

Background: Two polymorphic variants of rhodanese involved in mitochondrial H2S oxidation have been described.
Results: The variants exhibit higher thermal stability and differences in sulfur transfer kinetics from wild-type enzyme.
Conclusion: All three rhodanese preferentially catalyze sulfur transfer from GSSH to sulfite to form thiosulfate.
Significance: Differences between the rhodanese variants might be pertinent to disease susceptibility.
Keywords: enzyme, enzyme kinetics, hydrogen sulfide, single-nucleotide polymorphism (SNP), sulfur, cyanide, rhodanese, sulfide oxidation
Abstract
Rhodanese is a component of the mitochondrial H2S oxidation pathway. Rhodanese catalyzes the transfer of sulfane sulfur from glutathione persulfide (GSSH) to sulfite generating thiosulfate and from thiosulfate to cyanide generating thiocyanate. Two polymorphic variations have been identified in the rhodanese coding sequence in the French Caucasian population. The first, 306A→C, has an allelic frequency of 1% and results in an E102D substitution in the encoded protein. The second polymorphism, 853C→G, has an allelic frequency of 5% and leads to a P285A substitution. In this study, we have examined differences in the stability between wild-type rhodanese and the E102D and P285A variants and in the kinetics of the sulfur transfer reactions. The Asp-102 and Ala-285 variants are more stable than wild-type rhodanese and exhibit kcat/Km,CN values that are 17- and 1.6-fold higher, respectively. All three rhodanese forms preferentially catalyze sulfur transfer from GSSH to sulfite, generating thiosulfate and glutathione. The kcat/Km,sulfite values for the variants in the sulfur transfer reaction from GSSH to sulfite were 1.6- (Asp-102) and 4-fold (Ala-285) lower than for wild-type rhodanese, whereas the kcat/Km,GSSH values were similar for all three enzymes. Thiosulfate-dependent H2S production in murine liver lysate is low, consistent with a role for rhodanese in sulfide oxidation. Our studies show that polymorphic variations that are distant from the active site differentially modulate the sulfurtransferase activity of human rhodanese to cyanide versus sulfite and might be important in differences in susceptibility to diseases where rhodanese dysfunction has been implicated, e.g. inflammatory bowel diseases.
Introduction
Members of the sulfurtransferase superfamily are widely distributed in all three kingdoms of life and are characterized by the presence of a tertiary structure module, the rhodanese domain. The latter can be present singly, in tandem repeats, or fused to other protein modules (1, 2). Rhodanese is perhaps the most extensively characterized sulfurtransferase and initially was described as a thiosulfate:cyanide sulfurtransferase (3). Bovine rhodanese, for which a crystal structure is available (4, 5), has a tandem repeat of two globular rhodanese domains each ∼120 amino acids in length connected by a long linker region (Fig. 1, A and B). Each rhodanese domain has an α/β topology with α-helices surrounding a central five-stranded β-sheet. The active site is walled in by residues from both domains and resides in the interdomain cleft (6). The catalytically active cysteine residue, Cys-247, is embedded in a CRXGX(R/T) sequence motif in the C-terminal domain, in which X represents any amino acid, and the parentheses denote alternate residues. The sequence of the conserved active site loop distinguishes rhodanese from other sulfurtransferase family members (1, 2).
FIGURE 1.
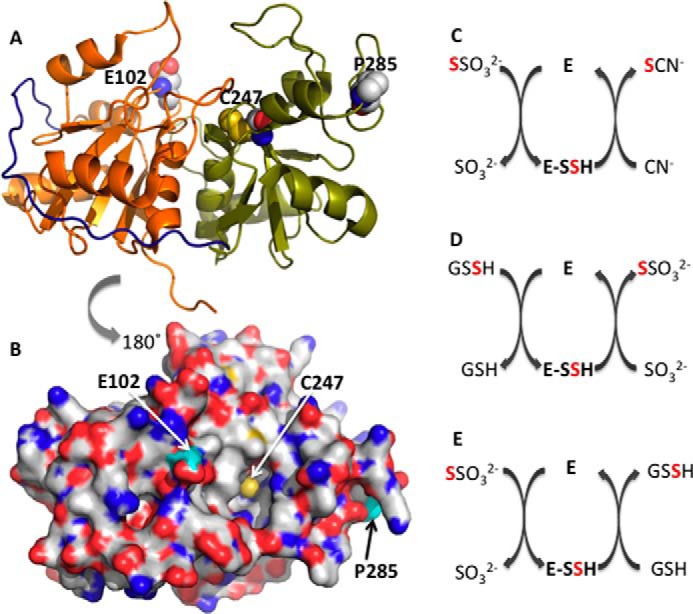
Structure and reactions catalyzed by rhodanese. A and B, crystal structure of bovine rhodanese (Protein Data Bank code 1BOH). The N- and C-terminal domains are in orange and green, respectively, whereas the interdomain loop is in navy blue. The active site cysteine, Cys-247, and the polymorphic loci, Asp-102 and Pro-285, are shown in sphere representation. C–E, representative reactions catalyzed by rhodanese. In C and E, thiosulfate is the sulfane sulfur donor, whereas in D, GSSH is the sulfur donor. In each case, an enzyme-bound persulfide intermediate (E-SSH) is formed. Cyanide, sulfite, and GSH are the sulfur acceptors in C–E, respectively.
The sulfurtransferase activity of rhodanese proceeds via two half-reactions (Fig. 1, C–E). In the first, the sulfane sulfur is transferred from the substrate to the active site cysteine to form a persulfide intermediate. In the second half-reaction, a thiophilic acceptor attacks the enzyme-bound persulfide intermediate forming product and regenerating the resting form of the enzyme (7, 8). Historically, the biological importance of rhodanese was ascribed to cyanide detoxification via transfer of the sulfane sulfur from thiosulfate to cyanide to generate thiocyanate and sulfite (Fig. 1C). This reaction explains the rationale for utilizing thiosulfate as an antidote against cyanide poisoning. An alternate role for rhodanese in physiology is, however, indicated by the high Km for cyanide, which is toxic, and the fact that dietary sources of cyanide are largely restricted to certain seeds or plants that contain cyanogenic glycosides. Environmentally, elevated cyanide is associated with tobacco smoke (9) and pyrolysis of nitrile polymers or plastic.
More recently, the role of rhodanese in the mitochondrial sulfide oxidation pathway has been recognized, where it catalyzes the transfer of sulfane sulfur from glutathione persulfide (GSSH)3 to sulfite to form thiosulfate (Fig. 1D) (10, 11). It has also been proposed that the role of rhodanese or another thiol sulfurtransferase in the mitochondrial sulfide oxidation pathway is to utilize thiosulfate, forming GSSH instead (Fig. 1E) (12, 13). However, the efficiency of thiosulfate production by wild-type rhodanese is much greater than that for thiosulfate utilization (11). Kinetic data combined with simulations at physiologically relevant substrate concentrations have established that the first enzyme in the mitochondrial sulfide oxidation pathway, sulfide quinone oxidoreductase, predominantly catalyzes the synthesis of GSSH, which is a substrate for rhodanese (11). Rhodanese is abundant in liver and kidney (14) and also in colon, where it is important in the detoxification of H2S produced by sulfate-reducing microbiota (15).
The TST gene encoding human rhodanese is located on chromosome 22 (16), and two single nucleotide polymorphisms, c.306A→C and c.853C→G leading to amino acid substitutions, E102D and P285A, have been reported (17). The allelic frequencies for the single nucleotide polymorphisms in the French Caucasian population are 1% (E102D) and 5% (P285A), respectively. The variant residues are located on loops in the N- and C-terminal domains and are ∼19 and 17 Å away from the active site of rhodanese (Fig. 1, A and B). Kinetic analysis of the heterologously expressed human rhodanese variants in yeast revealed a 2-fold lower kcat/Km(thiosulfate) value for the P285A variant, whereas the E102D variant was indistinguishable from wild-type rhodanese in the thiosulfate:cyanide sulfurtransferase assay (17). However, possible differences in protein expression were not taken into account in this study. Glu-102 resides at the lip of a cavity leading to the active site cysteine and could, in principle, influence substrate access and binding (Fig. 1B). In contrast, the role of the distant and solvent-exposed Pro-285 on enzyme activity is more difficult to rationalize.
Changes in sulfide metabolism have been reported in ulceritive colitis (18), whereas changes in cyanide metabolism have been seen in motor neuron disease (19). The contribution, if any, of single nucleotide polymorphisms in rhodanese to disease susceptibility remains to be determined. For these reasons, we have evaluated the effects of the E102D and P285A polymorphic variants on protein stability and on the kinetic parameters in sulfurtransferase assays in the context of cyanide detoxification and the mitochondrial sulfide oxidation pathway. Our results suggest a significant increase in protein stability in the two polymorphic variants and differential effects on the catalytic efficiency of the sulfurtransferase activities. Like wild-type rhodanese, the preferred reaction catalyzed by the polymorphic variants is the production rather than consumption of thiosulfate. The E102D variant is significantly more efficient than wild-type or P285A rhodanese in converting cyanide to thiocyanate.
Experimental Procedures
Materials
Sodium sulfite, sodium thiosulfate, l-cysteine, dl-homocysteine, and GSH were purchased from Sigma-Aldrich. Monobromobimane (FluoroPure grade) was purchased from Life Technologies.
Expression and Purification of Rhodanese Polymorphic Variants
The polymorphic variants were generated by site-directed mutagenesis using a QuikChange kit (Stratagene) using as template the wild-type human rhodanese expression construct, which has been described previously (11). The following primers were used: E102D, 5′-GTTGTCTATGATGGCGACCATCTGGGTTCT-3′; and P285A, 5′-CGCCGTGCCCCGGCGGAATCCCGTGTCAGC-3′. The mutagenic codons are underlined, and the reverse sequences were utilized in the 3′ → 5′ direction. The rhodanese variants were expressed in Escherichia coli and purified as described previously (11) with the following modifications. The protein eluted from the nickel-nitrilotriacetic acid-agarose column, were concentrated to 15–20 ml and dialyzed for 3 h and then overnight against 4 liters of 50 mm Tris buffer, pH 7.0. The dialyzed protein was loaded on to a 20-ml SP Sepharose High Performance cation exchange column (GE Heathcare), pre-equilibrated with 50 mm Tris buffer, pH 7.0. The proteins were eluted with 200 ml of the equilibration buffer with a linear gradient ranging from 0 to 1 m KCl. Fractions containing rhodanese were identified by SDS-PAGE analysis, pooled, concentrated, and applied to a HiLoad 16/60 Superdex 200 gel filtration column (GE Healthcare). The column was eluted with 100 mm HEPES, pH 7.4, containing 150 mm NaCl. Rhodanese-containing fractions were pooled, concentrated, and stored at −80 °C until use. Protein concentration was determined using the Bradford reagent (Bio-Rad) with bovine serum albumin as a standard.
Thermal Denaturation Assays
Thermal denaturation was followed by protein turbidity measurement at 600 nm. Briefly, a cuvette containing 200 μl of 0.5 mg/ml wild-type, E102D, or P285A rhodanese in 100 mm HEPES, pH 7.4, containing 150 mm NaCl, was placed in a temperature-controlled cuvette holder in a Cary 100 Bio UV-visible spectrophotometer equipped with a temperature-controlled water bath. The temperature was increased in 5 °C increments from 20 to 65 °C, and the sample was allowed to equilibrate for 5 min before recording protein turbidity.
Rhodanese Activity Assays
The activity of rhodanese was measured in the following assays.
Thiosulfate:Cyanide Sulfurtransferase Assay
The ability to detoxify cyanide was assayed using the method described (20). The reaction mixture contained cyanide (1–300 mm) and thiosulfate (1–300 mm) in 0.25 ml of 300 mm HEPES buffer, pH 7.4, containing 150 mm NaCl at 25 °C. The reaction was initiated by the addition of 0.5 μg of wild-type rhodanese or 0.1 μg of E102D and P285A variants. The reactions were incubated at room temperature for 5 min and terminated by adding 0.25 ml of 15% (w/v) formaldehyde. The reaction mixture was centrifuged for 5 min at 10,000 × g to remove protein. To the supernatant, 0.5 ml of ferric nitrate solution was added. The reaction of thiocyanate with ferric ion produces ferric thiocyanate, which is measured colorimetrically at 460 nm. Control reactions containing the same concentrations of cyanide and thiosulfate used in each reaction, but lacking rhodanese, were run in parallel. A calibration curve was generated using standard thiocyanate samples ranging from 0.1–5 μmol. One unit of enzyme activity catalyzed the formation of 1 μmol of thiocyanate min−1 at 25 °C.
H2S Production in the Thiosulfate:Thiol Sulfurtransferase Reaction
Formation of H2S was measured in a turbidometric lead acetate assay as described previously (11). Briefly, the reaction mixture containing thiosulfate (0.1–20 mm) as sulfur donor and either 50 mm GSH or 50 mm l-cysteine or 50 mm l-homocysteine and 0.4 mm lead acetate in 300 mm HEPES, pH 7.4, and 150 mm NaCl in a final volume of 1 ml was preincubated at 37 °C for 4 min. The reaction was started by the addition of 10 μg of rhodanese. The Km values for GSH, l-cysteine, and l-homocysteine were determined in the presence of 3 mm sodium thiosulfate, and the Km for sodium thiosulfate was determined in the presence of 50 mm GSH, cysteine, or homocysteine. A molar extinction coefficient of 5,500 m−1 cm−1 at 390 nm was used to calculate the lead sulfide concentration.
GSSH:Sulfite Sulfurtransferase Assay
The rate of thiosulfate formation was determined in the presence of sulfite and GSSH using an HPLC assay as described (11). Briefly, the reaction mixture contained in a final volume of 200 μl, sulfite (0.1–1 mm), and GSSH (0.1–2 mm) and 1 μg of rhodanese in 100 mm HEPES, pH 7.4, and 150 mm NaCl. The reactions were incubated for 5 min at 25 °C. Monobromobimane was added to a final concentration of 1 mm and incubated in the dark for 10 min at room temperature. The samples were then acidified with 100 μl of 0.2 mm sodium citrate, pH 2.0. The derivatized samples were centrifuged at 10,000 × g for 10 min at 4 °C, and 50 μl of the supernatants were injected onto a C8 reverse phase HPLC column (4.6 × 150 cm, 3 μm packing; Phenomenex), pre-equilibrated with 80% solvent A (10% methanol and 2.5% acetic acid) and 20% solvent B (90% of methanol and 0.25% acetic acid). A control reaction lacking rhodanese was prepared in parallel. The bimane adducts were detected by fluorescence excitation at 340 nm and emission at 450 nm. The concentration of GSSH was fixed at 2 mm to determine the Km for sulfite. The concentration of sulfite was fixed at 1 mm to determine the Km for GSSH.
Estimation of Rhodanese Levels in Murine Liver
Livers from 7–10-week-old male Balb/c mice (The Jackson Laboratory, Bar Harbor, ME) were homogenized in lysis buffer (100 mm HEPES, pH 7.4, 25 mm KCl, 0.5% Nonidet P40, 20 μg/ml phenylmethylsulfonyl fluoride, 25 μg/ml tosyl-l-lysyl-chloromethane hydrochloride, 1% protease inhibitor mixture (Sigma)). The homogenate was incubated on ice for 10 min and centrifuged at 13,000 × g for 10 min at 4 οC. The supernatant was collected, and the protein concentration was determined using the Bradford assay. Varying amounts of tissue homogenate along with known amounts of purified recombinant human rhodanese were separated by 12% SDS-PAGE and transferred to polyvinylidene fluoride membranes. Monoclonal anti-rhodanese antibody (Proteintech Group Inc.; 1:40,000 dilution) was used for detection, and signals were quantified using ImageJ software. The amount of rhodanese in the liver samples was quantified using a calibration curve generated using purified recombinant rhodanese.
Thiosulfate-dependent H2S Production by Murine Liver Lysate
Production of persulfide products by rhodanese in the thiosulfate:thiol sulfurtransferase reaction, leads to H2S production. Hence, we assessed thiosulfate-dependent H2S generation in liver tissue by a gas chromatography-based method described previously (21). Briefly, the reactions were performed at 37 οC in 25-ml polypropylene syringes with a three-way stopcock in HEPES buffer, pH 7.4, containing 10 mm GSH and 1 mm thiosulfate in a final volume of 0.5 ml. Control reactions in which liver lysate was omitted were run in parallel. The samples were made anaerobic by flushing the syringe with nitrogen and then sealed using a small sleeve stopper septum. After 20 min, 0.2-ml aliquots were collected from the headspace using a gas tight syringe and injected into an HP 6890 gas chromatography column. The concentration of H2S were estimated using a calibration curve as described previously (21). Specific activity is expressed as nmol of H2S formed per gram of tissue min−1 at 37 °C.
Results
Purification and Stability of Rhodanese
Wild-type and variant human rhodanese were expressed in E. coli and purified as described previously (11). E102D and P285A rhodanese were obtained in >90% purity as assessed by SDS-PAGE analysis (Fig. 2A). Based on size exclusion chromatography, the molecular mass of the two variants was estimated to be ∼33 kDa, consistent with them being monomers in solution like wild-type rhodanese.
FIGURE 2.
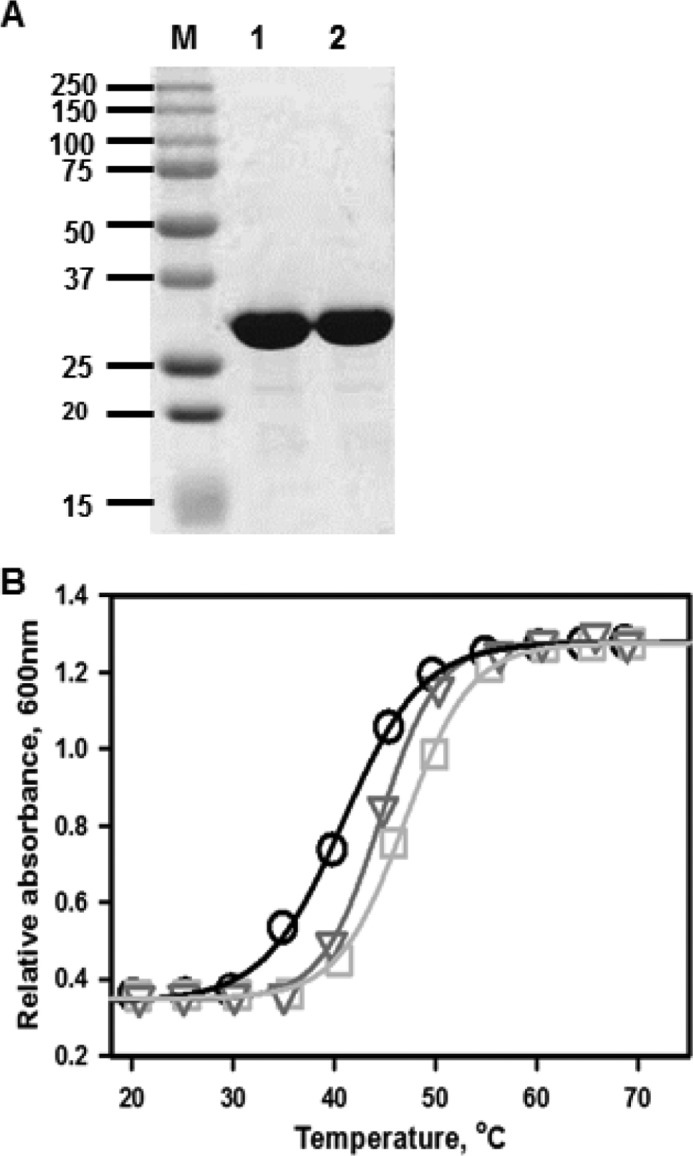
Purity and stabilities of the rhodanese variants. A, SDS-PAGE analysis of E102D (lane 1) and P285A (lane 2) variants of human rhodanese. The molecular mass markers are shown in the lane labeled M. B, thermal melting curves for wild-type rhodanese (circles), E102D (squares), and P285A (triangles) were monitored by turbidometric analysis at 600 nm of protein solutions (0.5 mg/ml) in 100 mm HEPES, pH 7.4, containing 150 mm NaCl buffer as described under “Experimental Procedures.” The data are representative of three independent experiments.
During purification, the E102D and P285A variants were observed to be more stable as evidenced by lower precipitation during protein concentration, which resulted in higher yields. Thermal denaturation profiles confirmed that the E102D and P285A variants are indeed more stable with Tm values of 47.3 ± 0.2 °C (E102D) and 44.4 ± 0.3 °C (P285A) compared with 40.8 ± 0.2 °C for wild-type rhodanese (Fig. 2B).
Thiosulfate:Cyanide Sulfurtransferase Activity of Rhodanese Variants
The activity of the rhodanese variants in sulfur transfer from thiosulfate to cyanide (Fig. 1C) was compared with wild-type enzyme. The specific activity of wild-type human rhodanese in the presence of cyanide and thiosulfate was 1636 ± 44 μmol min−1 mg−1 protein at 25 °C. The Km values for cyanide and thiosulfate were 29 ± 4 and 39.5 ± 2.5 mm, respectively (Fig. 3 and Table 1). By comparison, the E102D and P285A variants exhibited 4- and 2.7-fold higher specific activities. The Km for cyanide was lower for the E102D variant (7.0 ± 0.5 mm) but higher for the P285A (51 ± 3 mm) variant. The Km for thiosulfate was higher for E102D (88.7 ± 11.6 mm) but lower for P285A (24 ± 3 mm) rhodanese compared with wild-type enzyme (Table 1). Overall, the catalytic efficiency (kcat/Km(CN)) in the cyanide detoxification assay was 17- and 1.6-fold higher for the E102D and P285A variants, respectively, compared with wild-type rhodanese.
FIGURE 3.

Kinetic analysis of the thiosulfate:cyanide sulfurtransferase reaction catalyzed by rhodanese and its polymorphic variants. A, C, and E, dependence of the reaction velocity on cyanide concentration in the presence of 100 mm thiosulfate (A, wild type; C, E102D; and E, P285A rhodanese). B, D, and F, dependence of the reaction velocity on thiosulfate concentration in the presence of saturating (100 mm) cyanide (B, wild type; D, E102D; and F, P285A rhodanese). The reaction conditions are described under “Experimental Procedures,” and the data are representative of three independent experiments.
TABLE 1.
Comparison of kinetic parameters for the cyanide detoxification activity of the rhodanese variants
| Rhodanese | Km(TS) | Km(KCN) | Vmaxa | kcat | kcat/Km(TS) | kcat/Km(KCN) |
|---|---|---|---|---|---|---|
| mm | mm | μmol min−1 mg−1 | s−1 | m−1 s−1 | m−1 s−1 | |
| Wild-type | 39.5 ± 2.5 | 29 ± 4 | 1636 ± 44 | 910 | 23 × 103 | 31 × 103 |
| E102D | 88.7 ± 11.6 | 7.0 ± 0.5 | 6762 ± 59 | 3736 | 42 × 103 | 534 × 103 |
| P285A | 24 ± 3 | 51 ± 3 | 4412 ± 81 | 2437 | 101 × 103 | 48 × 103 |
a Vmax was estimated from graphs in which the concentration of cyanide was varied. The Vmax estimates obtained from graphs in which the concentration of thiosulfate was varied are: wild-type (1846 ± 49 μmol min−1 mg−1, kcat = 1020 s−1), E102D (7287 ± 59 μmol min−1 mg−1, kcat = 4026 s−1), and P285A (4204 ± 92 μmol min−1 mg−1, kcat = 2322 s−1).
GSSH:Sulfite Sulfurtransferase Activity of Rhodanese
The activity of the polymorphic variants was compared with wild-type rhodanese in the sulfur transfer reaction from GSSH to sulfite (Fig. 1D) (11). The Km for sulfite was 2.8- (E102D) and 4.7-fold (P285A) higher than for wild-type rhodanese, whereas the Km for GSSH was 1.8-fold higher for the E102D variant but comparable to that for wild-type enzyme for the P285A variant (Fig. 4 and Table 2).
FIGURE 4.

Kinetic analysis of the GSSH:sulfite sulfurtransferase reaction catalyzed by the rhodanese variants. A and C, dependence of the reaction velocity for sulfur transfer on the concentration of GSSH in the presence of 1 mm sulfite for E102D (A) and P285A (C) rhodanese. B and D, dependence of the reaction velocity on sulfite concentration in the presence of 2 mm GSSH for E102D (B) and P285A (D) rhodanese. The reaction conditions are described under “Experimental Procedures,” and the data are representative of three independent experiments.
TABLE 2.
Comparison of kinetic parameters for the sulfur transferase activities of rhodanese variants
| Rhodanese | Donor | Acc | Km,Donor | Km,Acca | Vmaxb | kcat | n | kcat/Km,Donor | kcat/Km,Acc |
|---|---|---|---|---|---|---|---|---|---|
| mm | mm | μmol min−1 mg−1 | s−1 | m−1 s−1 | m−1 s−1 | ||||
| Wild-typec | GSSH | SO32− | 0.450 ± 0.004 | 0.06 ± 0.01 | 609 ± 25 | 389 | 0.86 × 106 | 6.5 × 106 | |
| S2O32− | Cys | 0.35 ± 0.06 | 20.0 ± 0.5 | 13.0 ± 0.3 | 7.4 | 2.3 ± 0.1 | 21 × 103 | 0.37 × 103 | |
| S2O32− | Hcy | 0.3 ± 0.1 | 20.5 ± 1.9 | 15.5 ± 0.9 | 8.7 | 2.0 ± 0.1 | 29 × 103 | 0.4 × 103 | |
| S2O32− | GSH | 0.34 ± 0.05 | 21.0 ± 0.4 | 1.2 ± 0.02 | 0.67 | 2.3 ± 0.2 | 2.0 × 103 | 0.03 × 103 | |
| E102D | GSSH | SO32− | 0.81 ± 0.07 | 0.17 ± 0.02 | 1302 ± 62 | 719 | 0.88 × 106 | 4.2 × 106 | |
| S2O32− | Cys | 0.50 ± 0.08 | 16.0 ± 0.7 | 11.3 ± 0.3 | 6.3 | 2.1 ± 0.1 | 13 × 103 | 0.4 × 103 | |
| S2O32− | Hcy | 0.46 ± 0.04 | 19 ± 1 | 13.1 ± 0.5 | 7.3 | 1.8 ± 0.1 | 16 × 103 | 0.4 × 103 | |
| S2O32− | GSH | 0.56 ± 0.06 | 18.0 ± 0.8 | 1.10 ± 0.03 | 0.61 | 2.1 ± 0.2 | 1.1 × 103 | 0.03 × 103 | |
| P285A | GSSH | SO32− | 0.43 ± 0.04 | 0.28 ± 0.03 | 746 ± 35 | 412 | 0.96 × 106 | 1.5 × 106 | |
| S2O32− | Cys | 0.5 ± 0.2 | 19 ± 1 | 11.2 ± 0.4 | 6.2 | 1.9 ± 0.1 | 12 × 103 | 0.3 × 103 | |
| S2O32− | Hcy | 0.46 ± 0.03 | 20 ± 2 | 13.3 ± 0.9 | 7.4 | 2.2 ± 0.1 | 16 × 103 | 0.37 × 103 | |
| S2O32− | GSH | 0.52 ± 0.06 | 15.0 ± 0.8 | 0.93 ± 0.03 | 0.51 | 2.3 ± 0.2 | 1 × 103 | 0.03 × 103 |
a Km,Acc denotes the Km for the sulfane sulfur acceptor.
b Vmax was estimated from data sets in which the acceptor concentration was varied. Hcy is homocysteine.
c The values for wild-type rhodanese are from Ref. 11, and n is the Hill coefficient. S2O32− and SO32− denote thiosulfate and sulfite, respectively.
Thiosulfate:Thiol Sulfurtransferase Activity of Rhodanese
Next, we compared the efficiency of the rhodanese variants in utilizing thiosulfate as a sulfur donor (Fig. 1E) in the presence of the thiol acceptors, glutathione, l-cysteine, and l-homocysteine (Figs. 5 and 6). Like wild-type rhodanese, the variants exhibited marked substrate inhibition at concentrations >2–3 mm thiosulfate (Fig. 6). For this reason, the dependence of the reaction velocity at varying thiol concentrations was assessed at 3 mm thiosulfate. Overall, the kinetic parameters for the two variants were very similar and comparable to that for wild-type rhodanese (Table 2). Because of the high Km values for cysteine and homocysteine compared with their cellular concentrations (11), these thiols are unlikely to be significant acceptors in the rhodanese reaction. On the other hand, intracellular glutathione concentrations can vary from 1 to 10 mm depending on the cell type, and therefore, it is likely to be a physiologically relevant acceptor (11). Based on a comparison of the kcat/Km(sulfite) versus kcat/Km(GSH) values, the variants exhibited a 140,000-fold (E102D) and 50,000-fold (P285A) preference for sulfite versus glutathione as an acceptor (Table 2). Hence, like wild-type rhodanese, the variants show a marked preference for thiosulfate synthesis from GSSH and sulfite.
FIGURE 5.

Kinetic analysis of the thiosulfate:thiol sulfurtransferase reactions catalyzed by the rhodanese polymorphic variants. A–F, dependence of the reaction velocity of sulfur transfer from 3 mm thiosulfate on varying concentrations of GSH (A and D), cysteine (B and E), or homocysteine (C and F) for E102D (A, C, and E) and for P285A (B, D, and F) rhodanese. The reaction conditions are described under “Experimental Procedures,” and the data are representative of three independent experiments.
FIGURE 6.

Dependence of the thiosulfate:thiol sulfurtransferase reactions catalyzed by the rhodanese polymorphic variants on thiosulfate concentration. A–F, the kinetics of sulfur transfer from varying concentrations of thiosulfate in the presence of 50 mm GSH (A and B), 50 mm l-cysteine (C and D), or 50 mm l-homocysteine (E and F) for E102D (A, C, and E) and P285A (B, D, and F) rhodanese. The reaction conditions are described under “Experimental Procedures.” The data are representative of three independent experiments.
Thiosulfate-dependent H2S Production by Liver Lysate
Based on semiquantitative Western blot analysis, the amount of rhodanese in murine liver was estimated to be ∼1.8 ± 0.3 μg mg−1 protein (Fig. 7). By comparison, our estimates of the transsulfuration enzymes in murine liver were 0.13 ± 0.03 μg mg−1 protein for cystathionine β-synthase and 8.0 ± 0.4 μg mg−1 protein for γ-cystathionase (22). The capacity for thiosulfate-dependent H2S production by liver homogenate in the presence of 1 mm thiosulfate and 10 mm GSH was estimated to be ∼10.7 ± 1.3 nmol of H2S formed min−1 g tissue−1 at 37 °C and pH 7.4. By comparison, the capacity for (cysteine + homocysteine)-dependent H2S production by murine liver, representing the combined contributions of cystathionine β-synthase and γ-cystathionase, was reported to be 1.2 μmol min−1 g tissue−1 (22), which is >100-fold higher than H2S production from thiosulfate in the presence of GSH.
FIGURE 7.
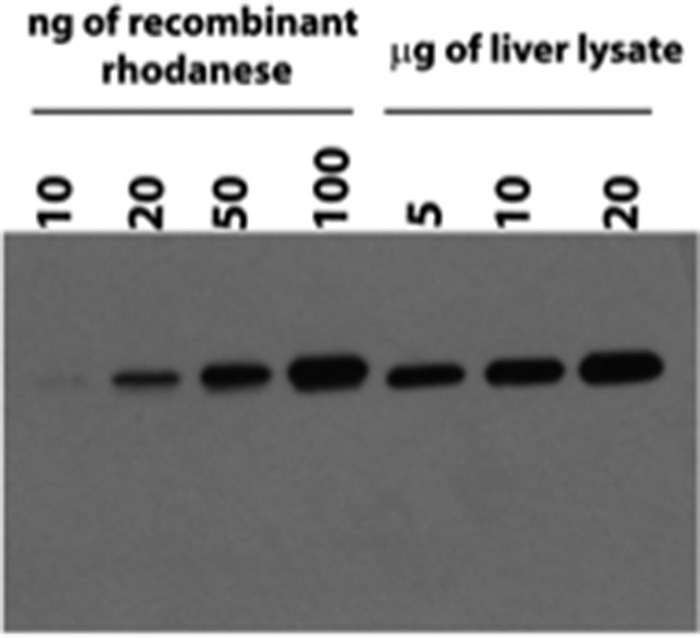
Semiquantitative Western blot analysis of rhodanese in murine liver tissue. Shown is representative Western blot analysis of purified recombinant human rhodanese compared with rhodanese present in murine liver lysate. The lanes contained the indicated amounts of purified human rhodanese or liver extract.
Discussion
Over the years, many functions have been ascribed to rhodanese including roles in iron-sulfur cluster biogenesis (23), modulation of mitochondrial respiratory activity (24), and cyanide detoxification (7). In E. coli, rhodanese is subject to catabolite repression and, based on expression studies, was concluded to play a role in aerobic energy metabolism (25). More recently, a role for rhodanese in the mitochondrial sulfide oxidation pathway has been established where it catalyzes the conversion of GSSH and sulfite to thiosulfate (10, 11). H2S levels are high in the lumen of the colon, where it is a product of commensal sulfate reducing bacteria (26). Colonic rhodanese levels and activity are down-regulated in colitis (27), and dysregulation of H2S homeostasis at the host-microbe interface could be important in the etiology of inflammatory bowel disease and colorectal cancer (18, 27–30). Identification of single nucleotide polymorphisms correlated with disease resistance or susceptibility is an area of intense current interest. In light of the possible correlations between rhodanese activity and disease susceptibility, it is important to understand possible differences in the stability and kinetics of the rhodanese polymorphic variants (17).
The polymorphic variations in rhodanese are located at surface-exposed residues, and both are correlated with significantly higher thermal stability compared with the wild-type protein (Fig. 2B). Rhodanese is a monomeric enzyme with a highly hydrophobic interdomain interface. It has served as an important model for studying bacterial GroEL-GroES chaperone-mediated folding of bovine rhodanese (31, 32). However, the mechanism of refolding human rhodanese following its relocation into the mitochondrion is not known. In addition to its role in sulfide oxidation, rhodanese is important in mammals for the mitochondrial import of 5S rRNA where the latter supports translation (33). The association between 5S rRNA and rhodanese occurs co-translationally and is followed by their concerted localization to the mitochondrion. The possible influence of melting temperatures that vary over an ∼6 °C range in the polymorphic variants, on mitochondrial import of rhodanese, and on 5S rRNA binding is not known.
The E102D variation resides at the entrance to the active site (Fig. 1B), and the presence of a shorter aspartate versus glutamate residue increases the Km for thiosulfate and decreases the Km for cyanide while increasing kcat ∼4-fold in the thiosulfate:cyanide sulfurtransferase assay (Table 1). The P285A variation on the other hand is quite remote from the active site, but it too increases kcat (∼2.7-fold). It exhibits an increased Km value for cyanide and decreased Km for thiosulfate compared with wild-type rhodanese. Because the E102D variant is more efficient than wild-type or P285A rhodanese in catalyzing the thiosulfate:cyanide sulfur transfer reaction, individuals with the E102D variation might be at an advantage to detoxify cyanide, which is elevated in smokers and in motor neuron disease (19).
Surprisingly, when thiosulfate is used as a sulfane sulfur donor in combination with thiols versus cyanide as acceptor, a change in the kinetic behavior of the enzyme is seen. Thus, the dependence of the reaction velocity on thiosulfate concentration changes from simple Michaelis-Menten behavior (Fig. 3) to behavior exhibiting marked substrate inhibition (Fig. 6). Inhibition by thiosulfate of the human thioltransferase, TSTD1, and of human rhodanese have been reported, albeit at much higher concentrations and at alkaline pH values (13, 34).
A second difference in the kinetic behavior of rhodanese is observed when the concentration of cyanide versus thiols is varied. In the presence of cyanide as an acceptor, the reaction exhibits hyperbolic behavior (Fig. 3). In contrast, in the presence of thiols, sigmoidal kinetics are observed (Fig. 5). Because thiosulfate is the sulfur donor in both sets of reactions, the molecular mechanism for the observed difference in kinetic behavior is unclear. The sigmoidal kinetics is accompanied by a decrease in the thiosulfate Km value by almost two orders of magnitude in the presence of thiols versus cyanide. It is possible that the noncatalytic N-terminal domain of rhodanese houses an allosteric site for thiols explaining both the sigmoidal kinetics and the influence on the thiosulfate Km. An earlier steady-state kinetic analysis of the thiosulfate:cyanide sulfurtransferase reaction catalyzed by human rhodanese yielded a set of intersecting lines, suggesting a ping-pong mechanism (34). In contrast, the initial velocity data for human TSTD1 in the thiosulfate:glutathione sulfurtransferase reaction yielded intersecting lines consistent with a ternary mechanism (13). It is possible that the kinetic mechanism of human rhodanese is influenced by the nature of the sulfur acceptor, i.e. thiols versus cyanide (13, 35).
Our kinetic analyses reveal that the polymorphic variants like wild-type rhodanese catalyze sulfur transfer from GSSH (Fig. 1D) much more efficiently than from thiosulfate (Fig. 1E). This reaction preference is consistent with the role of rhodanese in generating thiosulfate in the mitochondrial sulfide oxidation pathway (Fig. 8A) (10, 11). In this scheme, GSSH is produced in the first step of the sulfide oxidation pathway by sulfide quinone oxidoreductase and is a substrate for both the persulfide dioxygenase and for rhodanese. The kcat/KMGSSH for rhodanese (∼0.9 × 106 m−1 s−1 at pH 7.4 and 25 °C) is ∼2-fold higher than for human persulfide dioxygenase (0.4 × 106 m−1 s−1 at pH 7.4 and 22 °C) (36). Competition for GSSH between the two enzymes would be influenced by their relative abundance, as well as the concentrations of their co-substrates, namely O2 (for the dioxygenase) and sulfite (for rhodanese). Furthermore, interaction between the persulfide dioxygenase and rhodanese could increase the effective concentration of sulfite, allowing this reactive intermediate to channel between the two active sites. Natural fusions of rhodanese and persulfide dioxygenase are found in bacteria, suggesting that a functional interaction between them might exist in other organisms (13, 35, 37).
FIGURE 8.
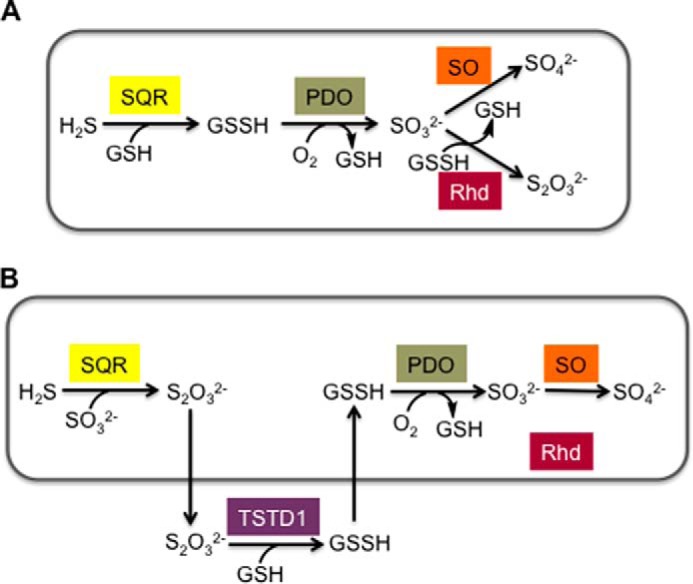
Alternative models of the mitochondrial sulfide oxidation pathway. A, scheme showing the organization of the mitochondrial sulfide oxidation pathway and the role of rhodanese in thiosulfate generation. For simplicity, only some of the co-substrates and products are shown in the scheme. B, an alternative model proposed for the organization of the mitochondrial sulfide oxidation pathway in which the conversion of thiosulfate to GSSH occurs outside the mitochondrion and is catalyzed by TSTD1. No role for the mitochondrially localized rhodanese is proposed in this model. SQR, sulfide quinone oxidoreductase; PDO, persulfide dioxygenase; SO, sulfite oxidase; Rhd, rhodanese. The boxes in A and B denote the mitochondrion.
An alternative model for the mitochondrial sulfide oxidation pathway has been proposed (12) in which the primary role of rhodanese is to utilize thiosulfate (Fig. 8B). There are several significant differences between the two models. Thiosulfate rather than GSSH is predicted to be the physiological product of the first step catalyzed by sulfide quinone oxidoreductase. Hence, to generate GSSH, required for sulfite production by persulfide dioxygenase, an additional step catalyzed by TSTD1, a single rhodanese domain thioltransferase was invoked (13). TSTD1, which is cytoplasmic in location, was shown to convert thiosulfate to GSSH, at the alkaline pH of 9.0, at which the reaction kinetics were characterized (13). The drawback of this model is that it necessitates the intercompartmental crossing of the highly reactive GSSH intermediate for operation of the sulfide oxidation pathway and does not address the role of rhodanese in it. The kinetics of the GSSH:sulfite sulfur transfer reaction by TSTD1 were not examined (13).
Our results are consistent with a role for rhodanese in generating thiosulfate, which is the major product of sulfide oxidation in colon where rhodanese levels are high. Compared with wild-type rhodanese, the P285A variant shows a 4-fold lower catalytic efficiency (kcat/Km,sulfite), whereas the E102D variant is only marginally less efficient (Table 2). The differences in catalytic efficiency stem from multiple sources because both Km for sulfite and kcat are affected by the polymorphic variations. Based on the comparison of their kcat/Km,sulfite values, individuals with the P285A variation might be expected to be more susceptible to disease under conditions of high H2S flux, for instance in the colon.
Interestingly, the kcat/Km,sulfite for rhodanese (1.5–6.5 × 106 m−1 s−1 at pH 7.4 and 25 °C) is comparable to the value reported for human sulfite oxidase (2.4 × 106 m−1 s−1 at pH 8.0 and 25 °C) (38). However, unlike rhodanese, sulfite oxidase is present in the mitochondrial intermembrane space (39). The relative amounts of rhodanese versus sulfite oxidase, the concentrations of their co-substrates (O2 for sulfite oxidase and GSSH for rhodanese), and the efficiency of sulfite transport into the intermembrane space likely govern utilization of sulfite by these two enzymes.
Finally, our assay used to monitor sulfur transfer from thiosulfate to thiols followed H2S production from the resulting persulfide product (Reactions 1 and 2), raising the possibility that rhodanese might be a source of H2S in vivo. If this reaction were to occur at a significant rate under physiological conditions, rhodanese could contribute to H2S formation rather than to its clearance, which is another argument against TST-dependent thiosulfate utilization as shown in Fig. 8B.
 |
To assess the production of H2S from thiosulfate in liver,the amount of rhodanese in murine liver was first determined. The amount of rhodanese is ∼14-fold higher than of cystathionine β-synthase and 4-fold lower than of γ-cystathionase (22), the transsulfuration pathway enzymes that generate H2S (40, 41). The capacity of rhodanese in murine liver lysate to produce H2S from thiosulfate in the presence of GSH was ∼1% of the capacity of cystathionine β-synthase and γ-cystathionase to produce H2S from cysteine and homocysteine, consistent with the primary role of rhodanese in H2S clearance (Fig. 8A) rather than H2S production.
Conclusions
In summary, we have shown that the polymorphic variants of rhodanese described in the French population influence the thermal stability and kinetic properties of the enzyme despite their distal locations from the active site. The conservative E102D variant is located in the noncatalytic N-terminal domain at the entrance to the active site, and the difference in a single methylene group in a protein with a molecular mass of 33 kDa leads to a 6.5 °C increase in thermal stability, an ∼2-fold increase in kcat for the physiologically relevant GSSH:sulfite sulfur transfer reaction, and an ∼4-fold increase in kcat in the thiosulfate:cyanide sulfur transfer reaction. The nonconservative P285A variation is located on a flexible loop in the catalytic C-terminal domain, and this substitution also has pleotropic effects, albeit primarily in the thiosulfate:cyanide sulfur transfer reaction (2.7-fold increase in kcat). In view of the kinetic and stability differences, correlations, if any, between these polymorphic variations and rhodanese-related disease susceptibility will be interesting to examine. Our results also demonstrate that thiosulfate-dependent H2S production by TSTs is insignificant in murine liver lysate, consistent with the role of rhodanese in H2S clearance.
Author Contributions
M. L. and A. S. designed, performed, and analyzed the experiments and wrote parts of the manuscript. R. B. helped conceive the experiments, analyzed the data, and co-wrote the manuscript. All authors approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM112455 (to R. B.). The authors declare that they have no conflicts of interest with the contents of this article.
- GSSH
- glutathione persulfide.
References
- 1. Bordo D., Bork P. (2002) The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 3, 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cipollone R., Ascenzi P., Visca P. (2007) Common themes and variations in the rhodanese superfamily. IUBMB Life 59, 51–59 [DOI] [PubMed] [Google Scholar]
- 3. Sörbo B. H. (1955) Rhodanese. Methods Enzymol. 2, 334–337 [Google Scholar]
- 4. Ploegman J. H., Drent G., Kalk K. H., Hol W. G. (1978) Structure of bovine liver rhodanese: I. structure determination at 2.5 A resolution and a comparison of the conformation and sequence of its two domains. J. Mol. Biol. 123, 557–594 [PubMed] [Google Scholar]
- 5. Trevino R. J., Gliubich F., Berni R., Cianci M., Chirgwin J. M., Zanotti G., Horowitz P. M. (1999) NH2-terminal sequence truncation decreases the stability of bovine rhodanese, minimally perturbs its crystal structure, and enhances interaction with GroEL under native conditions. J. Biol. Chem. 274, 13938–13947 [DOI] [PubMed] [Google Scholar]
- 6. Ploegman J. H., Drent G., Kalk K. H., Hol W. G., Heinrikson R. L., Keim P., Weng L., Russell J. (1978) The covalent and tertiary structure of bovine liver rhodanese. Nature 273, 124–129 [DOI] [PubMed] [Google Scholar]
- 7. Westley J., Adler H., Westley L., Nishida C. (1983) The sulfurtransferases. Fundam. Appl. Toxicol. 3, 377–382 [DOI] [PubMed] [Google Scholar]
- 8. Schlesinger P., Westley J. (1974) An expanded mechanism for rhodanese catalysis. J. Biol. Chem. 249, 780–788 [PubMed] [Google Scholar]
- 9. Tsuge K., Kataoka M., Seto Y. (2000) Cyanide and thiocyanate levels in blood and saliva of healthy adult volunteers. J. Health Sci. 46, 343–350 [Google Scholar]
- 10. Hildebrandt T. M., Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS. J 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 11. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., Banerjee R. (2014) Organization of the human mitochondrial H2S oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jackson M. R., Melideo S. L., Jorns M. S. (2012) Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 [DOI] [PubMed] [Google Scholar]
- 13. Melideo S. L., Jackson M. R., Jorns M. S. (2014) Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry 53, 4739–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aminlari M., Gilanpour H. (1991) Comparative studies on the distribution of rhodanese in different tissues of domestic animals. Comp. Biochem. Physiol. B 99, 673–677 [DOI] [PubMed] [Google Scholar]
- 15. Furne J., Springfield J., Koenig T., DeMaster E., Levitt M. D. (2001) Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: a specialized function of the colonic mucosa. Biochem. Pharmacol. 62, 255–259 [DOI] [PubMed] [Google Scholar]
- 16. Aita N., Ishii K., Akamatsu Y., Ogasawara Y., Tanabe S. (1997) Cloning and expression of human liver rhodanese cDNA. Biochem. Biophys. Res. Commun. 231, 56–60 [DOI] [PubMed] [Google Scholar]
- 17. Billaut-Laden I., Allorge D., Crunelle-Thibaut A., Rat E., Cauffiez C., Chevalier D., Houdret N., Lo-Guidice J. M., Broly F. (2006) Evidence for a functional genetic polymorphism of the human thiosulfate sulfurtransferase (Rhodanese), a cyanide and H2S detoxification enzyme. Toxicology 225, 1–11 [DOI] [PubMed] [Google Scholar]
- 18. Levine J., Ellis C. J., Furne J. K., Springfield J., Levitt M. D. (1998) Fecal hydrogen sulfide production in ulcerative colitis. Am. J. Gastroenterol. 93, 83–87 [DOI] [PubMed] [Google Scholar]
- 19. Kato T., Kameyama M., Nakamura S., Inada M., Sugiyama H. (1985) Cyanide metabolism in motor neuron disease. Acta. Neurol. Scand. 72, 151–156 [DOI] [PubMed] [Google Scholar]
- 20. Banerjee R., Chiku T., Kabil O., Libiad M., Motl N., Yadav P. K. (2015) Assay methods for H2S biogenesis and catabolism enzymes. Methods Enzymol. 554, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vitvitsky V., Banerjee R. (2015) H2S analysis in biological samples using gas chromatography with sulfur chemiluminescence detection. Methods Enzymol. 554, 111–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabil O., Vitvitsky V., Xie P., Banerjee R. (2011) The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 15, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ogata K., Volini M. (1990) Mitochondrial rhodanese: membrane-bound and complexed activity. J. Biol. Chem. 265, 8087–8093 [PubMed] [Google Scholar]
- 24. Ogata K., Dai X., Volini M. (1989) Bovine mitochondrial rhodanese is a phosphoprotein. J. Biol. Chem. 264, 2718–2725 [PubMed] [Google Scholar]
- 25. Alexander K., Volini M. (1987) Properties of an Escherichia coli rhodanese. J. Biol. Chem. 262, 6595–6604 [PubMed] [Google Scholar]
- 26. Suarez F., Furne J., Springfield J., Levitt M. (1998) Production and elimination of sulfur-containing gases in the rat colon. Am. J. Physiol. 274, G727–G733 [DOI] [PubMed] [Google Scholar]
- 27. Taniguchi E., Matsunami M., Kimura T., Yonezawa D., Ishiki T., Sekiguchi F., Nishikawa H., Maeda Y., Ishikura H., Kawabata A. (2009) Rhodanese, but not cystathionine-γ-lyase, is associated with dextran sulfate sodium-evoked colitis in mice: a sign of impaired colonic sulfide detoxification? Toxicology 264, 96–103 [DOI] [PubMed] [Google Scholar]
- 28. Picton R., Eggo M. C., Merrill G. A., Langman M. J., Singh S. (2002) Mucosal protection against sulphide: importance of the enzyme rhodanese. Gut 50, 201–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levitt M. D., Furne J., Springfield J., Suarez F., DeMaster E. (1999) Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J. Clin. Invest. 104, 1107–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramasamy S., Singh S., Taniere P., Langman M. J., Eggo M. C. (2006) Sulfide-detoxifying enzymes in the human colon are decreased in cancer and upregulated in differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G288–G296 [DOI] [PubMed] [Google Scholar]
- 31. Bhattacharyya A. M., Horowitz P. M. (2002) Rhodanese can partially refold in its GroEL-GroES-ADP complex and can be released to give a homogeneous product. Biochemistry 41, 2421–2428 [DOI] [PubMed] [Google Scholar]
- 32. Bhattacharyya A. M., Horowitz P. M. (2002) Isolation and characterization of rhodanese intermediates during thermal inactivation and their implications for the mechanism of protein aggregation. Biochemistry 41, 422–429 [DOI] [PubMed] [Google Scholar]
- 33. Smirnov A., Comte C., Mager-Heckel A. M., Addis V., Krasheninnikov I. A., Martin R. P., Entelis N., Tarassov I. (2010) Mitochondrial enzyme rhodanese is essential for 5 S ribosomal RNA import into human mitochondria. J. Biol. Chem. 285, 30792–30803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jarabak R., Westley J. (1974) Human liver rhodanese: nonlinear kinetic behavior in a double displacement mechanism. Biochemistry 13, 3233–3236 [DOI] [PubMed] [Google Scholar]
- 35. Luebke J. L., Shen J., Bruce K. E., Kehl-Fie T. E., Peng H., Skaar E. P., Giedroc D. P. (2014) The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol. Microbiol. 94, 1343–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kabil O., Banerjee R. (2012) Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J. Biol. Chem. 287, 44561–44567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen J., Keithly M. E., Armstrong R. N., Higgins K. A., Edmonds K. A., Giedroc D. P. (2015) Staphylococcus aureus CstB is a novel multidomain persulfide dioxygenase-sulfurtransferase involved in hydrogen sulfide detoxification. Biochemistry 54, 4542–4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson-Winters K., Nordstrom A. R., Emesh S., Astashkin A. V., Rajapakshe A., Berry R. E., Tollin G., Enemark J. H. (2010) Effects of interdomain tether length and flexibility on the kinetics of intramolecular electron transfer in human sulfite oxidase. Biochemistry 49, 1290–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cohen H. J., Betcher-Lange S., Kessler D. L., Rajagopalan K. V. (1972) Hepatic sulfite oxidase: congruency in mitochondria of prosthetic groups and activity. J. Biol. Chem. 247, 7759–7766 [PubMed] [Google Scholar]
- 40. Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., Banerjee R. (2009) H2S biogenesis by cystathionine γ-lyase leads to the novel sulfur metabolites, lanthionine and homolanthionine, and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 284, 11601–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. (2009) Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]