosis in a manner that can be inhibited by DpC, DIDS, SITS, or DNDS; by knockdown of VDAC1; or by ectopic expression of Bcl-2.
The results presented here, together with previous reports, clearly demonstrate that oligomeric VDAC1 is critical for cytochrome c release (34–37). It is thus important to understand how VDAC1 oligomerization is regulated in the cell. We observed that VDAC1 levels are increased by cyathin-R treatment, providing the basis for subsequent oligomerization. Indeed, several other apoptosis inducers were found to increase VDAC1 levels (40–42, 55, 56). By extending these observations, we have proposed that apoptosis-inducing agents act by increasing [Ca2+]i and that this in turn leads to up-regulation of VDAC1 expression (43). The increased level of monomeric VDAC1 shifts the equilibrium toward VDAC1 oligomerization, resulting in changes in OMM permeability allowing for cytochrome c release, leading to cell death (34–37). Moreover, the cellular level of VDAC1 was shown to be a crucial factor in the process of mitochondria-mediated apoptosis, with exogenous overexpression of VDAC1 resulting in cell death, regardless of the cell type or the origin of VDAC used (36, 57–60). Importantly, the causal relationship between VDAC1 levels and drug sensitivity was emphasized in several studies. A prostate cancer cell line resistant to G3139-induced apoptosis expresses low levels of VDAC1 relative to the G3139-sensitive cell line (61), whereas overexpression of VDAC1 sensitized carcinoma cells to apoptosis as induced by cisplatin and its derivatives (43). These observations suggest that the activity of numerous anti-cancer drugs and treatments is mediated via elevating VDAC1 levels.
Here, tight correlation between increased VDAC1 levels, VDAC1 oligomerization, and apoptosis was clearly demonstrated in cells lacking Bax/Bak, because all three processes were inhibited by the VDAC1-interacting molecules DpC and DIDS, the ROS scavenger NAC, or by ectopic expression of Bcl-2. Additionally, cyathin-R effectively attenuated tumor growth in Bax−/−/Bak−/− MEF and Bax−/− HCT116 cells and in a colon cancer xenograft model.
The finding that cyathin-R-triggered oxidative stress is associated with increased VDAC1 levels and oligomerization is in agreement with a previous report showing that ROS induced VDAC1 up-regulation that could be prevented by a ROS chelator (56). H2O2 was found to elevate intracellular Ca2+ levels, up-regulate VDAC1 expression, and induce VDAC1 oligomerization and apoptosis (36). These H2O2-mediated effects were prevented by BAPTA, suggesting their Ca2+ dependence (36), as also shown here for cyathin-R inducing increased intracellular Ca2+ and inhibition of its pro-apoptotic activity by BAPTA (Fig. 6). Several potential Ca2+-dependent steps could contribute to the enhanced expression of VDAC1, including mRNA transcription, elongation, splicing, stability, and/or translation.
Another possible mechanism may be linked to the proposed role of VDAC1 in mediating ROS release from mitochondria to the cytosol (62). VDAC1 could be subjected to oxidative modifications that would reduce its degradation, thereby leading to its accumulation and subsequent oligomerization.
Finally, we found that treatment with cyathin-R led to down-regulation of Bcl-2 levels. Thus, cyathin-R not only induces apoptosis via promoting VDAC1 oligomerization but also antagonizes the anti-apoptotic effects of Bcl-2 and Bcl-xL by decreasing their levels. Reduced Bcl-2 levels alter either mitochondrial oxidative stress, because Bcl-2 was previously shown to regulate ROS and cellular GSH (63), or its interaction with VDAC1 (25, 48, 50, 51), thereby releasing VDAC1 for subsequent oligomerization.
The findings that the Bcl-2/Bcl-xL inhibitor ABT737 (64) failed to induce apoptosis in Bax-/Bax-deficient cells and to prevent the Bcl-2-mediated protection against cyathin-R-induced apoptosis further highlight the critical role that VDAC1 plays in apoptosis as well as the need for new compounds able to induce apoptosis in such cells, such as cyathin-R. Because VDAC1, adenine nucleotide translocase, and cyclophilin D are proposed components of the mPTP (53, 54), it is possible that cyathin-R induces cytochrome c release via the mPTP. However, pretreatment of Bax−/−/Bak−/− MEF cells with cyclosporin A, a specific inhibitor of cyclophilin D, failed to inhibit apoptosis, and cyathin-R did not affect PTP opening. Hence, it is unlikely that the mPTP mediates cyathin-R-induced cytochrome c release and apoptosis.
In this respect, the interaction of cyathin-R and DpC with VDAC1, the inhibition of channel conductance, and the effects of these compounds in apoptosis induction and inhibition, respectively, are not directly related, because the VDAC1 pore, mediating transport of small molecules (up to 5 kDa), is not the pore mediating the release of cytochrome c (34–37). Indeed, some reagents, such as As2O3 and Koenig's polyanion, were shown to reduce channel conductance but induce apoptosis, whereas others inhibited both processes (for a review, see Ref. 65). As shown here, DpC and cyathin-R both decreased VDAC1 conductance, whereas DpC inhibited VDAC1 oligomerization and cyathin-R enhanced oligomerization. It seems that most interacting sites for small molecules are in the pore, and their interaction there reduced the channel conductance of current-conducting small ions. This was also shown for tubulin (66), with its tail being inserted into the pore. Other proteins interacting with VDAC1 and reducing its conductance, such as HK, Bcl2, and Bcl-xL (29, 48, 50, 60), interact from the cytoplasmic face and block channel conductance.
The model proposed here (Fig. 10F), in which cyathin-R enhances VDAC1 protein levels, thereby promoting VDAC1 oligomerization and apoptosis, involves a novel mechanism highlighting cyathin-R as a potential lead for an effective anti-cancer drug.
Many cancer cells develop cell survival strategies that implicate anti-apoptotic defense mechanisms. As vital regulators of apoptosis, deficiency of pro-apoptotic Bax (13–15, 52) or overexpression of anti-apoptotic Bcl-2 (6) renders cancer cells more resistant to most chemotherapies. It is thus important to develop effective drugs targeting different apoptotic pathways, able to eliminate cells with abnormal Bax or Bcl-2 expression. Our work has identified a VDAC1-dependent pathway specifically targeted by a novel pharmacological tool capable of inducing apoptosis in tumor cells, independently of Bax and Bak. Thus, small molecules that target VDAC1 to induce apoptosis and decrease anti-apoptotic Bcl-2 and Bcl-xL levels (like cyathin-R) can be considered as acting at the crossroads between apoptosis induction and antagonizing the anti-apoptotic effects of Bcl-2 and Bcl-xL and thus represent an emerging cancer drug class. Clinically, cyathin-R, inducing VDAC1-dependent apoptosis, together with compounds targeting different apoptotic steps, could provide enhanced anti-tumor potential for therapeutic treatment.
In summary, our studies demonstrate that cyathin-R is a potent inducer of apoptosis, acting via VDAC1 yet independently of Bax and Bak. Thus, identifying cyathin-R and VDAC1 as its novel target reveals a route to circumvent a common resistance mechanism of tumor cells.
Author Contributions
L. H., V. S.-B., and Q. C. designed the research; L. H., J. H., B. L., Z. C., L. H., Y. W., and Y. Y. performed the research; L. H., H. L., L. L., Y. Z., D. B.-H., V. S.-B., and Q. C. analyzed the data; and L. H., H. L., V. S.-B., and Q. C. wrote the paper.
Acknowledgments
We thank Professor Weixing Zong for providing E1A/K-RAS transformed Bax−/−/Bak−/− MEF cells and Jing Wang for operating the flow cytometer.
This work was supported by key projects of the Natural Science Foundation of China (Grants 91213304 and 8113004) and the Ministry of Science and Technology of China (Grant 2014CB138304) and by Israel Science Foundation Grant 307/13 (to V. S. B.). The authors declare that they have no conflicts of interest with the contents of this article.
CCDC 881686 contains supplementary crystallographic data for cyathin-R. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk/data_request/cif).
- OMM
- outer mitochondrial membrane
- Bcl-2
- B-cell lymphoma/leukemia-2
- Bax
- Bcl-2-associated X protein
- DIDS
- 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt
- DNDS
- 4,4′-dinitrostilbene-2,2′-disulfonic acid
- DpC
- N-phenylanthranilic acid
- EGS
- ethylene glycolbis(succinimidyl succinate)
- ΔΨm
- mitochondrial membrane potential
- MOMP
- mitochondrial outer membrane permeabilization
- PTP
- permeability transition pore
- mPTP
- mitochondrial permeability transition pore
- MEF
- mouse embryonic fibroblast
- NAC
- N-acetylcysteine
- PI
- propidium iodide
- PAO
- phenylarsine oxide
- ROS
- reactive oxygen species
- SITS
- 4-acetamido-4′-isothiocyanato-stilbene-2,2′-disulfonic acid
- VDAC
- voltage-dependent anion channel
- rVDAC1
- recombinant voltage-dependent anion channel 1
- H2DCFDA
- 2′,7′-dichlorodihydrofluorescein diacetate
- TMRM
- tetramethylrhodamine, methyl ester
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid.
References
- 1. Kroemer G., Galluzzi L., Brenner C. (2007) Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163 [DOI] [PubMed] [Google Scholar]
- 2. Riedl S. J., Salvesen G. S. (2007) The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 8, 405–413 [DOI] [PubMed] [Google Scholar]
- 3. Shoshan-Barmatz V., De Pinto V., Zweckstetter M., Raviv Z., Keinan N., Arbel N. (2010) VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285 [DOI] [PubMed] [Google Scholar]
- 4. Antignani A., Youle R. J. (2006) How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 18, 685–689 [DOI] [PubMed] [Google Scholar]
- 5. Crompton M. (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341, 233–249 [PMC free article] [PubMed] [Google Scholar]
- 6. Harris M. H., Thompson C. B. (2000) The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 7, 1182–1191 [DOI] [PubMed] [Google Scholar]
- 7. Brustovetsky T., Li T., Yang Y., Zhang J. T., Antonsson B., Brustovetsky N. (2010) BAX insertion, oligomerization, and outer membrane permeabilization in brain mitochondria: role of permeability transition and SH-redox regulation. Biochim. Biophys. Acta 1797, 1795–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dewson G., Kluck R. M. (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci. 122, 2801–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim H., Tu H. C., Ren D., Takeuchi O., Jeffers J. R., Zambetti G. P., Hsieh J. J., Cheng E. H. (2009) Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Claveria C., Martinez-A C., Torres M. (2004) A Bax/Bak-independent mitochondrial death pathway triggered by Drosophila Grim GH3 domain in mammalian cells. J. Biol. Chem. 279, 1368–1375 [DOI] [PubMed] [Google Scholar]
- 11. Mizuta T., Shimizu S., Matsuoka Y., Nakagawa T., Tsujimoto Y. (2007) A Bax/Bak-independent mechanism of cytochrome c release. J. Biol. Chem. 282, 16623–16630 [DOI] [PubMed] [Google Scholar]
- 12. Wan K. F., Chan S. L., Sukumaran S. K., Lee M. C., Yu V. C. (2008) Chelerythrine induces apoptosis through a Bax/Bak-independent mitochondrial mechanism. J. Biol. Chem. 283, 8423–8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ionov Y., Yamamoto H., Krajewski S., Reed J. C., Perucho M. (2000) Mutational inactivation of the proapoptotic gene BAX confers selective advantage during tumor clonal evolution. Proc. Natl. Acad. Sci. U.S.A. 97, 10872–10877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. LeBlanc H., Lawrence D., Varfolomeev E., Totpal K., Morlan J., Schow P., Fong S., Schwall R., Sinicropi D., Ashkenazi A. (2002) Tumor-cell resistance to death receptor-induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat. Med. 8, 274–281 [DOI] [PubMed] [Google Scholar]
- 15. Wang G. Q., Gastman B. R., Wieckowski E., Goldstein L. A., Gambotto A., Kim T. H., Fang B., Rabinovitz A., Yin X. M., Rabinowich H. (2001) A role for mitochondrial Bak in apoptotic response to anticancer drugs. J. Biol. Chem. 276, 34307–34317 [DOI] [PubMed] [Google Scholar]
- 16. Maldonado E. N., Lemasters J. J. (2012) Warburg revisited: regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J. Pharmacol. Exp. Ther. 342, 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shoshan-Barmatz V., Mizrachi D. (2012) VDAC1: from structure to cancer therapy. Front. Oncol. 2, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lemasters J. J., Holmuhamedov E. (2006) Voltage-dependent anion channel (VDAC) as mitochondrial governator: thinking outside the box. Biochim. Biophys. Acta 1762, 181–190 [DOI] [PubMed] [Google Scholar]
- 19. Martel C., Wang Z., Brenner C. (2014) VDAC phosphorylation, a lipid sensor influencing the cell fate. Mitochondrion 19, 69–77 [DOI] [PubMed] [Google Scholar]
- 20. Shimizu S., Narita M., Tsujimoto Y. (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487 [DOI] [PubMed] [Google Scholar]
- 21. Tsujimoto Y., Shimizu S. (2000) VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 7, 1174–1181 [DOI] [PubMed] [Google Scholar]
- 22. Tajeddine N., Galluzzi L., Kepp O., Hangen E., Morselli E., Senovilla L., Araujo N., Pinna G., Larochette N., Zamzami N., Modjtahedi N., Harel-Bellan A., Kroemer G. (2008) Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 27, 4221–4232 [DOI] [PubMed] [Google Scholar]
- 23. Yuan S., Fu Y., Wang X., Shi H., Huang Y., Song X., Li L., Song N., Luo Y. (2008) Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. 22, 2809–2820 [DOI] [PubMed] [Google Scholar]
- 24. Chacko A. D., Liberante F., Paul I., Longley D. B., Fennell D. A. (2010) Voltage dependent anion channel-1 regulates death receptor mediated apoptosis by enabling cleavage of caspase-8. BMC Cancer 10, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng Y., Shi Y., Tian C., Jiang C., Jin H., Chen J., Almasan A., Tang H., Chen Q. (2004) Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 23, 1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimizu S., Matsuoka Y., Shinohara Y., Yoneda Y., Tsujimoto Y. (2001) Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J. Cell Biol. 152, 237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Madesh M., Hajnóczky G. (2001) VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 155, 1003–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCommis K. S., Baines C. P. (2012) The role of VDAC in cell death: friend or foe? Biochim. Biophys. Acta 1818, 1444–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abu-Hamad S., Arbel N., Calo D., Arzoine L., Israelson A., Keinan N., Ben-Romano R., Friedman O., Shoshan-Barmatz V. (2009) The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 122, 1906–1916 [DOI] [PubMed] [Google Scholar]
- 30. Bayrhuber M., Meins T., Habeck M., Becker S., Giller K., Villinger S., Vonrhein C., Griesinger C., Zweckstetter M., Zeth K. (2008) Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. U.S.A. 105, 15370–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Betaneli V., Petrov E. P., Schwille P. (2012) The role of lipids in VDAC oligomerization. Biophys. J. 102, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geula S., Ben-Hail D., Shoshan-Barmatz V. (2012) Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 444, 475–485 [DOI] [PubMed] [Google Scholar]
- 33. Geula S., Naveed H., Liang J., Shoshan-Barmatz V. (2012) Structure-based analysis of VDAC1 protein: defining oligomer contact sites. J. Biol. Chem. 287, 2179–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keinan N., Tyomkin D., Shoshan-Barmatz V. (2010) Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 30, 5698–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shoshan-Barmatz V., Mizrachi D., Keinan N. (2013) Oligomerization of the mitochondrial protein VDAC1: from structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 117, 303–334 [DOI] [PubMed] [Google Scholar]
- 36. Weisthal S., Keinan N., Ben-Hail D., Arif T., Shoshan-Barmatz V. (2014) Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta 1843, 2270–2281 [DOI] [PubMed] [Google Scholar]
- 37. Zalk R., Israelson A., Garty E. S., Azoulay-Zohar H., Shoshan-Barmatz V. (2005) Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 386, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vaseva A. V., Marchenko N. D., Ji K., Tsirka S. E., Holzmann S., Moll U. M. (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolff S., Erster S., Palacios G., Moll U. M. (2008) p53's mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res. 18, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cheng S. L., Liu R. H., Sheu J. N., Chen S. T., Sinchaikul S., Tsay G. J. (2007) Toxicogenomics of A375 human malignant melanoma cells treated with arbutin. J. Biomed. Sci. 14, 87–105 [DOI] [PubMed] [Google Scholar]
- 41. Jiang N., Kham S. K., Koh G. S., Suang Lim J. Y., Ariffin H., Chew F. T., Yeoh A. E. (2011) Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). J. Proteomics 74, 843–857 [DOI] [PubMed] [Google Scholar]
- 42. Voehringer D. W., Hirschberg D. L., Xiao J., Lu Q., Roederer M., Lock C. B., Herzenberg L. A., Steinman L., Herzenberg L. A. (2000) Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 2680–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sharaf el dein O., Gallerne C., Brenner C., Lemaire C. (2012) Increased expression of VDAC1 sensitizes carcinoma cells to apoptosis induced by DNA cross-linking agents. Biochem. Pharmacol. 83, 1172–1182 [DOI] [PubMed] [Google Scholar]
- 44. Lei X., Chen Y., Du G., Yu W., Wang X., Qu H., Xia B., He H., Mao J., Zong W., Liao X., Mehrpour M., Hao X., Chen Q. (2006) Gossypol induces Bax/Bak-independent activation of apoptosis and cytochrome c release via a conformational change in Bcl-2. FASEB J. 20, 2147–2149 [DOI] [PubMed] [Google Scholar]
- 45. Liu J., Mu C., Yue W., Li J., Ma B., Zhao L., Liu L., Chen Q., Yan C., Liu H., Hao X., Zhu Y. (2013) A diterpenoid derivate compound targets selenocysteine of thioredoxin reductases and induces Bax/Bak-independent apoptosis. Free Radic. Biol. Med. 63, 485–494 [DOI] [PubMed] [Google Scholar]
- 46. Zhao L., He F., Liu H., Zhu Y., Tian W., Gao P., He H., Yue W., Lei X., Ni B., Wang X., Jin H., Hao X., Lin J., Chen Q. (2012) Natural diterpenoid compound elevates expression of Bim protein, which interacts with antiapoptotic protein Bcl-2, converting it to proapoptotic Bax-like molecule. J. Biol. Chem. 287, 1054–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han J., Chen Y., Bao L., Yang X., Liu D., Li S., Zhao F., Liu H. (2013) Anti-inflammatory and cytotoxic cyathane diterpenoids from the medicinal fungus Cyathus africanus. Fitoterapia 84, 22–31 [DOI] [PubMed] [Google Scholar]
- 48. Arbel N., Ben-Hail D., Shoshan-Barmatz V. (2012) Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 287, 23152–23161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shoshan-Barmatz V., Hadad N., Feng W., Shafir I., Orr I., Varsanyi M., Heilmeyer L. M. (1996) VDAC/porin is present in sarcoplasmic reticulum from skeletal muscle. FEBS Lett. 386, 205–210 [DOI] [PubMed] [Google Scholar]
- 50. Arbel N., Shoshan-Barmatz V. (2010) Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 285, 6053–6062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Malia T. J., Wagner G. (2007) NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry 46, 514–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCurrach M. E., Connor T. M., Knudson C. M., Korsmeyer S. J., Lowe S. W. (1997) bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc. Natl. Acad. Sci. U.S.A. 94, 2345–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Halestrap A. P. (2009) What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46, 821–831 [DOI] [PubMed] [Google Scholar]
- 54. Zoratti M., Szabò I. (1995) The mitochondrial permeability transition. Biochim. Biophys. Acta 1241, 139–176 [DOI] [PubMed] [Google Scholar]
- 55. Castagna A., Antonioli P., Astner H., Hamdan M., Righetti S. C., Perego P., Zunino F., Righetti P. G. (2004) A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics 4, 3246–3267 [DOI] [PubMed] [Google Scholar]
- 56. Jung J. Y., Han C. R., Jeong Y. J., Kim H. J., Lim H. S., Lee K. H., Park H. O., Oh W. M., Kim S. H., Kim W. J. (2007) Epigallocatechin gallate inhibits nitric oxide-induced apoptosis in rat PC12 cells. Neurosci. Lett. 411, 222–227 [DOI] [PubMed] [Google Scholar]
- 57. Ghosh T., Pandey N., Maitra A., Brahmachari S. K., Pillai B. (2007) A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS One 2, e1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Godbole A., Varghese J., Sarin A., Mathew M. K. (2003) VDAC is a conserved element of death pathways in plant and animal systems. Biochim. Biophys. Acta 1642, 87–96 [DOI] [PubMed] [Google Scholar]
- 59. Lü A. J., Dong C. W., Du C. S., Zhang Q. Y. (2007) Characterization and expression analysis of Paralichthys olivaceus voltage-dependent anion channel (VDAC) gene in response to virus infection. Fish Shellfish Immunol. 23, 601–613 [DOI] [PubMed] [Google Scholar]
- 60. Zaid H., Abu-Hamad S., Israelson A., Nathan I., Shoshan-Barmatz V. (2005) The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 12, 751–760 [DOI] [PubMed] [Google Scholar]
- 61. Lai J. C., Tan W., Benimetskaya L., Miller P., Colombini M., Stein C. A. (2006) A pharmacologic target of G3139 in melanoma cells may be the mitochondrial VDAC. Proc. Natl. Acad. Sci. U.S.A. 103, 7494–7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Han D., Antunes F., Canali R., Rettori D., Cadenas E. (2003) Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563 [DOI] [PubMed] [Google Scholar]
- 63. Hockenbery D. M., Oltvai Z. N., Yin X. M., Milliman C. L., Korsmeyer S. J. (1993) Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75, 241–251 [DOI] [PubMed] [Google Scholar]
- 64. van Delft M. F., Wei A. H., Mason K. D., Vandenberg C. J., Chen L., Czabotar P. E., Willis S. N., Scott C. L., Day C. L., Cory S., Adams J. M., Roberts A. W., Huang D. C. (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shoshan-Barmatz V., Israelson A., Brdiczka D., Sheu S. S. (2006) The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 12, 2249–2270 [DOI] [PubMed] [Google Scholar]
- 66. Rostovtseva T. K., Bezrukov S. M. (2012) VDAC inhibition by tubulin and its physiological implications. Biochim. Biophys. Acta 1818, 1526–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]