

Background: Ca2+ binding and/or permeation via CaV1.1 in skeletal muscle activates CaMKII.
Results: Mice with a Ca2+ binding/permeation defect in CaV1.1 have increased body fat, reduced fatty acid metabolism, and altered CD36 distribution.
Conclusion: CaV1.1 regulates CD36 distribution and fatty acid metabolism.
Significance: New therapeutic targets are identified to increase skeletal muscle energy expenditure.
Keywords: Ca2+/calmodulin-dependent protein kinase II (CaMKII), calcium channel, metabolism, mitochondria, nitric-oxide synthase, S-nitrosylation, skeletal muscle, CD36, mitochondrial beta oxidation, Ca2+ channel, CaV1.1, CaM kinase II, metabolic rate
Abstract
Ca2+ permeation and/or binding to the skeletal muscle L-type Ca2+ channel (CaV1.1) facilitates activation of Ca2+/calmodulin kinase type II (CaMKII) and Ca2+ store refilling to reduce muscle fatigue and atrophy (Lee, C. S., Dagnino-Acosta, A., Yarotskyy, V., Hanna, A., Lyfenko, A., Knoblauch, M., Georgiou, D. K., Poché, R. A., Swank, M. W., Long, C., Ismailov, I. I., Lanner, J., Tran, T., Dong, K., Rodney, G. G., Dickinson, M. E., Beeton, C., Zhang, P., Dirksen, R. T., and Hamilton, S. L. (2015) Skelet. Muscle 5, 4). Mice with a mutation (E1014K) in the Cacna1s (α1 subunit of CaV1.1) gene that abolishes Ca2+ binding within the CaV1.1 pore gain more body weight and fat on a chow diet than control mice, without changes in food intake or activity, suggesting that CaV1.1-mediated CaMKII activation impacts muscle energy expenditure. We delineate a pathway (Cav1.1→ CaMKII→ NOS) in normal skeletal muscle that regulates the intracellular distribution of the fatty acid transport protein, CD36, altering fatty acid metabolism. The consequences of blocking this pathway are decreased mitochondrial β-oxidation and decreased energy expenditure. This study delineates a previously uncharacterized CaV1.1-mediated pathway that regulates energy utilization in skeletal muscle.
Introduction
Excess body fat, in particular, abdominal/visceral obesity, increases the risk of a wide variety of human diseases, including type 2 diabetes (2), cancer (3, 4), dementia (5), and cardiovascular disease (6). Decreased metabolism of fat relative to carbohydrates, fatty acid overload, and/or incomplete fatty acid metabolism in skeletal muscle are risk factors for insulin resistance, metabolic syndrome, and obesity (7, 8). Altered muscle metabolism contributes to the development of insulin resistance and type II diabetes (9, 10). The underlying mechanisms regulating skeletal muscle fuel selection and metabolic rate include differences in mitochondrial biogenesis, fatty acid uptake into muscle and mitochondria, and both carnitine palmitoyltransferase 1 (CPT1)3-mediated (11, 12) and Sirt3-mediated pyruvate dehydrogenase activities (13).
CaV1.1, the skeletal muscle voltage sensor, is also a voltage-dependent Ca2+ channel that allows a small Ca2+ influx into the mammalian skeletal muscle fibers. However, Ca2+ influx through this channel is not required for excitation–contraction coupling (14). Consistent with this, in skeletal muscle of teleost fish (15), Ca2+ influx through the CaV1.1 channel is absent. One of the differences between teleost CaV1.1 and mammalian CaV1.1 is the sequence within the pore. The teleost pore prevents Ca2+ permeation due to the presence of a negatively charged aspartate in repeat II above Ca2+ binding glutamate (15). This negative charge is likely to increase the affinity of the pore for Ca2+, preventing Ca2+ from moving through the channel. Based on these findings, Schredelseker et al. (15) suggested the possibility that Ca2+ influx into mammalian skeletal muscle fibers might be vestigial. We created a mouse model in which glutamate in the repeat III of the pore was mutated to a positively charged lysine designed to decrease or abolish Ca2+ binding within the pore. These mice, designated E1014K or EK, displayed decreased activation of CaMKII, decreased Ca2+ influx, reduced refilling of sarcoplasmic reticulum Ca2+ stores, and decreased protein synthesis. The net result of activation of this CaV1.1-mediated pathway is increased fatigue and decreased muscle fiber size (1). A possible interpretation of these findings is that Ca2+ binding within the pore or transition through the pore alters CaV1.1 conformation to modulate intracellular signal transduction events.
Skeletal muscle plays a major role in regulating metabolic rate in adult humans (8). We now demonstrate that the EK mice display altered metabolic function. They exhibit increased body fat, decreased energy expenditure, and impaired glucose tolerance. In the current study, we elucidate a novel role for Ca2+ permeation/binding to the CaV1.1 pore in the regulation of whole body energy expenditure via effects on muscle mitochondrial function.
Experimental Procedures
Animals
CaV1.1 E1014K knock-in mice (EK) were generated as described previously (1). In this study, 6–25-week-old EK and WT male mice were used, unless otherwise indicated. All mice were housed at room temperature with a 12:12-h light-dark cycle and provided with food and water ad libitum. Mice were fed a chow diet consisting of 24% protein, 16% fat, and 60% carbohydrates. All procedures were approved by the Animal Care Committee at Baylor College of Medicine (BCM).
Materials
Autocamtide 2-related inhibitory peptide (AIP), l-NG-nitroarginine methyl ester (l-NAME), and l-carnitine, sodium palmitate, coenzyme A, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), antimycin A, oligomycin A, KN 92, KN-93, nifedipine, BayK8644, and sodium pyruvate were purchased from Sigma-Aldrich. N-Benzyl-p-toluene sulfonamide was purchased from Tocris Biosciences. Tetramethylrhodamine, ethyl ester (TMRE), and the nitric oxide sensor 4-amino-5-methylamino-2′,7′-difluorescein (DAF-FM) were purchased from Invitrogen. Ultra fatty acid-free BSA was purchased from Roche Applied Science.
Magnetic Resonance Imaging
For live imaging, animals were anesthetized with 5% isoflurane in O2 and maintained with 2% isoflurane in O2. Animals were placed headfirst in a prone position on a custom-built holder while in the magnet. Respiration was monitored using a pressure pad placed beneath the animal, and temperature was monitored using a rectal probe (SA Instruments). Temperature was maintained at 37 °C using an air heating system (SA Instruments). The Model 1025 Small Animal Monitoring & Gating System software was used to monitor respiratory rates and temperature (SA Instruments). All images were acquired on a 9.4-tesla, Bruker Avance BioSpec spectrometer, 21-cm bore horizontal scanner with 35-mm volume resonator using the ParaVision 5.0 software (Bruker Biospin). Initial pilot scans were performed to correctly position the mice before scanning for subcutaneous, visceral, epididymal, and muscle fat. For subcutaneous, visceral, and epididymal fat, a modified multi-slice multi-echo scan protocol was used with the following parameters: repetition time = 800 ms, echo time = 10.21 ms, field of view = 3.00 cm, slice thickness = 2.00 mm (axial scans) or 1.5–2.0 mm (sagittal scans), matrix = 128 × 128, number of excitations = 2, number of cycles = 1, and acquisition time = 3 min 24 s 800 ms. A rapid acquisition with refocused echoes and variable repetition time (RAREVTR) scan protocol was used to acquire spin lattice (T1) weighted images of the liver or muscle and had the following parameters: repetition time = 7500 ms, echo time = 6.57 ms, field of view = 3.00 cm, slice thickness = 1.0 mm, matrix = 128 × 128, number of excitations = 1, number of cycles = 1, and acquisition time = 10 min 28 s 405 ms. Analyses for all liver and muscle RAREVTR images were performed with ParaVision 5.0. For liver and muscle, seven or five regions of interest were chosen at random, respectively, within the RAREVTR slice. The image sequence analysis tool within ParaVision was then used to calculate the T1 relaxation time of each regions of interest. The T1 relaxation times were averaged to give the overall T1 relaxation time of the tissue. Signal intensities were normalized to a water phantom.
Body Composition
Scans were performed in the mouse phenotyping core at Baylor College of Medicine with a Lunar PIXI densitometer and were analyzed with the Lunar PIXImus 2.10 software. Mice were anesthetized with 2% isoflurane.
Indirect Calorimetry (IDC)
To obtain the respiratory exchange ratio (VCO2/VO2), 24 h of IDC was performed in the mouse phenotyping core at BCM with a VO2/VCO2 Oxymax system (Columbus Instruments). For the cold challenge, mice were placed in the Comprehensive Laboratory Animal Monitoring System (CLAM System, Columbus Instruments) and exposed to 4 °C for up to 12 h. Mouse body temperature was recorded via telemetry probes implanted subcutaneously in the abdominal area. The VO2 and VCO2 were recorded for metabolic rate evaluation.
Insulin Tolerance Tests, Glucose Tolerance Tests, and Insulin Levels
Measurements of insulin and glucose were performed in the mouse metabolism core at BCM. For the insulin tolerance tests, mice were given an intraperitoneal injection of insulin (0.75 units/kg of body weight) after 4 h of fasting, and for the glucose tolerance tests, mice were given an I.P injection of glucose (1.5 mg/g of body weight) after 6 h of fasting (16).
Activity Monitoring and Food Intake
Food intake and activity were assessed at 25 °C with a CLAM System in the mouse phenotyping core at BCM. For food intake monitoring, the chow diet was ground to fine powder. Mice, acclimated in the cages for 24 h before the experiment, were monitored for a period of 10 days, with a less than 24 h break after the first 5 days. Activity measurements included horizontal, vertical (Z), and grooming movements.
Co-immunoprecipitation (CoIP)
Muscles (gastrocnemius) were homogenized in IP buffer (10 mm Tris, pH 7.4, 1% Nonidet P-40, 0.5% sodium deoxycholate containing protease and phosphatase inhibitors). 600 μg of muscle lysates with a final volume of 400 μl were then incubated with 4 μg of either eNOS or CaV1.1 antibody at 4 °C overnight followed by a 90-min incubation with protein G-agarose beads (Roche Applied Science). As a negative control, the same amount of protein was incubated with non-immunized IgG as above. After incubation, beads were washed three times with IP buffer, and the final immune complex was eluted by boiling in the SDS sample buffer.
S-Nitrosylation
Proteins from diaphragm muscles of WT and EK mice were assessed using the iodoTMTTM reagent for labeling (PierceTM S-nitrosylation Western blot kit, Thermo Fisher Scientific). Total protein was stained with the Swift membrane stain (Geno Technology).
Western Blotting, Isolation of Flexor Digitorum Brevis (FDB) Fibers, Single Fiber Immunocytochemistry, and Proximity Ligation Assay (PLA)
These procedures were performed as described previously (1). Additional antibodies used are shown in Table 1. To allow the use of data from multiple Western blots, the intensity of each band within a single Western blot was first normalized to GAPDH as a loading control and then calculated as %WT average from that specific Western blot. Data were then pooled and calculated as %WT ± S.E. For PLA, FDB fibers were treated with 50 μm l-NAME, 1 μm KN93, or 1 μm AIP. The culture medium of FDB muscle fibers was then changed to Tyrode's solution with 50 μm l-NAME or 1 μm AIP in the presence of 20 μm of N-benzyl-p-toluene sulfonamide for an additional hour in 95% O2, 5% CO2 incubator at 37 °C for 30 min. The fibers were electrically stimulated with 20 trains (100 Hz, 250 ms, 0.17 duty cycle) using an RC-37FS chamber, with a constant flow of 1 ml/min of Tyrode's solution during the stimulation. Fibers were then fixed with 2% paraformaldehyde in 0.1 m phosphate buffer and incubated with the primary antibodies for PLA.
TABLE 1.
List of antibodies used
WB, Western blot; IHC, immunohistochemistry; MnSOD, manganese superoxide dismutase; ANT, adenine nucleotide translocator; TMT, Tandem Mass Tag.
| Antibody | Company | WB | IHC | PLA | CoIP | Catalog no. | |
|---|---|---|---|---|---|---|---|
| 1 | ACSL1 | Thermo Scientific | 1:50 | PA5-17136 | |||
| 2 | Cav1.1a | Thermo Scientific | 1:1000 | 1:200 | 1:100 | MA3-920 | |
| 3 | Cav1.1a | abcam | 1:200 | ab58552 | |||
| 4 | Cav1.1b | Santa Cruz Biotechnology | 1:30 | sc-15970 | |||
| 5 | CD36 | NOVUS BIOLOGICALS | 1:1000 | 1:200 | 1:50 | NB110-59724 | |
| 6 | CD36 | Santa Cruz Biotechnology | 1:30 | sc-9154 | |||
| 7 | eNOS | abcam | 1:1000 | 1:100 | 1:50 | 1:100 | ab66127 |
| 8 | GAPDH | Santa Cruz Biotechnology | 1:500 | sc-20357 | |||
| 9 | Lamin A/C | Santa Cruz Biotechnology | 1:500 | sc-20681 | |||
| 10 | nNOS | Santa Cruz Biotechnology | 1:500 | sc-5302 | |||
| 11 | nNOS | Thermo Scientific | 1:400 | PA3-032A | |||
| 12 | Phospho-eNOS (Ser-1177) | Cell Signaling | 1:1000 | 1:200 | 9571 | ||
| 13 | Phospho-nNOS (Ser-1416) | abcam | 1:1000 | ab5583 | |||
| 14 | RyR1 | Thermo Scientific | 1:2000 | MA3-925 | |||
| 15 | VDAC | Cell Signaling | 1:1000 | 4866 | |||
| 16 | UCP1 | abcam | 1:2000 | ab10983 | |||
| 17 | UCP3 | abcam | 1:1000 | ab3477 | |||
| 18 | SIRT3 | Cell Signaling | 1:1000 | 2627 | |||
| 19 | COXIV | Cell Signaling | 1:1000 | 4844 | |||
| 20 | MnSOD | Santa Cruz Biotechnology | 1:500 | sc-133254 | |||
| 21 | CPT1 | Santa Cruz Biotechnology | 1:500 | 1:30 | sc-31128 | ||
| 22 | ANT | Santa Cruz Biotechnology | 1:500 | sc-9299 | |||
| 23 | Ubiquitin | Santa Cruz Biotechnology | 1:500 | sc-8017 | |||
| 24 | TMT | Thermo Scientific | 1:1000 | 90075 | |||
| 25 | ACC | Cell Signaling | 1:1000 | 3662 | |||
| 26 | Phospho-ACC (Ser-79) | Cell Signaling | 1:1000 | 3661 | |||
| 27 | AMPK | Cell Signaling | 1:2000 | 2793 | |||
| 28 | Phospho-AMPK (Thr-172) | Cell Signaling | 1:2000 | 2531 |
Analysis of Complex I-IV Activity
Mitochondrial OXPHOS complex activity assay was performed according to Mitsuhashi et al. (17). Briefly, isolated mitochondria were solubilized with NativePAGE sample buffer (Invitrogen) containing 0.3% n-dodecyl-β-d-maltoside (Sigma-Aldrich). Fifty micrograms of protein were run on a 3–12% NativePAGE Bis-Tris gel (Invitrogen) in the NativePAGE buffer system, anode buffer is NativePAGE buffer and cathode buffer contains 0.02% n-dodecyl-β-d-maltoside and 0.05% deoxycholate. For in-gel catalytic activity assays of OXPHOS complex, gels were incubated in the following solutions: Complex I, 5 mm Tris-HCl, pH 7.4, 140 mm NADH, and 3 mm nitrotetrazolium blue; Complex II, 5 mm Tris-HCl, pH 7.4, 20 mm succinate, 3 mm nitro tetrazolium blue, and 200 mm phenazine methosulfate; Complex III, 50 mm sodium phosphate buffer, pH 7.2, and 0.5 mg/ml diaminobenzidine; Complex IV, 50 mm sodium phosphate buffer (pH 7.2), 0.5 mg/ml diaminobenzidine, and 5 mm cytochrome c.
Subcellular Fractionation
Diaphragms excised from WT and EK mice were used for fractionation of cytoplasm, nuclei, and plasma membrane using a MinuteTM plasma membrane protein isolation kit (Invent Biotechnologies). Mitochondria were purified following the user protocol in the mitochondria extraction kit (Boster Biological Technology).
mRNA Levels of Mitochondrial Proteins
RNA was isolated from fresh, snap frozen muscles with the RNAzol®RT (Sigma) according to manufacturer's protocol. Diaphragm and soleus muscles were used. Reverse transcription was done with random primers (Invitrogen) and Superscript II reverse transcriptase (Invitrogen). The RT-PCR was performed (in triplicates) in an AB ViiA7 RT-PCR system with SYBR Green (Bio-Rad). Primer sequences are from PrimerBank (and validated). Relative expressions were calculated according to the Livak method (51). GAPDH was used as a control. Data are presented as %WT.
Ubiquitination of Mitochondrial Proteins
The mitochondrial fraction isolated as described above was lysed in radioimmunoprecipitation assay buffer. The same amounts (60 μg) of mitochondrial lysates from EK and WT hindlimb muscle were loaded on a 5–15% SDS-PAGE gel (Bio-Rad), separated, and transferred to PVDF membrane. This blot was treated with anti-ubiquitin antibody (Santa Cruz Biotechnology). Western blots were performed as described previously (1).
Imaging of TMRE-loaded FDB Fibers
Images were acquired with a Plan-Apochromat 63 × 1.4 oil differential interference contrast lens and a Zeiss LSM 510 META confocal microscope. Images (xy) were acquired setting the optical slice in the z axis to 1 μm using the 543-nm excitation laser and the BP 560–615 emission filter. After starting the perfusion with 25 nm TMRE, images were acquired for up to 5 min.
TMRE Fluorescence to Indirectly Measure Fatty Acid β-Oxidation by Changes in Mitochondrial Membrane Potential
The effect of BSA-conjugated palmitate on relative changes in mitochondrial membrane potential was monitored with TMRE in non-quench mode (18). Palmitate was conjugated to BSA at a 6:1 molar ratio. To monitor changes in mitochondrial membrane potential, pre-plated FDB fibers were perfused (1 ml/min) with 5 ml of Tyrode's (includes 5.5 mm glucose) containing 25 nm TMRE in the presence of 0.5 mm carnitine and either 100 μm BSA-conjugated palmitate or 100 μm BSA control. When indicated, 1 μm AIP was included in the solution 15 min before starting the perfusion and then maintained throughout the experiment. During FCCP (2 μm) incubation, to avoid variations in voltage, antimycin A (1 μm) was used to block proton pumping by Complex III, and oligomycin (1 μm) was used to inhibit complex V from depleting ATP pools. Additionally, the effects of 25 mm glucose and 10 mm sodium pyruvate were monitored with the 5.5 mm glucose in Tyrode's as control. Fibers permeabilized with saponin as described previously (19) contained in “internal” solution (in mm): 140 KCl, 10 HEPES, 0.5 EGTA, 5 phosphocreatine (di-Tris), 3 Mg-ATP, and 0.114 CaCl2, pH 7.0, with KOH. Briefly, pre-plated FDB fibers were exposed to 50 μg/ml saponin for 30 s followed by complete washout with internal solution. Permeabilized cells were perfused as indicated in the previous section in the presence of 0.5 mm carnitine and 100 nm palmitate-CoA or 1 mm palmitate-carnitine as indicated (20). Fluorescence data were captured through a Nikon S. Fluor objective (×20, 0.75 numerical aperture) coupled to an inverted microscope (Nikon Eclipse TE-200) and digitized using a CCD Rolera MGi-Plus camera (QImaging) with 510 × 252-pixel field size (using 6 × 6 binning). Data were collected and stored using the MetaFluor software (version 6.2) for further analysis.
Nitric Oxide Production Stimulated with Electrical Field Stimulation (EFS)
Single isolated FDB fibers plated on coverslips and mounted in a recording chamber (RC-21BDW, Warner Instruments) were incubated for 1 h with 10 μm DAF-FM at room temperature, followed by washout with fresh DMEM. Nitric oxide production was stimulated with three cycles of EFS using two platinum wires placed ∼100 μm apart on both sides of the fiber. Each cycle of EFS was composed of 2 min of continuous application of trains every 1.5 s (100 Hz, 250-ms duration, 0.17 duty cycle) followed by 1 min of rest between cycles. Nitric oxide production was evaluated before and after the EFS protocol. The reactive nitrogen species sensor DAF-FM loaded into FDB fibers was excited at 0.5 Hz with the 500-nm filter placed in a DG4 system used as a light source. Emitted florescence was collected through a Nikon S. Fluor objective (20×, 0.75 NA) coupled to an inverted microscope (eclipse TE-200, Nikon) and digitized using a CCD Rolera MGi-Plus camera (QImaging) with 510 × 256-pixel field size using 6 × 6 binning. Data were collected and stored using the MetaFluor software (version 6.2) for further analysis. The drugs tested (10 μm BayK8644, 10 μm nifedipine, 50 μm l-NAME, 1 μm KN92 or KN93) were kept in the solution throughout the experiment.
Statistical Analyses
Statistical analyses between two groups were conducted using Student's t test or analysis of variance. p < 0.05 was considered statistically significant. The effect of two independent variables was analyzed with two-way analysis of variance. Analysis of covariance was performed on metabolic rate with body weight or lean muscle as covariance (21).
Results
Effects of the EK Mutation in CaV1.1 on Energy Expenditure
We previously demonstrated that a permeation defect in CaV1.1 (EK) leads to increased fatigue and decreased Ca2+ store refilling, protein synthesis, muscle fiber diameter, and type IIb/x fiber content (1). Because skeletal muscle represents ∼40% of total body mass in mammals and contributes about 30% of the resting metabolic rate in adult humans (22), we hypothesized that changes in Ca2+ signaling in the muscle of EK mice would lead to altered muscle energy utilization. We found that EK mice gained more weight than age-matched WT mice (Fig. 1A) and had a higher percentage of body weight due to fat (Fig. 1B). Food intake and cage activity did not differ between EK and WT mice (Fig. 1, C and D). Fat in EK mice accumulated in epididymal, abdominal, visceral, and subcutaneous areas (Fig. 1, E and F) with no detectable changes in either hepatic or intramuscular fat when compared with WT mice as assessed from spin-lattice (T1) relaxation time constants using MRI (Fig. 1G). VO2 was reduced (Fig. 1H) and the respiratory exchange ratio was increased during both day and night in EK mice when compared with WT mice, indicating a preference for carbohydrate over lipid metabolism (Fig. 1I). Although young EK mice (6–8 weeks) did not differ from WT mice in glucose or insulin tolerance (data not shown), by 16–18 weeks of age, EK mice developed glucose intolerance and insulin resistance (Fig. 1, J and K) relative to WT. Consistent with the insulin resistance of the older EK mice, plasma insulin levels increased with age in the EK mice and were significantly higher than that of the WT mice at 1 year of age (Fig. 1L).
FIGURE 1.

Metabolic changes associated with the EK mutation. A, body weight. B, percentage of fat. C, food intake. D, activity in WT and EK mice. Food intake and activity are the average values for the 10 days of recordings in the CLAM system. X TOTAL is the total average activity of mice in the x-y level and includes ambulatory movements, indicated as X AMP, and grooming movements (not shown). Z TOTAL is the total average up activity. E, representative axial MRI images (matching serial sections) from a three-dimensional dataset showing the distribution of fat (white regions) in EK and WT mice (20 weeks old). F, representative sagittal MRI images (matching serial sections) from a three-dimensional dataset showing the distribution of fat (white regions) in EK and WT mice (20 weeks old). G, T1 relaxation time (in ms) estimated with the MRI for liver and gastrocnemius muscle in 20–25-week-old littermates. H, VO2 from WT and EK mice measured day and night in 6-week-old WT and EK mice maintained on a chow diet and monitored by indirect calorimetry for 24 h. I, respiratory exchange ratio for day and night cycles in 6-week-old WT and EK mice maintained on a chow diet and monitored by indirect calorimetry for 24 h. J and K, glucose (J)and insulin tolerance (K) tests performed with 16–18-week-old mice fed a chow diet. L, plasma insulin levels of random fed EK and WT mice. Data are shown as mean ± S.E., numbers of mice are indicated, *, p < 0.05, **, p < 0.01, and ***, p < 0.001.
Using indirect calorimetry, we assessed heat production to evaluate differences in energy expenditure between WT and EK mice (21). A small decrease in metabolic rate was observed in the EK mice (Fig. 2A). Skeletal muscle and futile Ca2+ cycling by sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) contribute to non-shivering thermogenesis, and defects in this process are associated with obesity in mice (23). We assessed cold tolerance in the EK and WT mice by measuring body temperature with implanted telemetric probes and VCO2/VO2 with IDC upon exposure to 4 °C for up to 12 h. Despite a trend toward lower body temperature in the EK mice, these differences did not reach significance (Fig. 2B). However, heat production (measured during the 3rd to 8th hours of cold exposure) was significantly reduced in EK mice when compared with WT mice under both fed (Fig. 2C) and fasting (Fig. 2D) conditions. Taken together, these findings support a role for CaV1.1 in the control of basal metabolic rate and heat generation during cold challenge.
FIGURE 2.

Changes in energy expenditure in EK mice. A, metabolic rate (heat) as a function of lean mass determined from body scans of 6-week-old EK and WT littermate mice. R2 for WT regression is 0.55, and R2 for EK regression is 0.61. Data were analyzed with analysis of covariance. B, average drop in body temperature in EK and WT mice during the cold challenge at 4 °C is shown as the ratio of body temperature at the beginning of the experiment over the body temperature during the challenge. Data are plotted as mean ± S.E. C, heat production measured by IDC of WT and EK mice (Fed, 12 weeks old, averaged over the 3rd to 8th hours of cold exposure) exposed to 4 °C. Analysis of covariance was determined with body weight as covariance due to inability to measure lean muscle in mice bearing the telemetry probes. D, heat production measured by IDC of WT and EK mice (Food deprived, 12 weeks old, averaged over the 3rd to 8th hours of cold exposure) exposed to 4 °C. Analysis of covariance was determined with weight as covariant due to inability to measure lean muscle in mice bearing the telemetry probes. Numbers of mice are indicated.
Effect of EK Mutation on Mitochondrial Protein and mRNA Levels
We previously demonstrated that the soleus and diaphragm muscles of the EK mice are more affected than the extensor digitorum longus muscle (1), and we, therefore, concentrated on soleus and diaphragm in the current study. We first assessed mitochondrial to nuclear DNA levels and found no differences between EK and WT soleus or diaphragm muscles (Fig. 3A). We next assessed the protein and mRNA levels of a number of mitochondrial proteins in diaphragm (Fig. 3, B–E) and soleus muscles (Fig. 3, B, C, F, and G). Protein levels of COXIV, VDAC, SIRT3, and manganese superoxide dismutase (MnSOD) were reduced in both soleus and diaphragm muscles of EK when compared with WT mice. However, mRNA levels for these mitochondrial proteins were not decreased in the muscles of EK mice (Fig. 3, E and G). Thus, muscles of EK mice display reduced levels of some, but not all mitochondrial proteins in the absence of decreases in mRNAs for these proteins, suggesting that decreases in mitochondrial proteins are due to increased degradation. Because some mitochondrial membrane proteins are degraded by a ubiquitin-dependent pathway (24, 25), we purified mitochondria from diaphragms of EK and WT mice and assessed ubiquitination. We found a significant increase in the ubiquitination of mitochondrial proteins from the muscle of EK when compared with WT mice (Fig. 4, A and B). The decrease in COXIV protein levels suggests that OXPHOS may be altered in EK muscle. We assayed mitochondrial OXPHOS activity as described by Mitsuhashi et al. (17) and found a small, but significant decrease in Complex IV, but not Complex I–III activity (Fig. 4, C and D).
FIGURE 3.

Mitochondrial content. A, ratio of mitochondrial to nuclear DNA. B, representative Western blot images for SIRT3, UCP3, COXIV, VDAC, manganese superoxide dismutase (MnSOD), and GAPDH of whole muscle lysates in soleus and diaphragm muscles of EK and WT mice. C, representative Western blot images for CPT1, UCP1, adenine nucleotide translocator (ANT), and GAPDH in whole muscle lysates in soleus and diaphragm muscles of EK and WT mice. D, analysis of mitochondrial markers in diaphragm normalized to GAPDH. E, analysis of mRNA for mitochondrial proteins in diaphragm by quantitative RT-PCR. F, analysis of mitochondrial markers in soleus normalized to GAPDH. G, mRNA levels of mitochondrial markers in soleus. Values are shown as mean ± S.E. *, p < 0.05, and **, p < 0.01. n is the number of mice.
FIGURE 4.

Ubiquitination and OXPHOS activity. A, mitochondrial proteins from EK and WT muscle stained for total protein and ubiquitin. B, analysis of ubiquitinated mitochondrial proteins. C, mitochondrial Complex I–IV staining and activity. D, analysis of Complex I–IV activity. Values are shown as mean ± S.E. *, p < 0.05. n is the number of mice.
Alterations in Mitochondrial β-Oxidation in Response to Fatty Acids
We assessed mitochondrial function in FDB fibers indirectly by measuring changes in mitochondrial membrane potential using TMRE in non-quenching mode. TMRE fluorescence is frequently used to assess changes in mitochondrial membrane potential (26), but caution is required to avoid artifacts (18). Fig. 5A shows that, under the conditions used in this study, TMRE labels mitochondria (both subsarcolemmal and interfibrillar) in isolated intact FDB fibers, whereas sarcolemmal membrane partitioning is minimal. To evaluate perturbations in mitochondrial membrane potential driven by exogenously added fatty acids, we compared the effects of carnitine alone with BSA-conjugated palmitate plus carnitine. We found linear increases in TMRE fluorescence as a function of time, reflecting a change in mitochondrial membrane potential, in the presence of carnitine alone (in 5.5 mm glucose) in FDB fibers from both WT and EK mice (Fig. 5B). The initial rate of change in TMRE fluorescence was significantly increased by the addition of BSA-conjugated palmitate only in WT fibers (Fig. 5C). AIP, an inhibitor of CaMKII, and l-NAME, an inhibitor of NOS, blocked the palmitate-induced increase of the rate of change in TMRE fluorescence in the WT fibers (relative to carnitine alone) (Fig. 5, D and E). Both inhibitors slowed the rate of change of TMRE fluorescence in EK fibers treated with both carnitine and BSA-conjugated palmitate, but to a lesser extent (Fig. 5F). To demonstrate that changes in TMRE fluorescence were due to alterations in mitochondrial membrane potential, we used a mitochondrial uncoupler, FCCP, combined with oligomycin, to block complex V-dependent ATP depletion, and antimycin A, to block proton pumping by Complex III. The rate of change in the TMRE fluorescence was significantly less in both WT and EK fibers than in the presence of carnitine alone (Fig. 5F). The initial rates of change under all these conditions in intact FDB are summarized in Fig. 5F. These data suggest that the changes in mitochondrial function are downstream of alterations in CaV1.1, CaMKII, and NOS.
FIGURE 5.

Effects of the EK mutation on changes in mitochondrial membranes potential. A, representative confocal image of an FDB fiber loaded with TMRE. B, change of TMRE fluorescence in intact FDB fibers from WT and EK mice as a function of time after the addition of carnitine alone (0.5 mm). C, BSA-conjugated-palmitate (100 μm) plus carnitine. D, BSA-conjugated-palmitate (100 μm) plus carnitine plus AIP (1 μm). E, BSA-conjugated-palmitate (100 μm) plus carnitine plus l-NAME (50 μm). F, summary of the change of TMRE fluorescence as a function of time in intact fibers. Also shown is a slope with BSA-conjugated-palmitate (100 μm) plus carnitine plus a mix of FCCP (2 μm)/oligomycin (1 μm)/antimycin A (1 μm) (FCCP/O/A). G, summary of the initial rate of change in TMRE fluorescence in saponin permeabilized fibers. H, summary of initial rate in saponin permeabilized fibers. I, summary of initial rate in the presence of 5.5 mm glucose in Tyrode's (low glucose, LG), 10 mm sodium pyruvate (Pyr), or 25 mm glucose (high glucose, HG). Fibers are from at least three mice of each genotype. Data are shown as mean ± S.E., and numbers of fibers (n) are indicated. *, p < 0.05, **, p < 0.01, and ***, p < 0.001.
To determine whether the reduction in mitochondrial β-oxidation in response to palmitate in the EK fibers was due to alterations in the rate of fatty acid uptake into the muscle fiber, we assessed the rate of change in TMRE fluorescence in fibers permeabilized with saponin. Permeabilized WT fibers still responded to the addition of BSA-conjugated palmitate in the presence of carnitine with an increase in the rate of change in TMRE fluorescence (Fig. 5, G and H), but the rate change in TMRE fluorescence in permeabilized EK fibers was similar for carnitine alone and carnitine plus BSA-conjugated palmitate (Fig. 5G). Thus, differences in the rate of fatty acid uptake across the sarcolemma or t-tubules do not fully account for the differences in β-oxidation-driven changes in mitochondrial membrane potential between EK and WT fibers. To identify additional steps in β-oxidation altered by the EK mutation, we replaced the BSA-conjugated palmitate with either palmitoyl-CoA to bypass the fatty acyl-CoA synthetase (ACS) or palmitoyl-carnitine to bypass CPT1 (Fig. 5H). Both palmitoyl-CoA and palmitoyl-carnitine increased the initial rate of TMRE fluorescence (i.e. β-oxidation) above that of carnitine alone to a similar degree in both WT and EK fibers. These results suggest that the step that contributes to the decreased mitochondrial response to fatty acids in fibers from EK mice lies at or upstream of acyl-CoA synthetase.
The small decreases detected in COXIV activity in the EK muscle could be contributing to the change in the response of the mitochondrial membrane potential to fatty acids. To determine whether the difference in the rate of change in the mitochondrial membrane potential upon the addition of palmitate in EK when compared with WT fibers reflected the mitochondrial damage, we used different substrates to drive oxidative phosphorylation (5.5 mm glucose, 25 mm glucose, or 10 mm pyruvate). We did not detect differences in the rate of change in TMRE fluorescence between EK and WT fibers (Fig. 5I) with these substrates. We conclude that the fibers from EK display a selective decrease in fatty acid β-oxidation, which can be phenocopied in WT fibers by inhibition of either CaMKII or nitric-oxide synthase.
A Role for NOS
S-Nitrosylation of proteins has been shown to either slow (27) or enhance (28, 29) their ubiquitin-proteasome-mediated breakdown. In addition, the finding that l-NAME prevents the fatty acid-driven increases in TMRE fluorescence in WT fibers (Fig. 5E) suggests that NOS activity is contributing to the pathway by which CaV1.1 regulates mitochondrial function. NOS enzymes are regulated by muscle activity (30, 31), Ca2+/calmodulin (32), and CaMKII (33, 34). Reactive nitrogen species (RNS) generated by the NOS enzymes regulate both mitochondrial biogenesis and mitochondrial function (35). We evaluated the role of the CaV1.1/CaMKII pathway in activity-dependent RNS production in FDB fibers using DAF-FM. Activity-dependent changes in DAF fluorescence were significantly decreased in FDB fibers from EK when compared with WT mice (Fig. 6A). In WT fibers, RNS levels were enhanced by BayK8644 (an activator of CaV1.1), and inhibited by nifedipine (a CaV1.1 inhibitor), KN93 (CaMKII inhibitor), and l-NAME (NOS inhibitor) (Fig. 6A). BayK8644, nifedipine, and KN93 did not significantly alter RNS levels in FDB fibers from EK mice. On the other hand, l-NAME reduced RNS levels in fibers from both WT and EK mice, suggesting that the CaV1.1./CaMKII pathway enhances, but is not absolutely required for activity-dependent RNS production in muscle fibers.
FIGURE 6.

A role for NOS. A, effects of modulators of CaV1.1 and CaMKII on RNS generation in FDB fibers from WT and EK mice. RNS production was assessed by DAF fluorescence in the absence and presence of BayK8644 (1 μm), nifedipine (1 μm), KN93 (1 μm), or l-NAME (5 0 μm). B, representative Western blots for p-nNOS and total nNOS (top panel) and p-eNOS and total eNOS (bottom panel) in muscle homogenates of WT and EK mice. C, analysis of p-eNOS/eNOS. D, analysis of p-nNOS/nNOS. E, S-nitrosylation of membrane proteins assessed with iodoTMT switch technology. Representative blot of S-nitrosylation and total protein staining of membrane preparations from the muscle of WT and EK mice in the presence (+) or absence (−) of ascorbate is shown. F, representative blots of S-nitrosylation and total protein in mitochondrial preparations from muscle of WT and EK mice in the presence (+) or absence (−) of ascorbate. G, relative total S-nitrosylation of membrane proteins from muscle of WT and EK mice normalized to CaV1.1 and expressed as %WT. H, relative S-nitrosylation of mitochondrial proteins isolated from muscle of WT and EK mice normalized to total protein and expressed as %WT. I, summary diagram of NOS pathways altered by the EK mutation. S-NO, S-nitrosylated cysteine. Data are shown as mean ± S.E. *, p < 0.05, **, p < 0.01, and ***, p < 0.001. In A, n refers to the number of fibers. In B–H, n refers to the number of mice.
CaMKII phosphorylates eNOS at Ser-1177 (33) and nNOS at Ser-1416 (34). Using phospho-specific antibodies, we found decreased p-eNOS/eNOS and p-nNOS/nNOS levels in EK muscle (Fig. 6, B–D). Using iodoTMT and Swift membrane stain for total protein transferred to a PVDF membrane, we assessed S-nitrosylation of membrane (Fig. 6, E and G) and mitochondrial proteins (Fig. 6, F and H). S-Nitrosylation was decreased in both membrane and mitochondrial fractions in muscle from EK when compared with WT mice (Fig. 6, G and H). These data indicate that Ca2+ binding and/or permeation through CaV1.1 and CaMKII activation increase NOS activity and that this increase is prevented by the EK mutation in CaV1.1. Upstream and downstream pathways of NOS activity that are modulated by Ca2+ permeation or binding to CaV1.1 are summarized in Fig. 6I.
We previously showed that CaMKII was in close proximity to CaV1.1 in the t-tubule membrane (1). To determine whether NOS was in close proximity (<40 Å) to CaV1.1, we used both immunocytochemistry and PLA. As shown in Fig. 7, CaV1.1 and eNOS mostly co-localize in t-tubules of skeletal muscle fibers.
FIGURE 7.

CaV1.1 and eNOS are in close proximity in the transverse tubule membranes. A, representative immunocytochemistry images showing co-localization of eNOS and CaV1.1 in FDB fibers from WT and EK mice. B, line scan for immunocytochemistry of CaV1.1 and eNOS in FDB fibers of WT mice. C, line scans for immunocytochemistry of CaV1.1 and eNOS in FDB fibers of EK mice. D, representative PLA images for eNOS and CaV1.1 in fibers from WT and EK mice. E, analysis of spot density for eNOS and CaV1.1 co-localization by PLA. Fibers are from three different mice of each genotype. Data are shown as mean ± S.E., and numbers of fibers are indicated. n is number of fibers.
A Role for CD36
To further explore the mechanism by which the CaV1.1/CaMKII/eNOS pathway regulates mitochondrial function, we evaluated a potential role for the fatty acid (FA) transport protein CD36 (36), which regulates both FA uptake into muscle and the delivery of FAs to peroxisomes and mitochondria (37). Muscle activity promotes CD36 translocation between the t-tubules and mitochondria. In the t-tubules, CD36 increases FA uptake and, in the mitochondria, CD36 increases the rate of FA β-oxidation (38–41). Total CD36 levels were not different between muscle from EK and WT mice (Fig. 8, A and B). We assessed the subcellular distribution of CD36, eNOS, Cav1.1, RyR1, and VDAC in Western blots of subcellular fractions (cytosolic, nuclear, purified mitochondria, and membranes) from EK and WT muscle. CD36 was increased in the cytosolic fractions and decreased in the mitochondrial and membrane fractions from EK when compared with WT muscle (Fig. 8, C and D).
FIGURE 8.

The EK mutation alters CD36 distribution. A, representative Western blot of CD36 in homogenates from soleus, extensor digitorum longus (EDL), and diaphragm of WT and EK mice. B, analyses of CD36 in muscle homogenates. C, representative Western blots of CD36, eNOS, CaV1.1, RyR1, lamin A/C, GAPDH and VDAC from subfractions of muscle homogenates. Fractions: cytosolic (Cyto), nuclear (Nucl), purified mitochondria (Mito), and membranes (Memb). Lamin A/C, GAPDH, and VDAC were used as markers of nuclear, cytosolic, and mitochondrial fractions, respectively. D, analysis of CD36 in cytosolic fractions, mitochondria, and membranes. In A–D, n is the number of trials. E, PLA of CD36 and eNOS with and without AIP. F, analysis of PLA of CD36 and eNOS. G, representative Western blots of samples immunoprecipitated with eNOS antibody and blotted with CD36 or eNOS antibody. H, analysis of CD36 in eNOS IP. Fibers are from at least three mice of each genotype. Data are shown as mean ± S.E., and numbers of mice are indicated. *, p < 0.05, **, p < 0.01, and ***, p < 0.001.
Consistent with partial localization to the membrane fraction, some CD36 colocalized with eNOS (PLA) as shown in Fig. 8, E and F. The proximity of CD36 to eNOS was decreased in EK muscle and was decreased in WT muscle by an inhibitor of CaMKII, AIP. CD36 also immunoprecipitated with antibodies to eNOS (Fig. 8, G and H), and the amount of the CD36 in the IPs was decreased in EK muscle. These findings suggest that CD36 interacts with eNOS near the t-tubule membrane and that this interaction is modulated by the CaV1.1/CaMKII pathway.
Whether CD36 directly binds to mitochondria is controversial (42, 43). We examined the proximity (<40 Å) of CD36 to two additional proteins, CPT1, (an outer mitochondrial membrane protein) (44) and long-chain-fatty-acid-CoA ligase 1 (ACSL1). ACSL1 is found in plasma membranes, peroxisomes, and the outer mitochondrial membrane. The interactions (or proximity) of CD36 with CPT1 (Fig. 9, A and B) and CD36 with ACSL1 (Fig. 9, C and D) were markedly decreased in EK fibers. AIP and l-NAME decreased the interactions of CD36 with CPT1 and ACSL1. These findings again support a decreased interaction of CD36 with transverse tubules, mitochondria, and possibly other intracellular membranes and support a regulation of CD36 subcellular location by CaMKII and NOS regulated pathways.
FIGURE 9.

CD36 localizes in the proximity of mitochondrial proteins. A, representative PLA images for CD36 and CPT1 in FDB fibers from WT and EK mice in the absence and presence of AIP or l-NAME. Negative (−) control images are also shown. B, analysis of spot density for CD36 and CPT1 co-localization by PLA. C, representative PLA images for CD36 and ACSL1 in FDB fibers from WT and EK mice in the absence and presence of AIP or l-NAME. Negative (−) control images are also shown. D, analysis of spot density for CD36 and ASCL1 co-localization by PLA. Fibers are from at least three mice of each genotype. Data are shown as mean ± S.E., and numbers of fibers are indicated. *, p < 0.05, and ***, p < 0.001. n is number of fibers from at least 3 mice.
Another potential mechanism whereby altered CD36 subcellular localization could alter muscle metabolism is via its effects on AMPK. Recently, Samovski et al. (45) suggested that CD36 suppresses AMPK activation (assessed as p-AMPK/AMPK) but fatty acids relieve this inhibition. We assessed AMPK activation in muscle of EK and WT mice and found that AMPK activity is reduced in EK muscle (Fig. 10, A and B). AMPK phosphorylates acetyl CoA carboxylase (ACC) at Ser-79 (46), decreasing ACC activity. We found a decrease in the ratio of Ser-79 phosphorylated to total ACC in extensor digitorum longus and diaphragm (Fig. 10, C and D), suggesting increased ACC activity. ACC produces malonyl CoA, which inhibits FA β-oxidation by inhibiting CPT1 (47). Hence an additional mechanism whereby changes in CD36 within the muscle regulate FA β-oxidation is by altering AMPK activation.
FIGURE 10.

The EK mutation alters AMPK activation. A, representative Western blot images of pT172-AMPK and AMPK in homogenates of indicated muscles. EDL, extensor digitorum longus. B, analysis of p-AMPK/AMPK plotted as %WT. C, representative Western blot images of p-Ser-79-ACC and ACC in homogenates of the indicated WT and EK muscles. D, analysis of p-Ser-79 ACC/ACC plotted as %WT. Data are shown as mean ± S.E. *, p < 0.05, **, p < 0.01, and ***, p < 0.001. n is the number of mice.
Discussion
Ca2+ influx through the voltage-dependent Ca2+ channel is not required for excitation-contraction coupling, but Ca2+ does bind to and permeate the pore of this channel, raising the question of the role, if any, of Ca2+ binding/permeation through this channel to skeletal muscle function. We previously demonstrated that a mutation in the CaV1.1 pore, which blocked Ca2+ binding and permeation, decreased CaMKII activity, inhibited refilling of SR Ca2+ stores, and reduced protein synthesis (1). Collectively, these changes resulted in increased fatigue and decreased muscle fiber cross-sectional area. We now show that these mice (EK) also gain weight on a chow diet without increased food intake or decreased activity, suggesting decreased energy expenditure.
We demonstrate an accumulation of abdominal/visceral and epididymal fat in the EK mice and show that these mice generate less heat when exposed to colder temperatures. The EK mice also exhibit insulin and glucose intolerance by the time they reach 16 weeks of age. These animals display decreased energy expenditure (heat production) and lower respiratory quotient (respiratory exchange ratio), strongly supporting a significant defect in fatty acid utilization.
We also found that a pathway that requires Ca2+ binding to the pore of CaV1.1, as well as activation of both CaMKII and NOS, is required for normal mitochondrial β-oxidation (assessed as a BSA-conjugated palmitate-induced alteration in the mitochondrial membrane potential). The TMRE response to palmitate is reduced in EK when compared with WT fibers. This suggests that the mitochondria in the EK mice are not functioning optimally. Our data to support this include: (a) decreased mitochondrial proteins, (b) increased ubiquitination of mitochondrial proteins, and (c) a decreased mitochondrial membrane potential response to palmitate (but not glucose or pyruvate) in EK muscle.
We also found that the CaV1.1/CaMKII/NOS pathway regulates the intracellular distribution of CD36. In skeletal muscle, CD36 has been shown to bind to the outer mitochondrial membrane (36). Continuous cycling of CD36 between cell surface and intracellular compartments has been reported (48). Continuous cycling of CD36 between the plasma membrane, endosomes, and other intracellular organelles (i.e. mitochondria) has been reported (43, 49, 50). Yoshida et al. (12) showed that CD36 deficiency decreases β-oxidation in the absence of changes in mitochondrial number and enzyme content and proposed that CD36 is a key component in fuel selection both at rest and during exercise. Smith et al. (36) found that CD36 deficiency decreased palmitate-supported mitochondrial respiration by 34%, but did not alter palmitoyl-CoA-supported respiration, demonstrating that CD36 functions upstream of acyl-CoA synthetase. Mice lacking CD36 have similarities to the EK mice, including reduced FA oxidation in muscle and increased respiratory exchange ratio (47). We propose that one possible explanation of the activity dependence of CD36 function is the regulation of its intracellular distribution by the CaV1.1/CaMKII/NOS pathway. Even a transient contact between CD36 and CPT1 could be an important mechanism for fatty acid shuttling upon electrical stimulation and muscle contraction. Our data, however, cannot discriminate between a true mitochondrial localization for CD36 versus a transient contact with the mitochondria via nearby endosomes or plasma membrane.
Recently, CD36 was shown to negatively regulate AMPK (45), which phosphorylates and decreases ACC activity. Decreased AMPK and p-ACC raises the possibility that malonyl CoA, produced by ACC, plays a role in decreased FA oxidation in the muscle of EK mice.
In summary, our data suggest that Ca2+ binding/permeation of CaV1.1 sequentially activates CaMKII and eNOS to regulate CD36 distribution, thereby regulating FA β-oxidation. These effects on FA β-oxidation could arise from S-nitrosylation of mitochondrial proteins, increased interaction of CD36 with mitochondria, or decreased AMPK activity (Fig. 11). The relative contributions of these pathways remain to be determined. Our studies identify CaV1.1, CaMKII, and eNOS as potential new molecular targets for therapeutic interventions in metabolic syndrome. These findings also have important implications for understanding the mechanisms that underlie metabolic changes in obesity and adaptations to exercise.
FIGURE 11.
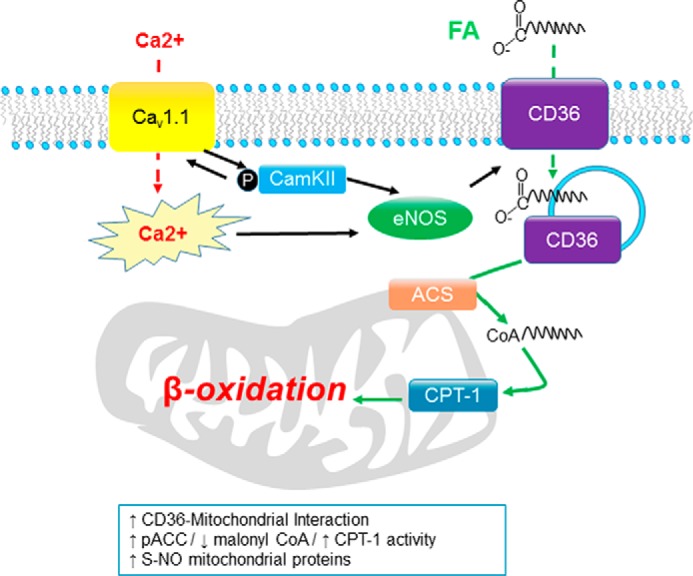
Model showing how the CaV1. 1/CaMKII/eNOS pathway regulates CD36 intracellular distribution and enhances FA β-oxidation. We propose that the pathway initiated by Ca2+ permeation through or binding to CaV1.1 activates CaMKII. Activated CaMKII, in turn, further increases Ca2+ influx to the fiber either through CaV1.1 itself (positive feedback loop) or through another influx pathway. Ca2+ and CaMKII activate NOS, which, in turn, alters the intracellular distribution of CD36 and S-nitrosylates mitochondrial proteins to slow their degradation through the ubiquitin-proteasome pathway. We also propose that this pathway regulates CD36 distribution and that CD36 directly or closely interacts with mitochondria to regulate β-oxidation. CD36 may also regulate AMPK activity and indirectly regulate mitochondrial FA β-oxidation through malonyl CoA inhibition of CPT1 (not shown in figure). ACS, acyl-CoA synthetase.
Author Contributions
D. K. G. conceived, performed, and analyzed the metabolic analyses in Figures 1 and 2, analyzed the PLA data, and helped to write the manuscript. A. D.-A. performed all of the TMRE studies in Figure 5 and the DAF experiments in Figure 6 and helped to write the manuscript. C. S. L. performed and analyzed the Western blotting experiments in Figures 3 and 6 and the experiments in Figures 4, 7, 8, 9, and 10, and helped write the manuscript. D. M. G. performed the MRI experiments in Figure 1. H. W. performed all of the quantitative RT-PCR experiments and helped to write the manuscript. W. R. L. helped to supervise and interpret the metabolic experiments and helped write the manuscript. R. G. P. supervised and analyzed the MRI studies and helped with manuscript preparation. R. T. D. helped in the supervision and interpretation of the TMRE and DAF studies and helped in preparing the manuscript. S. L. H. helped plan and supervise all experiments and wrote the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We thank the staff of the BCM core facilities (supported by National Institutes of Health Grant P30 DK079638).
This work is supported by National Institutes of Health Grants AR053349 (to S. L. H. and R. T. D.) and AR041802 (to S. L. H.), The authors declare that they have no conflicts of interest with the contents of this article.
- CPT1
- carnitine palmitoyltransferase 1
- CaMKII
- Ca2+/calmodulin kinase type II
- eNOS
- endothelial NOS
- nNOS
- neuronal NOS
- AMPK
- AMP-activated protein kinase
- ACC
- acetyl CoA carboxylase
- VDAC
- voltage-dependent anion channel
- AIP
- autocamtide 2-related inhibitory peptide
- l-NAME
- l-NG-nitroarginine methyl ester
- TMRE
- tetramethylrhodamine, ethyl ester
- DAF-FM
- 4-amino-5-methylamino-2′,7′-difluorescein
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- RAREVTR
- refocused echoes and variable repetition time
- IDC
- indirect calorimetry
- PLA
- proximity ligation assay(s)
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol
- EFS
- electrical field stimulation
- FDB
- flexor digitorum brevis
- OXPHOS
- oxidative phosphorylation
- RNS
- reactive nitrogen species
- ACSL1
- long-chain-fatty-acid-CoA ligase 1
- FA
- fatty acid
- p
- phospho
- IP
- immunoprecipitation.
References
- 1. Lee C. S., Dagnino-Acosta A., Yarotskyy V., Hanna A., Lyfenko A., Knoblauch M., Georgiou D. K., Poché R. A., Swank M. W., Long C., Ismailov I. I., Lanner J., Tran T., Dong K., Rodney G. G., Dickinson M. E., Beeton C., Zhang P., Dirksen R. T., Hamilton S. L. (2015) Ca2+ permeation and/or binding to CaV1.1 fine-tunes skeletal muscle Ca2+ signaling to sustain muscle function. Skelet. Muscle 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bays H. E. (2009) “Sick fat,” metabolic disease, and atherosclerosis. Am. J. Med. 122, S26–S37 [DOI] [PubMed] [Google Scholar]
- 3. Giovannucci E. (2007) Metabolic syndrome, hyperinsulinemia, and colon cancer: a review. Am. J. Clin. Nutr. 86, s836–s842 [DOI] [PubMed] [Google Scholar]
- 4. Xue F., Michels K. B. (2007) Diabetes, metabolic syndrome, and breast cancer: a review of the current evidence. Am. J. Clin. Nutr. 86, s823–S835 [DOI] [PubMed] [Google Scholar]
- 5. Whitmer R. A., Gustafson D. R., Barrett-Connor E., Haan M. N., Gunderson E. P., Yaffe K. (2008) Central obesity and increased risk of dementia more than three decades later. Neurology 71, 1057–1064 [DOI] [PubMed] [Google Scholar]
- 6. Haffner S. M. (2007) Abdominal adiposity and cardiometabolic risk: do we have all the answers? Am. J. Med. 120, Supplement 1, S10–S16; discussion S16–S17 [DOI] [PubMed] [Google Scholar]
- 7. Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R. B., Newgard C. B., Lopaschuk G. D., Muoio D. M. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56 [DOI] [PubMed] [Google Scholar]
- 8. Zurlo F., Larson K., Bogardus C., Ravussin E. (1990) Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Invest. 86, 1423–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patti M. E., Butte A. J., Crunkhorn S., Cusi K., Berria R., Kashyap S., Miyazaki Y., Kohane I., Costello M., Saccone R., Landaker E. J., Goldfine A. B., Mun E., DeFronzo R., Finlayson J., Kahn C. R., Mandarino L. J. (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. U.S.A. 100, 8466–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Petersen K. F., Shulman G. I. (2006) Etiology of insulin resistance. Am. J. Med. 119, S10–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith B. K., Bonen A., Holloway G. P. (2012) A dual mechanism of action for skeletal muscle FAT/CD36 during exercise. Exerc. Sport Sci. Rev. 40, 211–217 [DOI] [PubMed] [Google Scholar]
- 12. Yoshida Y., Jain S. S., McFarlan J. T., Snook L. A., Chabowski A., Bonen A. (2013) Exercise- and training-induced upregulation of skeletal muscle fatty acid oxidation are not solely dependent on mitochondrial machinery and biogenesis. J. Physiol. 591, 4415–4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jing E., O'Neill B. T., Rardin M. J., Kleinridders A., Ilkeyeva O. R., Ussar S., Bain J. R., Lee K. Y., Verdin E. M., Newgard C. B., Gibson B. W., Kahn C. R. (2013) Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62, 3404–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chandler W. K., Rakowski R. F., Schneider M. F. (1976) A non-linear voltage dependent charge movement in frog skeletal muscle. J. Physiol. 254, 245–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schredelseker J., Shrivastav M., Dayal A., Grabner M. (2010) Non-Ca2+-conducting Ca2+ channels in fish skeletal muscle excitation-contraction coupling. Proc. Natl. Acad. Sci. U.S.A. 107, 5658–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saha P. K., Kojima H., Martinez-Botas J., Sunehag A. L., Chan L. (2004) Metabolic adaptations in the absence of perilipin: increased β-oxidation and decreased hepatic glucose production associated with peripheral insulin resistance but normal glucose tolerance in perilipin-null mice. J. Biol. Chem. 279, 35150–35158 [DOI] [PubMed] [Google Scholar]
- 17. Mitsuhashi S., Hatakeyama H., Karahashi M., Koumura T., Nonaka I., Hayashi Y. K., Noguchi S., Sher R. B., Nakagawa Y., Manfredi G., Goto Y., Cox G. A., Nishino I. (2011) Muscle choline kinase β defect causes mitochondrial dysfunction and increased mitophagy. Hum. Mol. Genet. 20, 3841–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perry S. W., Norman J. P., Barbieri J., Brown E. B., Gelbard H. A. (2011) Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 50, 98–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Isaeva E. V., Shirokova N. (2003) Metabolic regulation of Ca2+ release in permeabilized mammalian skeletal muscle fibres. J. Physiol. 547, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tominaga H., Katoh H., Odagiri K., Takeuchi Y., Kawashima H., Saotome M., Urushida T., Satoh H., Hayashi H. (2008) Different effects of palmitoyl-l-carnitine and palmitoyl-CoA on mitochondrial function in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 295, H105–H112 [DOI] [PubMed] [Google Scholar]
- 21. Tschöp M. H., Speakman J. R., Arch J. R. S., Auwerx J., Brüning J. C., Chan L., Eckel R. H., Farese R. V. Jr., Galgani J. E., Hambly C., Herman M. A., Horvath T. L., Kahn B. B., Kozma S. C., Maratos-Flier E., Müller T. D., Münzberg H., Pfluger P. T., Plum L., Reitman M. L., Rahmouni K., Shulman G. I., Thomas G., Kahn C. R., Ravussin E. (2012) A guide to analysis of mouse energy metabolism. Nat. Methods 9, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Egan B., Zierath J. R. (2013) Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 17, 162–184 [DOI] [PubMed] [Google Scholar]
- 23. Bal N. C., Maurya S. K., Sopariwala D. H., Sahoo S. K., Gupta S. C., Shaikh S. A., Pant M., Rowland L. A., Bombardier E., Goonasekera S. A., Tupling A. R., Molkentin J. D., Periasamy M. (2012) Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat. Med. 18, 1575–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heo J. M., Rutter J. (2011) Ubiquitin-dependent mitochondrial protein degradation. Int. J. Biochem. Cell Biol. 43, 1422–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karbowski M., Youle R. J. (2011) Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 23, 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Reilly C. M., Fogarty K. E., Drummond R. M., Tuft R. A., Walsh J. V. (2003) Quantitative analysis of spontaneous mitochondrial depolarizations. Biophys. J. 85, 3350–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim S., Wing S. S., Ponka P. (2004) S-Nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell. Biol. 24, 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kwak Y. D., Ma T., Diao S., Zhang X., Chen Y., Hsu J., Lipton S. A., Masliah E., Xu H., Liao F. F. (2010) NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol. Neurodegener. 5, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Azad N., Vallyathan V., Wang L., Tantishaiyakul V., Stehlik C., Leonard S. S., Rojanasakul Y. (2006) S-Nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation: a novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 281, 34124–34134 [DOI] [PubMed] [Google Scholar]
- 30. Balon T. W., Nadler J. L. (1997) Evidence that nitric oxide increases glucose transport in skeletal muscle. J. Appl. Physiol. 82, 359–363 [DOI] [PubMed] [Google Scholar]
- 31. Tidball J. G., Lavergne E., Lau K. S., Spencer M. J., Stull J. T., Wehling M. (1998) Mechanical loading regulates NOS expression and activity in developing and adult skeletal muscle. Am. J. Physiol. 275, C260–C266 [DOI] [PubMed] [Google Scholar]
- 32. Nathan C., Xie Q.-w. (1994) Nitric oxide synthases: roles, tolls, and controls. Cell 78, 915–918 [DOI] [PubMed] [Google Scholar]
- 33. Fleming I., Fisslthaler B., Dimmeler S., Kemp B. E., Busse R. (2001) Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 88, E68–E75 [DOI] [PubMed] [Google Scholar]
- 34. Agostino P. V., Ferreyra G. A., Murad A. D., Watanabe Y., Golombek D. A. (2004) Diurnal, circadian and photic regulation of calcium/calmodulin-dependent kinase II and neuronal nitric oxide synthase in the hamster suprachiasmatic nuclei. Neurochem. Int. 44, 617–625 [DOI] [PubMed] [Google Scholar]
- 35. Nisoli E., Carruba M. O. (2006) Nitric oxide and mitochondrial biogenesis. J. Cell Sci. 119, 2855–2862 [DOI] [PubMed] [Google Scholar]
- 36. Smith B. K., Jain S. S., Rimbaud S., Dam A., Quadrilatero J., Ventura-Clapier R., Bonen A., Holloway G. P. (2011) FAT/CD36 is located on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem. J. 437, 125–134 [DOI] [PubMed] [Google Scholar]
- 37. Eaton S. (2002) Control of mitochondrial β-oxidation flux. Prog. Lipid Res. 41, 197–239 [DOI] [PubMed] [Google Scholar]
- 38. Jeppesen J., Albers P. H., Rose A. J., Birk J. B., Schjerling P., Dzamko N., Steinberg G. R., Kiens B. (2011) Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J. Lipid Res. 52, 699–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sebastián D., Guitart M., García-Martínez C., Mauvezin C., Orellana-Gavaldà J. M., Serra D., Gómez-Foix A. M., Hegardt F. G., Asins G. (2009) Novel role of FATP1 in mitochondrial fatty acid oxidation in skeletal muscle cells. J. Lipid Res. 50, 1789–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holloway G. P., Luiken J. J. F. P., Glatz J. F. C., Spriet L. L., Bonen A. (2008) Contribution of FAT/CD36 to the regulation of skeletal muscle fatty acid oxidation: an overview. Acta Physiol. (Oxf.) 194, 293–309 [DOI] [PubMed] [Google Scholar]
- 41. Coe N. R., Smith A. J., Frohnert B. I., Watkins P. A., Bernlohr D. A. (1999) The fatty acid transport protein (FATP1) is a very long chain acyl-CoA synthetase. J. Biol. Chem. 274, 36300–36304 [DOI] [PubMed] [Google Scholar]
- 42. Jeppesen J., Mogensen M., Prats C., Sahlin K., Madsen K., Kiens B. (2010) FAT/CD36 is localized in sarcolemma and in vesicle-like structures in subsarcolemma regions but not in mitochondria. J. Lipid Res. 51, 1504–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stefanyk L. E., Bonen A., Dyck D. J. (2012) Insulin and contraction-induced movement of fatty acid transport proteins to skeletal muscle transverse-tubules is distinctly different than to the sarcolemma. Metabolism. 61, 1518–1522 [DOI] [PubMed] [Google Scholar]
- 44. Lee K., Kerner J., Hoppel C. L. (2011) Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 286, 25655–25662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Samovski D., Sun J., Pietka T., Gross R. W., Eckel R. H., Su X., Stahl P. D., Abumrad N. A. (2015) Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes 64, 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Park S. H., Gammon S. R., Knippers J. D., Paulsen S. R., Rubink D. S., Winder W. W. (2002) Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J. Appl. Physiol. 92, 2475–2482 [DOI] [PubMed] [Google Scholar]
- 47. Saggerson D. (2008) Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 28, 253–272 [DOI] [PubMed] [Google Scholar]
- 48. Aguer C., Foretz M., Lantier L., Hebrard S., Viollet B., Mercier J., Kitzmann M. (2011) Increased FAT/CD36 cycling and lipid accumulation in myotubes derived from obese type 2 diabetic patients. PLoS One 6, e28981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Su X., Abumrad N. A. (2009) Cellular fatty acid uptake: a pathway under construction. Trends Endocrinol. Metab. 20, 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Steinbusch L. K., Schwenk R. W., Ouwens D. M., Diamant M., Glatz J. F., Luiken J. J. (2011) Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell. Mol. Life Sci. 68, 2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]