

Background: The fungus Aspergillus fumigatus causes human diseases that are treated with CYP51 azole inhibitors.
Results: We report crystal structures of A. fumigatus CYP51B complexes with two inhibitors, voriconazole and VNI.
Conclusion: The structures reveal fungus-specific features not observed in CYP51 enzymes from other biological kingdoms.
Significance: This molecular insight facilitates rational design of novel antifungals without inhibition of human enzymes.
Keywords: Aspergillus, cytochrome P450, drug design, enzyme inhibitor, sterol, x-ray crystallography, antifungal drugs, sterol 14alpha-demethylase (CYP51)
Abstract
Aspergillus fumigatus is the opportunistic fungal pathogen that predominantly affects the immunocompromised population and causes 600,000 deaths/year. The cytochrome P450 51 (CYP51) inhibitor voriconazole is currently the drug of choice, yet the treatment efficiency remains low, calling for rational development of more efficient agents. A. fumigatus has two CYP51 genes, CYP51A and CYP51B, which share 59% amino acid sequence identity. CYP51B is expressed constitutively, whereas gene CYP51A is reported to be inducible. We expressed, purified, and characterized A. fumigatus CYP51B, including determination of its substrate preferences, catalytic parameters, inhibition, and x-ray structure in complexes with voriconazole and the experimental inhibitor (R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide (VNI). The enzyme demethylated its natural substrate eburicol and the plant CYP51 substrate obtusifoliol at steady-state rates of 17 and 16 min−1, respectively, but did not metabolize lanosterol, and the topical antifungal drug miconazole was the strongest inhibitor that we identified. The x-ray crystal structures displayed high overall similarity of A. fumigatus CYP51B to CYP51 orthologs from other biological kingdoms but revealed phylum-specific differences relevant to enzyme catalysis and inhibition. The complex with voriconazole provides an explanation for the potency of this relatively small molecule, whereas the complex with VNI outlines a direction for further enhancement of the efficiency of this new inhibitory scaffold to treat humans afflicted with filamentous fungal infections.
Introduction
Fungal diseases represent an increasing global health burden and presently affect ∼1.2 billion people. The severity of these diseases ranges from topical superficial infections of skin, hair, and nails, which are relatively straightforward to cure (e.g. athlete's foot, ringworm of the scalp) to invasive infections that kill 1.5–2.0 million people annually, surpassing the mortality from tuberculosis or malaria (1). Aspergillus, Candida, and Cryptococcus are the three fungal genera responsible for most of this mortality, the incidence of which is growing steadily, particularly in clinically ill and immunocompromised populations (1–6).
Aspergillus is a genus of soil-dwelling saprophytic molds (filamentous fungi), consisting of ∼200 species. These molds are found throughout the world and are the most common type of fungi in the environment (see the Aspergillus Website). About 16 species of Aspergillus are known to cause disease in humans, Aspergillus fumigatus being responsible for >90% (7, 8). In immunocompetent patients, A. fumigatus can be involved in chronic pulmonary aspergillosis, also known as aspergilloma or “fungal ball,” which is a gradually destructive disease in the lung and often associated with tuberculosis, pulmonary emphysema, and sarcoidosis (>3 million people are estimated to be affected). A. fumigatus is also a ubiquitous aeroallergen. Severe asthma related to fungal sensitization affects up to 12 million people worldwide, and 100,000 people die from asthma annually. About 5 million people have allergic bronchopulmonary aspergillosis, whereas ∼12 million people are afflicted with fungal rhinosinusitis (9, 10). In immunocompromised individuals (cancer chemotherapy patients, those on steroids, solid organ and bone marrow transplant recipients, HIV/AIDS patients, and many others), A. fumigatus often manifests as invasive aspergillosis, the most dangerous form of infection, which spreads to multiple organs, is difficult to treat, and leads to ∼600,000 deaths annually. The treatment options are still very scarce. Overall, invasive aspergillosis has a ∼50% mortality rate if diagnosed and treated early, but if diagnosis is missed or delayed, then it is nearly 100% fatal (9).
Voriconazole remains the agent of choice for treatment (1), although the success rate is not particularly high, and the adverse side effects (visual disturbances, skin rushes, hepatotoxicity, vomiting, abdominal pain, etc.) require permanent therapeutic drug monitoring (11, 12). Other medications used clinically include itraconazole, posaconazole, amphotericin B, or caspofungin and micafungin (combination therapy), but aspergillosis is known to be insensitive to fluconazole (see the Aspergillus Website).
Voriconazole, posaconazole, itraconazole, and fluconazole (Fig. 1A) are inhibitors of sterol 14α-demethylase (CYP51, EC 1.14.13.70), the cytochrome P450 (CYP)2 enzyme that catalyzes the three-step reaction of oxidative removal of the 14α-methyl group from cyclized sterol precursors (Fig. 1, B and C) (13). The CYP51 reaction is required for sterol biosynthesis and therefore is conserved across biological kingdoms (14). Fungal CYP51 enzymes participate in the biosynthesis of ergosterol, which is an indispensable component of fungal membranes. Ergosterol depletion affects both the structure of the membrane and several of its functions. Ergosterol also has a hormone-like (“sparking”) role in fungal cells, which stimulates growth and proliferation. This function, however, may be disrupted only when ergosterol depletion is virtually complete (15). Azole-, pyridine-, or pyrimidine-based compounds block ergosterol biosynthesis and cause accumulation of the 14α-methylated precursors (16, 17). These drugs act as competitive CYP51 inhibitors that occupy the P450 active site, preventing substrate binding and oxidation. The basic heterocyclic nitrogen coordinates to the P450 heme iron (Fig. 1A), sharing its lone pair of electrons and blocking binding of molecular oxygen, whereas the non-ligated portion of the inhibitor molecule forms multiple contacts with the protein moiety, shaping the protein-ligand surface interface that largely defines the strength of the inhibition (18, 19).
FIGURE 1.

CYP51 inhibitors and catalytic reaction. A, structural formulas of the marketed drugs, 1,2,4-triazoles voriconazole (Vor), fluconazole (Fluc), and posaconazole (Poz), and an experimental CYP51 inhibitor, the 1,3-imidazole VNI. The heme-coordinating nitrogen atoms are circled. B, 14α-demethylation of eburicol. The reaction involves three consecutive P450 catalytic cycles, each consuming two electrons (provided by CPR, plus two protons) to reduce the catalytic heme iron and activate the molecular oxygen, resulting in the insertion of one oxygen atom into the substrate and reduction of the other oxygen atom to water. In this reaction, the sterol 14α-methyl group is first converted into the 14α-alcohol and then into the 14α-aldehyde (CHO-) intermediate and finally released as formic acid. C, HPLC profile of sterols extracted after a 5-min reaction of A. fumigatus CYP51B with eburicol (37 °C; P450, CPR, and eburicol concentrations were 0.5, 1, and 25 μm, respectively).
Because fungal CYP51 enzymes are very hydrophobic membrane-bound proteins (a feature that complicates their handling and assay in vitro), data on structure-function relationships and inhibition are limited and, when they exist, often controversial (20). As a result, although azoles represent the leading class of antimycotic agents both in medicine and agriculture (15, 21), each new compound has to be discovered and developed empirically by monitoring its action on fungal cell growth. Thus, 1200 fluconazole analogs were synthesized and tested at Pfizer before voriconazole was discovered, an inefficient and costly process (1).
Drug resistance (both intrinsic and acquired) represents another serious problem. Different mechanisms have been proposed, including activation of azole efflux pumps, CYP51 mutations/CYP51 gene overexpression, and the combination of pumps and P450 (reviewed in Refs. 22 and 23). Also, it has been suggested that A. fumigatus resistance to clinically used azoles can be acquired through long time use in the environment (24). Yeast, human, and other vertebrate genomes contain only one CYP51 gene; however, A. fumigatus and some other filamentous ascomycetes (25) have two CYP51 paralogs (CYP51A and CYP51B; Fig. 2), each present as a single copy (26) (chromosomes 4 and 7, respectively). Although both genes had been suggested to be active in A. fumigatus, it was later reported that the gene CYP51B encodes the enzyme primarily responsible for sterol 14α-demethylation. The CYP51B gene is expressed constitutively and found in all sequenced filamentous fungi, whereas the CYP51A gene appears in some fungal lineages (22, 25). The presence of two CYP51 genes implies a possibility for faster sterol biosynthesis in A. fumigatus as one of the reasons for high resistance of the pathogen to treatment.
FIGURE 2.

Sequence alignment of CYP51 proteins from A. fumigatus (A.fuB and A.fuA), human, and a protozoan pathogen, T. brucei (T.bru). The alignment was generated in ClustalW and processed in ESPript to add secondary structure information on A.fuB (top) and T.bru (bottom), using molecules A of Protein Data Bank files 4UYL and 3GW9, respectively. The amino acid sequence identity between A.fuB and A.fuA is 59%. Within the Aspergillus genus, CYP51B identities range from 78% (Aspergillus niger) to 99% (Aspergillus fischerianus), whereas CYP51A identities range from 69% (Aspergillus nidulans) to 96% (A. fischerianus) (not shown). The identities between A.fuB versus human and A.fuB versus T.bru CYP51 amino acids are 33 and 23%, respectively. The alignment shows that, regardless of low amino acid sequence identity, the length and location of the secondary structural elements in A.fuB and T.bru CYP51s match very well, except for the FG arm segment, which is longer in T.bru (in green), and the additional β-bundle in A.fuB (strands β5-1 and β5-2, in blue); this segment appears to be specific to fungal CYP51 and so far has not been seen in the structures of CYPs from any other families. The CYP51-specific helical turn in the SRS1 region (ηB″) is shown in brown. The heme-coordinated cysteine and the conserved CYP51-specific histidine (proton delivery, helix I) are colored in yellow; the five residues bonded with porphyrin propionates are marked with asterisks.
We report the expression and purification of A. fumigatus CYP51B and its substrate preferences, catalytic parameters, inhibition, and x-ray structures in complexes with voriconazole and (R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide (VNI), an experimental inhibitor of protozoan CYP51 enzymes. Overall, A. fumigatus CYP51B has a strong requirement for the C24-methylene group in sterols (eburicol and obtusifoliol) and has a relatively high catalytic efficiency but is rather unstable, particularly in the reduced ligand-free form. The x-ray structures confirm high three-dimensional similarity as the molecular basis for the CYP51 catalytic conservation across phyla, but some structural features appear to be fungus-specific and therefore potentially useful in antifungal drug development involving rational structure-based design of novel drugs.
Experimental Procedures
Chemicals
VNI was synthesized by the Chemical Synthesis Core Facility (Vanderbilt Institute of Chemical Biology) (27). Voriconazole, ketoconazole, itraconazole, and posaconazole were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX); fluconazole, clotrimazole, and miconazole were from ICN Biomedicals. Hydroxypropyl-β-cyclodextrin (HPCD) was purchased from Cyclodextrin Technology Development (Gainesville, FL). DEAE- and CM-Sepharose were from GE Healthcare, and Ni2+-nitrilotriacetate (NTA)-agarose was purchased from Qiagen.
Proteins
Trypanosoma brucei and Candida albicans CYP51 and T. brucei and rat NADPH-cytochrome P450 reductase (CPR) were expressed in Escherichia coli and purified as described previously (28). The A. fumigatus CYP51B gene (strain Af293), commercially synthesized by GeneCust Europe (Dudelange, Luxembourg) in pUC57 (20), was a gift from Dr. D. Kelly (University of Swansea). The gene encoding the full-length CYP51B protein (UniProt protein accession number Q96W81, 524 amino acid residues plus the His6 tag at the C terminus, 59,800 Da) was used for functional studies, including ligand binding, enzymatic activity, and inhibition. For crystallization purposes, the protein was truncated (477 amino acid residues, 54,000 Da) as follows. The 49-amino acid membrane anchor sequence at the N terminus (up to the conserved CYP51 proline Pro50 in A. fumigatus CYP51B) was replaced with the 5-amino acid sequence fragment MAKKT- (29), and 6 amino acid residues at the C terminus (-ESATKA) were removed, using the upstream primer 5′-CGCATATGGCTAAGAAAACCCCGCCGGTTGTTTTTCATTGGTTCCCG-3′ and the downstream primer 5′-CGCAAGCTTCTAGTGATGGTGATGGTGATGACGTTTTTCGAACTCGACG-3′. The PCR included 50 ng of genomic DNA, 1 μm each of the forward and reverse primers, and 0.5 μl of FailSafe PCR enzyme (Epicenter Biotechnologies, Madison, WI) in a final volume of 25 μl. FailSafe PCR 2× Premix E (25 μl) was added, and amplification was carried out by denaturation at 95 °C for 2 min, followed by 28 cycles of denaturation at 95 °C for 60 s, annealing at 52 °C for 30 s, and extension at 72 °C for 140 s. Terminal extension for 2 min at 72 °C completed the reaction. The products were purified from an agarose gel and subcloned into the pGEM-T Easy vector (Promega). The correctness of the insert was confirmed by nucleotide sequence analysis. For protein expression, both the full-length and truncated CYP51B genes were subcloned into the pCW expression vector using the NdeI (5′) and HindIII (3′) sites.
Expression of A. fumigatus CYP51B
E. coli HMS174 (DE3) (Novagen) competent cells were transformed with 100 ng of the A. fumigatus CYP51B plasmid (see above). A single colony of bacteria was used to inoculate 5 ml of LB medium containing 0.1 mg/ml ampicillin. An overnight culture was incubated at 37 °C and 250 rpm for 16 h (C25KC incubator, New Brunswick Scientific, Edison, NJ) and then diluted into 500 ml of Terrific Broth medium supplemented with 100 mm potassium phosphate buffer (pH 7.2) containing 0.1 mg/ml ampicillin and 125 μl of trace element salt solution (30). After incubation at 37 °C and 250 rpm for 5 h, the flasks were cooled to 26 °C, and the cultures were supplemented with 1 mm δ-aminolevulinic acid, 1 mm isopropyl 1-thio-β-d-galactopyranoside, and 0.05 mg/ml ampicillin (final concentrations) and incubated for 42 h at 26 °C and 180 rpm. The bacterial cells were harvested by centrifugation at 2851 × g for 20 min (Allegra X-15R centrifuge, Beckman Coulter). The pellet was resuspended in 100 mm Tris acetate buffer (pH 7.2) containing 0.5 mm EDTA and 250 mm sucrose. Lysozyme (1.25 mg/ml) was added, and the suspension was placed on ice for 10 min and then centrifuged at 4 °C and 2100 × g for 20 min to collect the pellet.
Purification of A. fumigatus CYP51B
All purification steps were done at 4 °C, and all buffers contained 0.1 mm phenylmethylsulfonyl fluoride and 0.1 mm dithiothreitol, which were added fresh daily. The pellet was homogenized in 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl, 0.5 mm EDTA, 10% glycerol (v/v), and 0.2% Triton X-100 (v/v) (20% glycerol (v/v) and 0.4% Triton X-100 (v/v) in the case of the full-length protein). The suspension was sonicated on ice (Sonic Dismembrator model 500, Fisher) and stirred at 4 °C for 4 h. The solubilized protein was separated from the insoluble material by centrifugation at 82,000 × g for 40 min (Optima L-80 ultracentrifuge, Beckman). The supernatant was frozen in liquid nitrogen and stored at −80 °C.
For enzymatic experiments, full-length A. fumigatus CYP51B was purified by affinity chromatography using Ni2+-NTA-agarose, followed by dialysis. The supernatant was thawed and applied to the Ni2+-NTA column equilibrated with 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl, 20% glycerol (v/v), 5 mm imidazole, and 0.4% Triton X-100 (v/v). The column was washed with 10 bed volumes of equilibration buffer and then with 50 bed volumes of 50 mm potassium phosphate buffer (pH 7.2) containing 500 mm NaCl, 20% glycerol (v/v), 20 mm imidazole, and 0.2% Triton X-100 (v/v). The protein was eluted with the same buffer containing 150 mm imidazole, concentrated to about 50 μm, dialyzed against 50 mm potassium phosphate buffer (pH 7.2) containing 500 mm NaCl, 20% glycerol (v/v), 0.1 mm EDTA, and 0.2% Triton X-100 (v/v) to remove imidazole, aliquoted, frozen in liquid nitrogen, and stored at −80 °C until use. The yield was about 100 nmol/liter of culture.
For crystallographic experiments, the truncated A. fumigatus CYP51B was purified in three steps, including anion exchange chromatography on DEAE-Sepharose, affinity chromatography on Ni2+-NTA-agarose, and cation exchange chromatography on CM-Sepharose. The thawed supernatant was diluted 2-fold with 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl, 10% glycerol (v/v), and 5 mm imidazole and applied to columns of DEAE-Sepharose and Ni2+-NTA-agarose linked in tandem, equilibrated with 50 mm potassium phosphate buffer (pH 7.2) containing 100 mm NaCl, 10% glycerol (v/v), 5 mm imidazole, and 0.1% Triton X-100 (v/v). The P450 did not bind to the DEAE column and was concentrated on the Ni2+-NTA column; the columns were disconnected, and the (NTA-) bound protein was washed with 10 bed volumes of equilibration buffer and then with 50 mm potassium phosphate buffer (pH 7.2) containing 500 mm NaCl, 10% glycerol (v/v), and 10 mm imidazole until the Triton X-100 was eliminated (as judged by A280 measurements). The P450 was eluted with a linear gradient of imidazole (20–150 mm), and the fractions with a spectrophotometric index (A425/A280) ≥ 1 were pooled and concentrated using an Amicon Ultra 50 K (Millipore) concentration device to a volume of 2–4 ml. At this stage, A. fumigatus CYP51B was co-purified with an inhibitor (VNI or voriconazole) as follows. The concentrated protein was diluted 10-fold with 20 mm potassium phosphate buffer (pH 7.2) containing 10% glycerol (v/v), 0.1 mm EDTA, and 10 μm inhibitor (CM-buffer), incubated for 30 min, and applied to a CM-Sepharose column (5-ml bed volume) equilibrated with CM-buffer containing 50 mm NaCl. The column was washed with 5 bed volumes of equilibration buffer and then 40 bed volumes of CM-buffer with an increasing linear gradient of NaCl (50–200 mm). The protein was eluted with CM-buffer containing 350 mm NaCl, pooled, concentrated to about 500 μm using an Amicon Ultra 50 K concentration device, aliquoted, frozen in liquid nitrogen, and stored at −80 °C until use. The yield was between 200 and 300 nmol/liter of culture. The purity was verified by SDS-PAGE.
Spectroscopic Measurements and Ligand Binding Assays
UV-visible absorption spectra were recorded using a dual-beam Shimadzu UV-2401PC spectrophotometer in 50 mm potassium phosphate buffer (pH 7.2) containing 10% glycerol (v/v) and 0.1% Triton X-100 (v/v). P450 concentrations were estimated from the Soret band intensity using ϵ417 = 117 mm−1 cm−1 for the low spin ferric form of the protein or Δϵ450–490 = 91 mm−1 cm−1 for the reduced carbon monoxide difference spectra (30, 31). The spin state of the P450 samples was estimated from the absolute absorbance spectra as the ratio (ΔA393 − A470/ΔA418 − A470), the values 0.4 and 2.0 corresponding to 100% low spin and 100% high spin iron, respectively (32).
To record the CO-binding spectra, chemical reduction was carried out with Na2S2O4, either in the presence of a 5-fold molar excess of substrate (two cuvettes, Na2S2O4 added before CO was introduced into the sample cuvette) or in the absence of substrate (single cuvette, Na2S2O4 added after CO was introduced into the cuvette; e.g. Fig. 3A). Enzymatic reduction with NADPH was performed at a 1:2:10 molar ratio (P450/CPR/substrate, two cuvettes, 100-fold molar excess of NADPH added before CO was passed through the sample cuvette).
FIGURE 3.
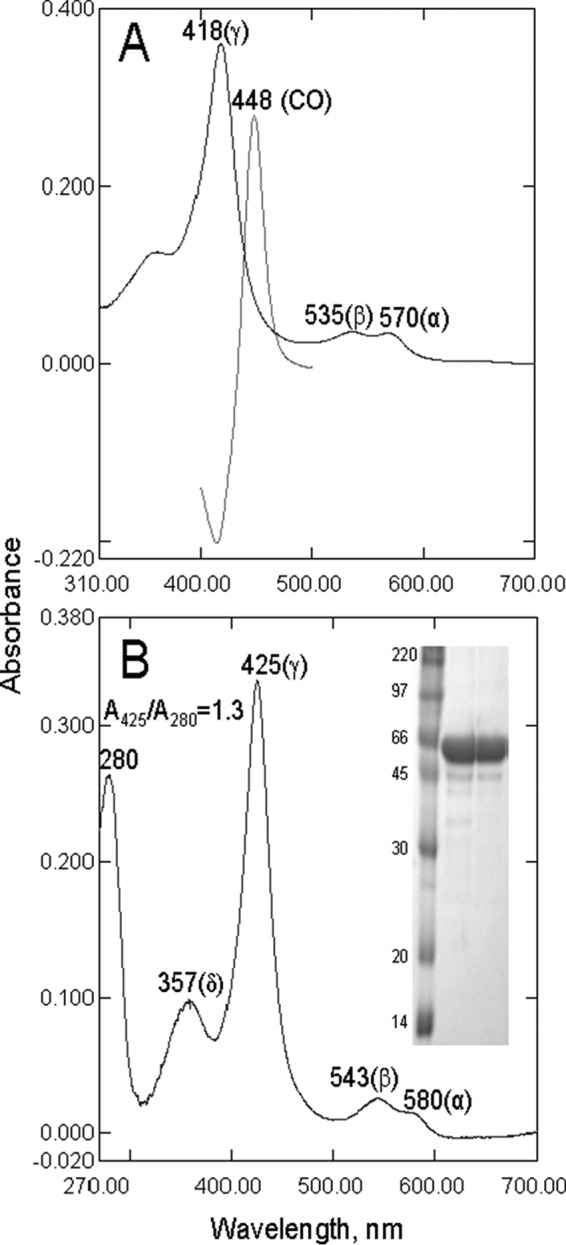
Spectral characteristics of A. fumigatus CYP51B. A, ligand-free full-length protein. Shown are the absolute absorbance spectrum of the ferric (Fe3+) state (Soret band maximum at 418 nm) and the difference absorbance spectrum of the reduced CO-bound state (Soret band maximum at 448 nm). The P450 concentration was 3.3 μm, and the ratio (ΔA393 − A470)/ΔA418 − A470) was 0.40. B, truncated protein co-purified with VNI and used for crystallization. Shown is the absolute absorbance spectrum; spectrophotometric index A425/A280 = 1.3. The P450 concentration was 2.8 μm. Inset, 12% (w/v) SDS-PAGE electrophoretogram. Left lane, rainbow marker; middle and right lanes, P450 after Ni2+-NTA-agarose and CM-Sepharose chromatography, respectively (54,000 Da).
Substrate binding was monitored as a “Type I” spectral response reflecting low to high spin transition of the ferric P450 heme iron as a result of displacement of the heme-coordinated water molecule (blue shift in the Soret band maximum from 418 to 393 nm) (33). Various aliquots of sterols (dissolved in 45% (w/v) HPCD) (28) were added to the sample cuvette (1-cm optical path length), and the same volume of HPCD was added to the reference cuvette. The P450 concentration in these experiments was 5 μm. The apparent dissociation constants of the enzyme-substrate complex (Kd) were calculated in Prism version 6 (GraphPad Software, La Jolla, CA) by fitting the data for the substrate-induced absorbance changes in the difference spectra Δ(A390 − A420) versus substrate concentration to a one-site total binding equation (binding-saturation). Correction was made for the equilibrium with HPCD by fitting to two equations, the equilibrium expressions for binding of the sterols to both the P450 and HPCD (34, 35), in the program DynaFit (36), with the assumption that the Kd value for cholesterol is similar to those of the sterols examined here.
Titration with azoles was carried out at 1 μm P450 concentration in 5-cm optical path length cuvettes, with inhibitor binding being monitored as a “Type II” spectral response reflecting coordination of the heterocyclic nitrogen to the P450 heme iron (red shift in the Soret band maximum from 418 to 421–427 nm) (27, 33). Difference spectra were generated by recording the P450 absorbance in a sample cuvette versus the absorbance in a reference cuvette, both containing the same amount of the protein. Aliquots of azoles (dissolved in (CH3)2SO) were added to the sample cuvette in the concentration range 0.1–2.0 μm, with each titration step being 0.1 μm. At each step, the corresponding volume of (CH3)2SO was added to the reference cuvette. The apparent dissociation constants of the enzyme-ligand complex (Kd) were calculated in GraphPad Prism version 6 by fitting the data for the ligand-induced absorbance changes in the difference spectra Δ(Amax − Amin) versus ligand concentration to quadratic Equation 1 (tight binding ligands) (18),
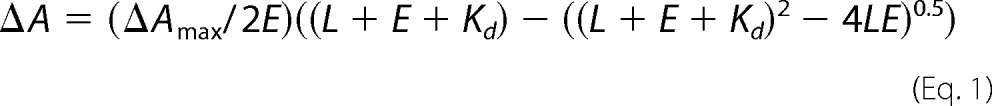 |
where L and E represent the total concentrations of ligand and enzyme used for the titration, respectively.
Reconstitution of Catalytic Activity, Kinetic Analysis, and CYP51 Inhibition Assays
The standard reaction mixture (28) contained 0.5 μm A. fumigatus CYP51B and 1.0 μm rat CPR, 100 μm l-α-1,2-dilauroyl-sn-glycerophosphocholine, 0.4 mg/ml isocitrate dehydrogenase, and 25 mm sodium isocitrate in 50 mm potassium phosphate buffer (pH 7.2) containing 10% glycerol (v/v) and 0.1% Triton X-100 (v/v). After the addition of the radiolabeled (3-3H) sterol substrates (eburicol, lanosterol, obtusifoliol, or C4-norlanosterol, ∼4,000 dpm/nmol; dissolved in 45% (w/v) HPCD) (37), the mixture was preincubated for 30 s at 37 °C in a shaking water bath, and the reaction was initiated by the addition of 100 μm NADPH and stopped by extraction of the sterols with 5 ml of ethyl acetate. The extracted sterols were dried, dissolved in CH3OH, and analyzed by a reversed-phase HPLC system (Waters) equipped with a β-RAM detector (INUS Systems, Inc.) using a NovaPak octadecylsilane (C18) column (particle size 4 μm, 3.9 × 150 mm) and a linear gradient H2O/CH3CN/CH3OH (1.0:4.5:4.5, v/v/v) (solvent A) to CH3OH (solvent B), increasing from 0 to 100% B for 30 min at a flow rate of 1.0 ml/min. Time course experiments were carried out at 50 μm concentrations of sterol substrates. For steady-state kinetic analysis, the reactions were run for 60 s at 37 °C, and the sterol concentration range was 6–50 μm. Michaelis-Menten parameters were calculated using GraphPad Prism, with the reaction rates (nmol of product formed/nmol of P450/min) being plotted against total substrate concentration. As in the Kd work, correction was made for the equilibrium with HPCD by fitting to two equations (34, 35) in the program DynaFit (36) (see above). The inhibitory potencies of VNI, VFV, and antifungal azoles on A. fumigatus CYP51 activity were compared on the basis of decreases in substrate conversion in 60-min reactions (29, 38, 39) at a substrate/enzyme/inhibitor molar ratio of 50:1:2 (18, 40), with the reaction temperature being decreased to 28 °C.
Crystallization, Data Collection, Structure Determination, and Analysis
The initial screening of crystallization conditions was performed using Hampton Research crystallization kits. The crystals were obtained by the hanging drop vapor diffusion technique. Crystals of A. fumigatus CYP51B with VNI were grown at 18 °C. Equal volumes of complex solution preincubated with 24.5 mm n-octyl-β-d-glucoside and 5.8 mm tris(carboxyethyl)phosphine were mixed with mother liquor (15% PEG 4000 (w/v) and 0.2 m lithium acetate (pH 7.4) and equilibrated against the reservoir solution. Crystals of A. fumigatus CYP51B with voriconazole were grown at 16 °C by mixing equal volumes of the P450-voriconazole complex solution preincubated with 11.5 mm cyclohexylpentanoyl-N-hydroxyethylglucamide (Anagrade) and 5.8 mm tris(carboxyethyl)phosphine with 15% PEG 3350 (w/v) and 0.2 m lithium acetate (pH 7.5). In both cases, crystals appeared after several days and were cryoprotected by soaking them in mother liquor with 40% glycerol (v/v) and flash-cooled in liquid nitrogen. All data were collected on the 21-ID-F beamline of the Life Sciences Collaborative Access Team at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL) at a wavelength of 0.9787 Å and using a MAR225 CCD detector.
The diffraction images were integrated using Mosflm (41) and scaled with Aimless (CCP4 Program Suite version 6.3.0) (42) in the hexagonal P31 space group to maximum resolutions of 2.81 Å (VNI) and 2.55 Å (voriconazole). Solvent content was estimated with the Matthews probability calculator in the CCP4 suite (42). Both structures were determined by molecular replacement. The structure of the A. fumigatus CYP51B-VNI complex was solved in Phenix (43) using a poly-Ala chain of ligand-free T. brucei CYP51 (3G1Q) to calculate the initial phases and model building with autobuild. Iterative models of the protein-inhibitor complexes were then built with Coot (44) and refined with Refmac5 in the CCP4 suite (42). The structure of the A. fumigatus CYP51B-voriconazole complex was determined by molecular replacement in Phaser (45), using a complex of A. fumigatus CYP51B with VNI as the search model, and refined in Refmac5. Voriconazole was inserted in Coot. Data collection and refinement statistics are shown in Table 1. Both structures have two monomers in the asymmetric unit. The protein chain was seen from Lys50 (KT- in the N-terminal MAKKT- sequence) to His519 (C-terminal His tag). The electron density for both VNI and voriconazole (the ligand Protein Data Bank ID VOR) was well defined, showing a single orientation of the inhibitor molecules within the enzyme binding cavity and full occupancy. (The 2Fo − Fc electron density maps for the inhibitor-containing area are shown in Figs. 11 and 12; see below.) The coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 4UYL (A. fumigatus CYP51B-VNI) and 4UYM (A. fumigatus CYP51-voriconazole). The accession codes of other CYP51 structures discussed in this work are 3G1Q (ligand-free T. brucei), 3GW9 (T. brucei-VNI), 3K1O (T. cruzi-posaconazole), and 3LD6 (human-ketoconazole); the Protein Data Bank code of CYP46-voriconazole is 3MDT.
TABLE 1.
Data collection and refinement statistics
| Complex | A. fumigatus CYP51B-VNI | A. fumigatus CYP51B- voriconazole |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.9787 | 0.9787 |
| Space group | P31 | P31 |
| Cell dimensions | ||
| a, b, c (Å) | 110.498, 110.498, 90.479 | 109.193, 109.193, 90.221 |
| α, β, γ (degrees) | 90.00, 90.00, 120.00 | 90.00, 90.00, 120.00 |
| Molecules per asymmetric unit | 2 (A/B) | 2 (A/B) |
| Solvent content (%) | 57.6 | 57.2 |
| Resolution (last shell) (Å) | 30–2.81 (2.91–2.81) | 30–2.55 (2.64–2.55) |
| No. of reflections (last shell) | 28,394 (1526) | 36,885 (1939) |
| Rmerge (last shell) | 0.047 (0.627) | 0.051 (0.615) |
| I/σ (last shell) | 17.5 (2.1) | 18.9 (2.1) |
| Completeness (last shell) (%) | 99 (100) | 99.8 (100) |
| Redundancy (last shell) | 4.8 (4.4) | 3.9 (4.2) |
| Refinement | ||
| Rwork | 0.237 | 0.209 |
| Rfree | 0.279 | 0.239 |
| Root mean square deviations from ideal geometry | ||
| Bond lengths (Å) | 0.002 | 0.001 |
| Bond angles (degrees) | 1.21 | 1.03 |
| Ramachandran plot | ||
| Residues in favorable/allowed regions (%) | 96.6/100 | 96.9/100 |
| Outliers (%) | 0.0 | 0.3 |
| Wilson B-factor (Å) | 85.6 | 68.3 |
| No. of atoms (mean B-factor (Å)) | 7790 (92.8) | 7803 (78.6) |
| No. of residues per molecule (A/B) | ||
| Protein (mean B-factor (Å)) | 470 (83.0)/470 (105.1) | 470 (77.0)/470 (81.7) |
| Heme (mean B-factor (Å)) | 1 (66.7)/1 (90.1) | 1 (56.0)/1 (65.1) |
| Ligand (mean B-factor (Å)) | VNI 1 (83.5)/1(91.1) | Vor 1 (63.8)/1 (77.1) |
| Water (mean B-factor (Å)) | 128 (77.4) | 163 (71.4) |
| Protein Data Bank code | 4UYL | 4UYM |
FIGURE 11.

Voriconazole binding mode. A, view of the A. fumigatus CYP51B active site illustrating interactions of the hemoprotein with the inhibitor. The residues located within van der Waals contacts (<4.5 Å) with voriconazole are depicted as wire models and labeled; the carbon atoms are colored in gray; and three reference secondary structural elements are shown as semitransparent blue ribbon. The carbon atoms of voriconazole and the heme (stick representations) are blue and orange, respectively. The hydrogen bonds are shown as green dashed lines. B, 2Fo − Fc omit electron density map of the active site area around voriconazole contoured at 1.5σ. C, superimposition of voriconazole complexes with A. fumigatus CYP51B and with CYP46 (Protein Data Bank code 3MDT). The heme and the drug are shown in a stick representation; the carbon atoms are blue and gray in A. fumigatus CYP51B and CYP46, respectively.
FIGURE 12.

VNI binding mode. A, 2Fo − Fc omit electron density map of the active site area around VNI contoured at 1.3 σ. B, superimposition of the VNI complexes with A. fumigatus CYP51B and T. brucei CYP51 (Protein Data Bank code 3GW9). The heme and VNI are shown in a stick representation; the carbon atoms are magenta and gray in the A. fumigatus and T. brucei enzymes, respectively. The hydrogen bonds are presented as green dashed lines. Two segments of the T. brucei CYP51 molecule that are connected by the hydrogen bond network with VNI (helices B′ and I) are outlined as gray ribbons. C, view of the A. fumigatus CYP51B active site illustrating its interactions with VNI. The residues located within van der Waals distances (<4.5 Å) with VNI are depicted as wire models and labeled, and the carbon atoms are colored in gray. Three reference secondary structural elements are seen as a semitransparent pink ribbon. The carbon atoms of VNI and the heme (stick representations) are colored magenta and orange, respectively.
Structure superpositions were done in LSQkab of the CCP4 suite. The substrate was positioned in the active sites of the superimposed CYP51 enzymes to adopt an orientation similar to that of 14α-methylenecyclopropyl-Δ7-24,25-dihydrolanosterol (MCP) in complex with T. brucei CYP51 (Protein Data Bank code 3P99) (46) using Coot. Molecular volumes and surface areas were calculated in Accelrys Discovery Studio Visualizer verison 2.5 (probe radius 1.4 Å). Figures were prepared with Accelrys and Chimera. A molecular model of A. fumigatus CYP51A was built in Modeler (CCP4 suite).
A. fumigatus Cellular Growth Inhibition Assays
A. fumigatus strain af293 (47) was maintained on Sabouraud dextrose agar at 37 °C. Sensitivity tests were carried out using RPMI 1640 medium (Sigma-Aldrich) buffered with 165 mm MOPS, pH 7.0. The spore inoculum was prepared in sterile phosphate-buffered saline containing 0.05% Tween 80 (v/v), and dilutions in RPMI 1640 were made to 2 × 103/ml. Treatments with VNI, itraconazole, and voriconazole were done in triplicate for 48 h at 37 °C (150 rpm), and the minimal inhibitory concentration was defined as the concentration of drug that produced no growth, as described previously (48). (CH3)2SO was used in the control experiments. Each independent experiment was repeated in triplicate.
Results
Spectral Characterization
Similar to other CYP51 orthologs (28, 30, 37), A. fumigatus CYP51B was obtained in the ligand-free ferric low spin form after purification from E. coli (Fig. 3A). The heme iron was readily reduced by sodium dithionite, and the complex with CO was formed very rapidly, with an absorbance maximum at 448 nm and no presence of (inactive) cytochrome P420. The ferrous form, however, was highly unstable and, particularly in the ligand-free state, displayed a rapid loss of heme absorbance. The binding of CO or another ligand stabilized the hemoprotein (both ferrous and ferric forms); therefore, for crystallization purposes, the last step of purification was performed in the presence of an azole inhibitor, which afforded a stable spectrophotometric index (A425/A280) of ∼1.3 (Fig. 3A).
Enzymatic reduction of A. fumigatus CYP51B in the presence of the natural fungal CYP51 substrate eburicol was at least 50-fold faster and more efficient than when the protein was reduced in the ligand-free form (Fig. 4). Titration of A. fumigatus CYP51B with the substrate eburicol produced a characteristic Type I spectral response, with the peak, isosbestic point, and trough in the difference spectra at 390, 408, and 420 nm, respectively, and an ∼18% low to high spin transition in the heme iron (Fig. 5A). Titration with lanosterol (the major natural substrate of mammalian and yeast CYP51 enzymes) did not produce any spectral changes (Fig. 5B). The C4-monomethylated lanosterol analog C4-norlanosterol (a natural CYP51 substrate in T. brucei and Leishmania (30)) also did not induce any spectral response (data not shown), whereas the response to the plant CYP51 substrate obtusifoliol (∼14% low to high spin transition in the heme iron) was quite similar to the response to eburicol, with the apparent Kd being 1.7 versus 1.3 μm (Fig. 5C), respectively, thus implying that in order to be productively bound to A. fumigatus CYP51B, the sterol substrates must contain a methylene group at the C24 position and/or not a C24=C25 double bond. Finally, titration of A. fumigatus CYP51B with azoles induced typical Type II spectral responses. Two are shown in Fig. 6 as examples, with the apparent Kd values for VNI and voriconazole being ∼200 and 60 nm, respectively.
FIGURE 4.

Time course of enzymatic reduction (detected as the CO complex) of A. fumigatus CYP51B in the presence and in the absence of substrate (1.5 μm P450, 3.0 μm CPR, 150 μm NADPH). Optical path length was 1 cm. Insets, difference CO-binding spectra, Δt = 2 min.
FIGURE 5.

Spectral responses of A. fumigatus CYP51B to the addition of sterols. A, eburicol. B, lanosterol. C, obtusifoliol. Absolute (top) and difference (bottom) absorbance spectra are shown. S, sample; R, reference. P450 concentration was 5 μm, and optical path length was 1 cm. The titration curves (obtained using Equation 1) are shown in the insets, and the Kd values are corrected for cyclodextrin binding. The C24-methylene group appears to be required for the proper binding of sterol substrates to A. fumigatus CYP51B.
FIGURE 6.

Spectral response of A. fumigatus CYP51B to the addition of the heme-coordinating ligands VNI and voriconazole. Absolute (top) and difference (bottom) absorbance spectra are shown. The P450 concentration was 1.0 μm, and the optical path length was 5 cm. The titration curves (obtained using Equation 1) are shown in the inset.
Catalytic Activity, Steady-state Kinetic Parameters, Substrate Preferences, and Inhibition
Preliminary time course studies (Fig. 7A) indicated that, under the standard CYP51 reaction conditions (0.5 μm P450, 1 μm CPR, 37 °C), A. fumigatus CYP51B had a relatively high initial turnover rate but was unstable and rapidly inactivated (no P450 could be detected in the reaction mixture after a 10-min reaction). Steady-state kinetic parameters were measured using 60-s reactions to minimize the influence of P450 inactivation (Fig. 7B). Under these conditions, A. fumigatus CYP51B displayed a slight substrate preference toward eburicol over obtusifoliol (1.8-fold higher catalytic efficiency, due to the lower Km value (Table 2)). Longer reaction times and various reaction temperatures were employed with 14α-demethylation of lanosterol and C4-norlanosterol, but no conversion of these C24-demethyl sterols was observed.
FIGURE 7.
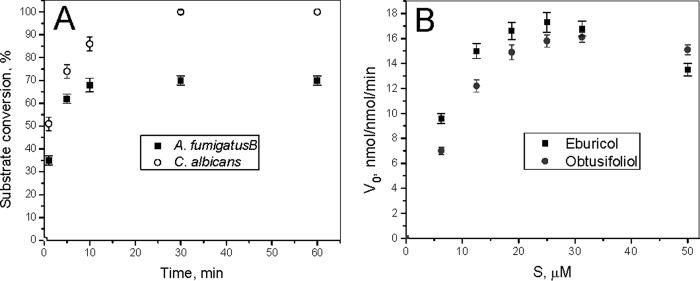
Enzymatic activity of A. fumigatus CYP51B. A, time course of substrate conversion at 37 °C (0.5 μm P450, 1.0 μm CPR, and 25 μm eburicol), shown in comparison with C. albicans CYP51. B, steady-state kinetics of A. fumigatus CYP51B with its natural substrate eburicol and obtusifoliol (a substrate for plant CYP51 enzymes). The P450 concentration was 0.5 μm (37 °C, 60-s reaction). The experiments were performed in triplicate, and results are presented as means ± S.E. (error bars).
TABLE 2.
Steady-state kinetic parameters for 14α-demethylation of eburicol and obtusifoliol by A. fumigatus CYP51B
| Sterol | Michaelis-Menten parameters |
||
|---|---|---|---|
| kcat | Kma | kcat/Km | |
| min−1 | μm | μm−1 min−1 | |
| Eburicol | 45 ± 3 | 1.8 ± 0.4 | 25 ± 5 |
| Lanosterol | —b | ||
| Obtusifoliol | 52 ± 5 | 3.7 ± 0.8 | 14 ± 4 |
| C4-norlanosterol | — | ||
a Km values are corrected for HPCD binding.
b —, the limit of detection was <0.05 min−1.
To compare the potencies of different inhibitors of A. fumigatus CYP51B, the reaction temperature was decreased to 28 °C to attenuate the rate of enzyme denaturation. We used a 60-min reaction (instead of a 1–5-min reaction) because a longer reaction time affords higher sensitivity in these assays (27, 38, 39). The initial molar ratio of enzyme/inhibitor/eburicol was 1:2:50, and under these conditions, ∼40% of the original amount of P450 was still detected in the CO complex of the control sample after a 60-min reaction (results not shown). Among all of the clinical antifungal azoles and experimental CYP51 inhibitors tested (Fig. 8), the topical antifungal drug miconazole was the only compound that caused complete inhibition of the A. fumigatus CYP51 activity. Voriconazole (6% substrate conversion), posaconazole (10%), and itraconazole (16%) were followed by VNI (20%), whereas VFV (presently the most potent inhibitor of protozoan CYP51 enzymes (49)) had 31% substrate conversion. The weakest inhibitory effect on A. fumigatus CYP51B was observed with fluconazole, in accordance with the lack of activity of this drug in cellular experiments with A. fumigatus and against aspergillosis in vivo (see the Aspergillus Website).
FIGURE 8.

Inhibitory effects of azoles on the activity of A. fumigatus CYP51B. The incubation time was 60 min (at 28 °C). The molar enzyme/inhibitor/substrate ratio was 1:2:50, with 0.5 μm P450. The experiments were performed in triplicate, and results are presented as means ± S.E. (error bars).
Crystal Structures of CYP51B
In both complexes (with VNI and with voriconazole), A. fumigatus CYP51B crystallized in the same trigonal space group (P31). The asymmetric unit consisted of two monomers that are related via a non-crystallographic 180° rotation axis and are positioned in such a way that the most hydrophobic, membrane-bound fragments of the molecule (the mouth of the CYP51 substrate access channel: helix A′, FG loop, and the β4-hairpin), are facing each other (Fig. 9A). The monomers exhibit high structural similarity (Fig. 9B), with the root mean square deviation for the Cα atom positions being 0.51 ± 0.04 and 0.55 ± 0.07 Å between the proteins in the VNI- and voriconazole-bound complexes, respectively (molecular breathing) and 0.73 ± 0.06 Å between the two complexes (ligand accommodation). This result supports the view (14) that structural rigidity of the active site cavity, previously observed in various complexes of protozoan CYP51 orthologs (18, 29, 30, 39, 40, 46), is a general feature of the CYP51 family.
FIGURE 9.

Overall view of the A. fumigatus CYP51 structures. A, view along the rotation axis that runs from top to bottom and relates two VNI (pink)-A. fumigatus CYP51B complexes that comprise the asymmetric unit. The secondary structural elements forming the mouth of the substrate access channel (helices A′ and F″ and the β4-hairpin) are marked. B, superimposition of four molecules of A. fumigatus CYP51B. Shown is a distal P450 view; x/y/z: 65/67/45 Å. Molecule A of the VNI complex is shown in a ribbon representation; the backbones of the other three molecules are presented as wires. VNI and voriconazole are deleted for clarity. In both panels, the protein backbone is presented in rainbow coloring from blue (N terminus) to red (C terminus). The heme is shown as a stick model, and the iron is depicted as an orange sphere. C, stereo view of B.
Overall, A. fumigatus CYP51 displayed the typical P450 fold (an upside-down triangular prism from the distal view (Fig. 9B)) with the heme prosthetic group positioned between helices I and L and the iron coordinated to Cys463 on the proximal side of the porphyrin plane. The structure has an α/β/coil ratio of 60:10:30 and consists of 22 helices (12 main ones plus 10 additional helices (denoted with a prime)) and five β-bundles (instead of four) formed by 14 β-strands. Because of the additional β5-bundle, the molecular volume of A. fumigatus CYP51 (63,500 ± 300 Å3) is slightly higher than the volumes of the protozoan (61,300 ± 400 Å3) and human (61,900 ± 500 Å3) CYP51 orthologs.
The active side cavity is formed by the distal surface of the porphyrin macrocycle, helix B″ and B′C loop (Val121–Tyr136, substrate recognition sequence 1 (SRS1)) (50), helix C (Gln146–Val150), helix F″ (Pro231–Met235, SRS2), the N-terminal portion of helix I (Met300–Ser311, SRS4), the Kβ1–4 loop (Pro372–Arg378, SRS5), and the β4-hairpin (Leu503–Ser505, SRS6) (Fig. 10A). SRS1 represents the major CYP51 substrate binding area. It carries the family signature 1 (13), the conserved or phylum-specific residues that are essential for enzyme function (37, 51, 52). In the T. brucei CYP51 complex with the substrate analog MCP, this area of the protein (shown as a yellow ribbon in Fig. 10A) was found to encircle the whole skeleton of the sterol molecule from its β-side (46). Helix C represents an SRS that thus far is unique for CYP51 (30, 46); SRS4 and SRS5 are slightly shifted (toward the protein N terminus) relative to their canonical locations in the sequences of other CYP families; and the P450 SRS3 (the residues preceding helix G) does not appear to be able to directly interact with a ligand because, as in other CYP51 structures, this region is shielded from the active site cavity by the B′ helix.
FIGURE 10.
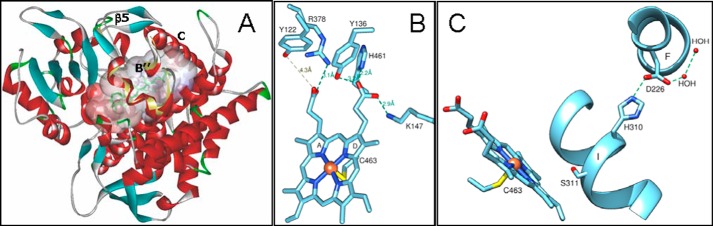
Family-specific structural features of A. fumigatus CYP51B. A, the active site cavity. Eburicol (green) was modeled in a position similar to the substrate analog MCP as described under “Experimental Procedures.” The protein backbone is colored by secondary structure (helices are red, β-strands are blue, loops are gray, and turns are green). The corresponding SRS1 area in T. brucei CYP51 is shown with a light yellow ribbon. The B′ helical turn, helix C, and the β5-bundle are marked. B, heme support from the protein moiety. Hydrogen bonds are shown as green dashes. C, proton transfer route. His310 is conserved across the CYP51 family and in all known CYP51 structures forms a hydrogen bond with an acidic residue (aspartate or glutamate) of the F helix. The length of the His310-Asp227 hydrogen bond in the four A. fumigatus CYP51B molecules is 2.8 ± 0.1 Å (mean ± S.E.). Ser311 corresponds to the conserved P450 threonine.
As with the CYP51 structures from other biological kingdoms, A. fumigatus CYP51B has five residues that can provide hydrogen bonds to the heme propionates (Fig. 10B). Arg378 and His461 are conserved in most CYP families, and Lys147 corresponds to Lys156 in the human CYP51 structure and is conserved in fungi and vertebrates, whereas in the protozoan CYP51 orthologs, the same role is played by the phylum-specific arginine that is located one turn upstream in the C helix (Arg124 in T. brucei CYP51). Tyr122 is invariant across the whole CYP51 family. In the ligand-free and sterol-bound structures, it forms the hydrogen bond with the heme ring A propionate, but binding of strong inhibitors often disrupts this hydrogen bond (29, 39, 49). The hydrogen bond between Tyr136 (phenylalanine in plant CYP51, tyrosine in all other phyla) was also found disrupted in several CYP51-inhibitor complexes (29, 40, 53).
His310 (Fig. 10C) is the invariant SRS4 residue (CYP51 signature 2 (-qHtSs-) (13)). It precedes the “conserved P450 threonine” (Ser311 in both A. fumigatus CYP51 sequences (see also Fig. 2)) and, being hydrogen-bonded with Asp226, lines the surface of a small channel (see Fig. 3 in Ref. 39) that is directed toward the water-soluble exterior of the protein. In the sequences of P450s from all other families, this position is always occupied by an acidic residue (aspartic or glutamic acid (13)). The exact mechanism of proton delivery in CYP51 has not been clarified, but the conservation of the His310/Asp226 salt bridge across the structures of CYP51s from different biological kingdoms strongly suggests that it is likely to be similar to the mechanism described for CYP101 (P450cam), where it was reported to be switched on upon the formation of the P450-electron donor complex, which disrupts the corresponding salt bridge (between Asp251 (I helix) and Lys178 (F helix) (54)) and thus activates the proton delivery. The charge of this salt bridge pair is reversed in the CYP51 family.
Complex with Voriconazole
Fluconazole is not efficient in treating A. fumigatus infections (see the Aspergillus Website), and for many years, it remained enigmatic why its close derivative voriconazole (see Fig. 1), also a rather small molecule (molecular volume 399 Å3 versus 348 Å3 for fluconazole (29)), has much higher potency. The structure of the voriconazole-A. fumigatus CYP51B complex suggests that the reason may be the formation of the hydrogen bonds between the 5-fluoropyrimidine ring of voriconazole (versus the substantially smaller triazole ring of fluconazole) and A. fumigatus CYP51 Tyr122 (Fig. 11A), the residue hydrogen-bonded with the heme ring A propionate. In addition to these hydrogen bonds with Tyr122 and the coordination of the N4-triazole nitrogen to the heme iron (the length of the coordination bond is 2.1 Å), voriconazole forms van der Waals interactions (distance <4.5 Å) with 14 more amino acid residues of A. fumigatus CYP51B (Fig. 11A). The contacting residues are mainly from the B′ helix/B′C loop (Tyr122, Leu125, Thr126, Phe130, Val135, and Tyr136); Phe229 is from the F″ helix; Ala303, Ala307, and Ser311 are from helix I; and Ile373, Ser375, and Ile376 are from the K helix/β1-4 loop and β1-4 strand, whereas Leu503 and Phe504 are from the β4-hairpin. Interestingly, the conformation of voriconazole in the active site of A. fumigatus CYP51 (Fig. 11, B and C) is very different from the conformation that the drug adopts within the active site of CYP46 (55), providing an additional example of the importance of experimental target-based structural information in the process of target-driven drug discovery.
Complex with VNI
The strong electron density within the active site of the A. fumigatus CYP51B co-crystallized with VNI (Fig. 12A) unambiguously demonstrates that the position of this inhibitor in the fungal CYP51 binding cavity is also quite different from its position in the complex with the protozoan CYP51 ortholog (Fig. 12B). The observed differences (i) validate the importance of the hydrogen bond network around the VNI carboxamide fragment for its high antiprotozoan potency and selectivity (27, 39, 49, 56), (ii) explain its weaker effect on the A. fumigatus CYP51 activity (see Fig. 8), and (iii) outline a potential strategy for further enhancement of the VNI scaffold antifungal activity. Thus, the structure suggests that the lack of the hydrogen bond network between the protein and the inhibitor may result from the altered orientation of the VNI three-ring arm, which in turn repositions (shifts down as it is seen in Fig. 12B) the dichlorinated β-phenyl ring of the inhibitor. Because the VNI derivative VFV contains the longer, β-biphenyl arm here (shown in Fig. 8), it is unlikely to acquire the VNI position in A. fumigatus CYP51B because its two-ring arm should not fit into the C-helix-directed portion of the enzyme binding cavity (not shown). This might be the reason why VFV, which to date is our most potent antiprotozoan drug candidate (49), is a weaker inhibitor of A. fumigatus CYP51B than VNI (Fig. 8).
Although no direct hydrogen bonds are formed with the protein, VNI makes van der Waals contacts (distance <4.5 Å) with 19 amino acid residues, including Tyr122, which is pushed by the inhibitor too far away from the heme ring A propionate (4.3 Å) to interact with its oxygen (Fig. 12C). The length of the coordination bond between the VNI N3 nitrogen and the heme iron is 2.0 Å. Interestingly, in the complex with VNI, some rearrangements in the side chain positioning of A. fumigatus CYP51B can be seen, especially around the substrate access channel (Fig. 13), when compared with the voriconazole-bound structure. Thus, the basic His310 shifts closer to the VNI carboxamide oxygen (the distance is 3.7 Å versus 4.5 Å in the complex with voriconazole), whereas the hydrophobic Phe504 moves about 5 Å away from this polar area of the inhibitor. Most relocations, however, occur around the substrate channel entrance. Particularly interesting is the movement of Phe234, the aromatic ring of which turns about 70° and moves toward the aromatic ring of VNI to form face-to-face (sandwich) π-p stacking interactions (∼4 Å), which are likely to be important in stabilizing both the position of the inhibitor and the entry into the enzyme substrate access channel. The side chains of Leu92 (the residue from the turn between the β1-1 and β1-2), Met235 (helix F′), and Leu503 (the β4-hairpin) must also shift toward the distal aromatic ring of VNI ∼3, 6, and 4 Å, respectively, to accomplish this change (Fig. 13).
FIGURE 13.

VNI-induced rearrangements in A. fumigatus CYP51B (semitransparent pink structure, except for Phe234 and VNI, which are colored in magenta). Superimposition with the voriconazole co-structure (semitransparent blue) is shown. The π-π stacking interactions between VNI and Phe234 are depicted as black dashed lines; the distances are marked. The hydrogen bonds are green. Helices A′ and F″ and the β4-hairpin are the elements forming the entrance into the CYP51 substrate access channel. The heme is seen as a gray sphere model. The other 4 residues in the superimposed complex with voriconazole (Leu92, Met235, His310, and Phe504), whose side chain locations differ substantially, are shown as semitransparent blue lines; the directions of the rearrangements are indicated with gray arrows.
A. fumigatus Cellular Growth Inhibition Assays
The potency of VNI against A. fumigatus cells was found to be comparable with the effects of clinical antifungals, with the minimal inhibitory concentration values for itraconazole, voriconazole, and VNI being 0.5, 0.7, and 0.5 μg/ml, respectively.
Discussion
When overlaid with the structures of its protozoan and mammalian orthologs, A. fumigatus CYP51 displays remarkable similarity in the spatial organization of the protein backbone. Although its amino acid sequence identity to T. brucei and human sterol 14α-demethylases is only 23 and 33%, respectively, the average root mean square deviation values for the Cα atom positions are only 1.8 and 1.4 Å (Fig. 14A). The residues that are conserved across the whole CYP51 family also superimpose very well; those located within the substrate binding cavity are shown in Fig. 14B. Thus, altogether, the fungal structures strongly support the proposal (14) that sterol 14α-demethylases from different biological kingdoms have preserved their conserved metabolic roles by maintaining high similarity at the secondary and tertiary structural levels, variations in their substrate preferences, catalytic parameters, and susceptibility to inhibition being fine tuned (as experimentally established for the protozoan CYP51 enzymes (30, 37, 57) by the phylum-specific residues that line the interior of the CYP51 binding cavity, thus defining the local topology of the active site.
FIGURE 14.

Superimposed structures of A. fumigatus (4UYL, pink), T. brucei (3GW9, blue), and human (3LD6, yellow) CYP51 enzymes. A, distal view. The protein backbone is presented as a semitransparent ribbon, and the area of the substrate-binding cavity is within the black square. B, enlarged view of the binding cavity. The heme and the residues invariant in all known CYP51 enzymes (>300 sequences) are depicted in a stick representation with T. brucei CYP51 numbering (see also Fig. 2).
In addition to the topological differences inside the binding cavity, which must be taken into account in CYP51 structure-based drug development, the A. fumigatus CYP51B structures reveal two very specific features, not seen among the CYP51 enzymes from other phyla, which are of functional importance.
Substrate Entrance
A. fumigatus CYP51 has a relatively shorter FG arm (Fig. 15A). A different position of the FG loop alters the shape of the enzyme substrate access channel, so that the ligand entry site looks more like a long cleft extended to the upper P450 surface with a “bridge” over it (Fig. 15B, 1 and 2) than like a small round opening, which in protozoan and human CYP51 enzymes is only seen from the distal side of the molecule (Fig. 15B, 3 and 4). The bridge in A. fumigatus CYP51B is formed by the hydrophobic interactions between Phe61 (helix A′) or Leu92 (the β1-1/β1-2 turn) and Met235 (helix F″). In the complex with voriconazole, the bridge appears to be more flexible (about to adopt an open conformation, Fig. 15B, 1), whereas the π-π stacking interactions between the aromatic rings of VNI and Phe234 of A. fumigatus CYP51 (shown in Fig. 13) keep it in the closed state (Fig. 15B, 2). In both complexes, the ligands can be seen from both the distal and the upper view of the fungal P450 molecule. This relatively wider opening of the substrate entry may well be the reason why A. fumigatus is not very sensitive to the small inhibitors, except for voriconazole (which forms hydrogen bonds with Tyr122), and “longer” structures (e.g. posaconazole) are required to restrict the motions of the channel entry and therefore strengthen the inhibition. Most important, this structural feature of A. fumigatus CYP51 implies that the FG loop fragment is likely to play a gating role in all CYP51 enzymes, opening briefly for the substrate (ligand) to enter the active site and closing when the complex acquires energetically optimal conformation.
FIGURE 15.
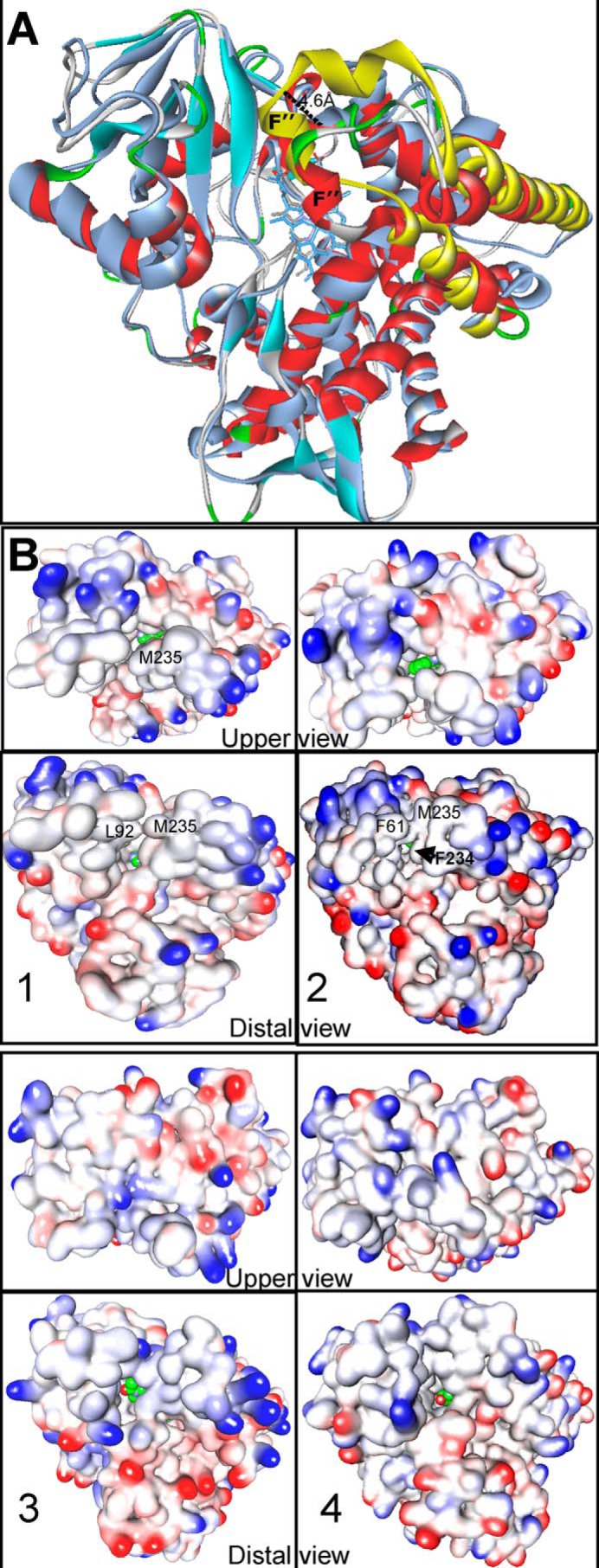
Substrate entry. A, superimposition of A. fumigatus (4UYL, colored according to secondary structure as in Fig. 10) with T. brucei CYP51 (3SW9, gray; the FG-arm is yellow). The distal view is shown. The length of the FG arm in A. fumigatus CYP51B is shorter (by ∼5 Å) than it is in the T. brucei enzyme. B, surface representation of CYP51s from different phyla. 1, voriconazole-bound A. fumigatus CYP51; 2, VNI-bound A. fumigatus CYP51; 3, ketoconazole-bound human CYP51; 4, MCP-bound T. brucei CYP51. The ligands are shown as spheres with green carbon atoms. There is no opening in the upper surface of the human or protozoan CYP51 structure.
β5-Bundle on the Proximal P450 Surface
Multiple sequence alignment of the CYP51 family proteins from different biological kingdoms reveals a 15–25-amino acid residue-long insert that is present in all fungal CYP51 sequences between the “meander” and the heme binding segment (also known as the heme bulge) (Fig. 16). In the A. fumigatus CYP51B structures, this post-meander insert forms an additional β-bundle (β5), which consists of two adjacent antiparallel β-strands (5-1 and 5-2 in Fig. 16A) and buries the heme bulge segment inside the protein globule, shielding it from the proximal surface. The function of the β5-bundle remains unclear, and, to our knowledge, no analogous structure has been observed so far in any other P450 families.
FIGURE 16.

Fungi-specific postmeander insert (β5-bundle). A, proximal view of the CYP51 molecule, colored by secondary structure as in Fig. 10. Insets, electrostatic potential mapped onto the proximal surface. Red, negative charge; blue, positive charge. The β5-area is circled. B, a fragment of multiple CYP51 sequence alignment.
We hypothesize that the β5-bundle may somehow be involved in regulation of the electron transfer process by influencing the interaction of the fungal CYP51 with the P450 reductase, modulating the heme environment (58, 59). The proximal surface of the P450 molecule, including the heme bulge area, is known to be involved in the electrostatic interaction with the negatively charged surface of the electron donor protein (54, 60, 61), and therefore, it must be electropositive (blue in the insets in Fig. 16A). Due to the β5-bundle, in the fungal CYP51 structures, the proximal surface appears to be electronegative (Fig. 16A, right inset, the β5-bundle region is circled). This discrepancy, however, can be easily resolved if the base of the β5-bundle is highly flexible (residues 427–432 and 450–454 (see Fig. 2) have the highest B-factor values in all four A. fumigatus CYP51B molecules) and the β5-bundle is likely to move aside upon CYP51-reductase complex formation. Such motion would expose the CYP51 heme bulge segment and alter the number of heme-contacting residues. Our structural analysis supports such a possibility, particularly because the β5-bundle pushes the upper portion of the heme bulge segment slightly away from the porphyrin ring plane, so that the number of heme-contacting residues in the fungal CYP51 structure is lower (i.e. 19 versus 24 in the protozoan CYP51 ortholog). This result, in turn, correlates with the experimentally observed elevated propensity of A. fumigatus CYP51B to form a destabilized ferrous species (results not shown).
The question of why A. fumigatus CYP51B does not oxidize C24-demethyl sterols (e.g. lanosterol) remains open. The structures suggest that it might be because of the relatively larger volume of this enzyme binding cavity, particularly in the area holding the sterol aliphatic arm (Fig. 17). If this is the case, the model of A. fumigatus CYP51A implies that this CYP51 enzyme might oxidize lanosterol, due to a single amino acid difference in the I-helix (Thr285 versus Ala303; Fig. 17B). More structure-functional characterization of A. fumigatus CYP51A is desirable before drawing any conclusions regarding both enzyme substrate preferences and CYP51A and CYP51B susceptibility to inhibition. The experiments on A. fumigatus CYP51A expression/purification in the P450 form are currently in progress.
FIGURE 17.

A. fumigatus CYP51B and CYP51A may differ in their substrate preferences. A, substrate binding cavities in A. fumigatus CYP51B and T. brucei CYP51. A. fumigatus CYP51B has more space in the area holding the distal portion of the sterol aliphatic arm (circled). B, a fragment of multiple sequence alignment of CYP51s A and B from filamentous fungi. A.fum, A. fumigatus; A.flav, Aspergillus flavus; A.lent, Aspergillus lentulus; A.clav, Aspergillus clavatus; N.fisch, Neosartorya fischeri; T.tons, Trichophyton tonsurans; F.gram, Fusarium graminearum; V.inaec, Venturia inaequalis; U.necator, Uncinula necator; C.neoform, Cryptococcus neoformans; U.maydis, Ustilago maydis. C, location of Thr285 (yellow), which aligns with Ala303 in A. fumigatus CYP51B (blue) in the molecular model of A. fumigatus CYP51A.
The stabilizing rearrangements in the region of the A. fumigatus CYP51B substrate entrance that we observed upon binding of VNI suggest that minor modifications around the aromatic ring of the VNI long arm might significantly increase its potency (and selectivity) as a fungal CYP51 inhibitor. This would be of special interest because of the VNI scaffold advantages (i.e. low toxicity, excellent cellular permeability, and weak influence on human drug-metabolizing P450s) and favorable phamacokinetics and tissue distribution (27, 49, 62).
To summarize, the A. fumigatus CYP51B structures support high overall three-dimensional similarity as the molecular basis for the CYP51 family catalytic conservation across phyla. They also reveal a few interesting differences, which we believe to be important for better understanding CYP51 family structure/function in general; to establish some phylum-specific features; and to help in distinguishing species-related peculiarities and their possible relevance to enzyme catalysis, inhibition, and structure-based design of more effective antifungal agents.
Author Contributions
G. I. L. conceived and coordinated the study. T. Y. H., Z. W., D. C. L., and G. I. L. performed the experiments. G. I. L. and F. P. G. analyzed the data and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Diane Kelly (University of Swansea) for the A. fumigatus CYP51 cDNA, Dr. Sandeep Goyal for the DynaFit calculations, and K. Trisler for assistance in preparation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM067871 (to G. I. L.). Vanderbilt University is a member institution of the Life Science Collaborative Team at Sector 21 of the Advanced Photon Source (APS) (Argonne, IL). Use of the APS at Argonne National Laboratory was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4UYL and 4UYM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CYP or P450
- cytochrome P450
- CYP51
- sterol 14α-demethylase
- CPR
- NADPH-cytochrome P450 reductase
- SRS
- substrate recognition sequence
- VNI
- (R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide
- VFV
- (R)-N-(1-(3,4′-difluorobiphenyl-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide
- MCP
- 14α-methylenecyclopropyl-Δ7-24,25-dihydrolanosterol
- HPCD
- hydroxypropyl-β-cyclodextrin
- NTA
- nitrilotriacetic acid.
References
- 1. Denning D. W., Bromley M. J. (2015) How to bolster the antifungal pipeline. Science 347, 1414–1416 [DOI] [PubMed] [Google Scholar]
- 2. Shapiro R. S., Robbins N., Cowen L. E. (2011) Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol. Mol. Biol. Rev. 75, 213–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gullo A. (2009) Invasive fungal infections. Drugs 69, 65–73 [DOI] [PubMed] [Google Scholar]
- 4. Xie J. L., Polvi E. J., Shekhar-Guturja T., Cowen L. E. (2014) Elucidating drug resistance in human fungal pathogens. Future Microbiol. 9, 523–542 [DOI] [PubMed] [Google Scholar]
- 5. Angarone M. (2014) Fungal infections in cancer patients. Cancer Treat. Res. 161, 129–155 [DOI] [PubMed] [Google Scholar]
- 6. Chen S. C. A., Playford E. G., Sorrell T. C. (2010) Antifungal therapy in invasive fungal infections. Cur. Opin. Pharmacol. 10, 522–530 [DOI] [PubMed] [Google Scholar]
- 7. Hope W. W., Walsh T. J., Denning D. W. (2005) The invasive and saprophytic syndromes due to Aspergillus spp. Med. Mycol. 43, S207–S238 [DOI] [PubMed] [Google Scholar]
- 8. Latgé J.-P. (1999) Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 12, 310–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown G. D., Denning D. W., Gow N. A. R., Levitz S. M., Netea M. G., White T. C. (2012) Hidden killers: human fungal infections. Sci. Trans. Med. 4, 165rv113. [DOI] [PubMed] [Google Scholar]
- 10. Denning D. W., Pleuvry A., Cole D. C. (2013) Global burden of allergic bronchopulmonary aspergillosis with asthma and its complication chronic pulmonary aspergillosis in adults. Med. Mycol. 51, 361–370 [DOI] [PubMed] [Google Scholar]
- 11. Johnson L. B., Kauffman C. A. (2003) Voriconazole: a new triazole antifungal agent. Clin. Infect. Dis. 36, 630–637 [DOI] [PubMed] [Google Scholar]
- 12. Ashbee H. R., Barnes R. A., Johnson E. M., Richardson M. D., Gorton R., Hope W. W. (2014) Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology. J. Antimicrob. Chemother. 69, 1162–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lepesheva G. I., Waterman M. R. (2007) Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 1770, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lepesheva G. I., Waterman M. R. (2011) Structural basis for conservation in the CYP51 family. Biochim. Biophys. Acta 1814, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sheehan D. J., Hitchcock C. A., Sibley C. M. (1999) Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 12, 40–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van den Bossche H., Willemsens G., Cools W., Cornelissen F., Lauwers W. F., van Cutsem J. M. (1980) In vitro and in vivo effects of the antimycotic drug ketoconazole on sterol synthesis. Antimicrob. Agents Chemother. 17, 922–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van den Bossche H. (ed) (1988) Mode of Action of Pyridine, Pyrimidine and Azole Antifungals, pp. 79–119, Ellis Horwood, Chichester, UK [Google Scholar]
- 18. Hargrove T. Y., Wawrzak Z., Alexander P. W., Chaplin J. H., Keenan M., Charman S. A., Perez C. J., Waterman M. R., Chatelain E., Lepesheva G. I. (2013) Complexes of Trypanosoma cruzi sterol 14α-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: structural basis for pathogen selectivity. J. Biol. Chem. 288, 31602–31615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Correia M. A., Ortiz de Montellano P. R. (2005) in Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd Ed (Ortiz de Montellano P. R., ed) pp. 246–322, Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 20. Warrilow A. G. S., Melo N., Martel C. M., Parker J. E., Nes W. D., Kelly S. L., Kelly D. E. (2010) Expression, purification, and characterization of Aspergillus fumigatus sterol 14-α demethylase (CYP51) isoenzymes A and B. Antimicrob. Agents Chemother. 54, 4225–4234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heeres J., Meerpoel L., Lewi P. (2010) Conazoles. Molecules 15, 4129–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan J., Urban M., Parker J. E., Brewer H. C., Kelly S. L., Hammond-Kosack K. E., Fraaije B. A., Liu X., Cools H. J. (2013) Characterization of the sterol 14α-demethylases of Fusarium graminearum identifies a novel genus-specific CYP51 function. New Phytol. 198, 821–835 [DOI] [PubMed] [Google Scholar]
- 23. Lupetti A., Danesi R., Campa M., Del Tacca M., Kelly S. (2002) Molecular basis of resistance to azole antifungals. Trends Mol. Med. 8, 76–81 [DOI] [PubMed] [Google Scholar]
- 24. van der Linden J. W. M., Camps S. M. T., Kampinga G. A., Arends J. P. A., Debets-Ossenkopp Y. J., Haas P. J. A., Rijnders B. J. A., Kuijper E. J., van Tiel F. H., Varga J., Karawajczyk A., Zoll J., Melchers W. J. G., Verweij P. E. (2013) Aspergillosis due to voriconazole highly resistant Aspergillus fumigatus and recovery of genetically related resistant isolates from domiciles. Clin. Infect. Dis. 57, 513–520 [DOI] [PubMed] [Google Scholar]
- 25. Hawkins N. J., Cools H. J., Sierotzki H., Shaw M. W., Knogge W., Kelly S. L., Kelly D. E., Fraaije B. A. (2014) Paralog re-emergence: a novel, historically contingent mechanism in the evolution of antimicrobial resistance. Mol. Biol. Evol. 31, 1793–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mellado E., Diaz-Guerra T. M., Cuenca-Estrella M., Rodriguez-Tudela J. L. (2001) Identification of two different 14α-sterol demethylase-related genes (cyp51A and cyp51B) in Aspergillus fumigatus and other Aspergillus species. J. Clin. Microbiol. 39, 2431–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hargrove T. Y., Kim K., de Nazaré Correia Soeiro M., da Silva C. F., Batista D. D., Batista M. M., Yazlovitskaya E. M., Waterman M. R., Sulikowski G. A., Lepesheva G. I. (2012) CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int. J. Parasitol. Drugs Drug Resist. 2, 178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lepesheva G. I., Nes W. D., Zhou W., Hill G. C., Waterman M. R. (2004) CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry 43, 10789–10799 [DOI] [PubMed] [Google Scholar]
- 29. Lepesheva G. I., Hargrove T. Y., Anderson S., Kleshchenko Y., Furtak V., Wawrzak Z., Villalta F., Waterman M. R. (2010) Structural insights into inhibition of sterol 14α-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 285, 25582–25590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hargrove T. Y., Wawrzak Z., Liu J., Nes W. D., Waterman M. R., Lepesheva G. I. (2011) Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase (CYP51) from Leishmania infantum. J. Biol. Chem. 286, 26838–26848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Omura T., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 32. Lepesheva G. I., Strushkevich N. V., Usanov S. A. (1999) Conformational dynamics and molecular interaction reactions of recombinant cytochrome P450scc (CYP11A1) detected by fluorescence energy transfer. Biochim. Biophys. Acta 1434, 31–43 [DOI] [PubMed] [Google Scholar]
- 33. Schenkman J. B., Remmer H., Estabrook R. W. (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 3, 113–123 [PubMed] [Google Scholar]
- 34. Shinkyo R., Guengerich F. P. (2011) Cytochrome P450 7A1 cholesterol 7α-hydroxylation: individual reaction steps in the catalytic cycle and rate-limiting ferric iron reduction. J. Biol. Chem. 286, 4632–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mast N., Pikuleva I. A. (2005) A simple and rapid method to measure cholesterol binding to P450s and other proteins. J. Lipid Res. 46, 1561–1568 [DOI] [PubMed] [Google Scholar]
- 36. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 37. Lepesheva G. I., Zaitseva N. G., Nes W. D., Zhou W., Arase M., Liu J., Hill G. C., Waterman M. R. (2006) CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B′ helix defines substrate preferences of sterol 14α-demethylase. J. Biol. Chem. 281, 3577–3585 [DOI] [PubMed] [Google Scholar]
- 38. Lepesheva G. I., Ott R. D., Hargrove T. Y., Kleshchenko Y. Y., Schuster I., Nes W. D., Hill G. C., Villalta F., Waterman M. R. (2007) Sterol 14α-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol. 14, 1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lepesheva G. I., Park H. W., Hargrove T. Y., Vanhollebeke B., Wawrzak Z., Harp J. M., Sundaramoorthy M., Nes W. D., Pays E., Chaudhuri M., Villalta F., Waterman M. R. (2010) Crystal structures of Trypanosoma brucei sterol 14α-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 285, 1773–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Friggeri L., Hargrove T. Y., Rachakonda G., Williams A. D., Wawrzak Z., Di Santo R., De Vita D., Waterman M. R., Tortorella S., Villalta F., Lepesheva G. I. (2014) Structural basis for rational design of inhibitors targeting Trypanosoma cruzi sterol 14α-demethylase: two regions of the enzyme molecule potentiate its inhibition. J. Med. Chem. 57, 6704–6717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leslie A. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 42. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hargrove T. Y., Wawrzak Z., Liu J., Waterman M. R., Nes W. D., Lepesheva G. I. (2012) Structural complex of sterol 14α-demethylase (CYP51) with 14α-methylenecyclopropyl-Δ7-24, 25-dihydrolanosterol. J. Lipid Res. 53, 311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nierman W. C., Pain A., Anderson M. J., Wortman J. R., Kim H. S., Arroyo J., Berriman M., Abe K., Archer D. B., Bermejo C., Bennett J., Bowyer P., Chen D., Collins M., Coulsen R., Davies R., Dyer P. S., Farman M., Fedorova N., Fedorova N., Feldblyum T. V., Fischer R., Fosker N., Fraser A., García J. L., García M. J., Goble A., Goldman G. H., Gomi K., Griffith-Jones S., Gwilliam R., Haas B., Haas H., Harris D., Horiuchi H., Huang J., Humphray S., Jiménez J., Keller N., Khouri H., Kitamoto K., Kobayashi T., Konzack S., Kulkarni R., Kumagai T., Lafon A., Latgé J.-P., Li W., Lord A., Lu C., Majoros W. H., May G. S., Miller B. L., Mohamoud Y., Molina M., Monod M., Mouyna I., Mulligan S., Murphy L., O'Neil S., Paulsen I., Penalva M. A., Pertea M., Price C., Pritchard B. L., Quail M. A., Rabbinowitsch E., Rawlins N., Rajandream M.-A., Reichard U., Renauld H., Robson G. D., de Cordoba S. R., Rodriguez-Pena J. M., Ronning C. M., Rutter S., Salzberg S. L., Sanchez M., Sanchez-Ferrero J. C., Saunders D., Seeger K., Squares R., Squares S., Takeuchi M., Tekaia F., Turner G., de Aldana C. R. V., Weidman J., White O., Woodward J., Yu J.-H., Fraser C., Galagan J. E., Asai K., Machida M., Hall N., Barrell B., Denning D. W. (2005) Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438, 1151–1156 [DOI] [PubMed] [Google Scholar]
- 48. Denning D. W., Venkateswarlu K., Oakley K. L., Anderson M. J., Manning N. J., Stevens D. A., Warnock D. W., Kelly S. L. (1997) Itraconazole resistance in Aspergillus fumigatus. Antimicrob. Agents Chemother. 41, 1364–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lepesheva G. I., Hargrove T. Y., Rachakonda G., Wawrzak Z., Pomel S., Cojean S., Nde P. N., Nes W. D., Locuson C. W., Calcutt M. W., Waterman M. R., Daniels J. S., Loiseau P. M., Villalta F. (2015) VFV as a new effective CYP51 structure-derived drug candidate for Chagas disease and visceral leishmaniasis. J. Infect. Dis. 10.1093/infdis/jiv228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gotoh O. (1992) Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 267, 83–90 [PubMed] [Google Scholar]
- 51. Lepesheva G. I., Virus C., Waterman M. R. (2003) Conservation in the CYP51 family: role of the B′ helix/BC loop and helices F and G in enzymatic function. Biochemistry 42, 9091–9101 [DOI] [PubMed] [Google Scholar]
- 52. Lepesheva G. I., Seliskar M., Knutson C. G., Stourman N. V., Rozman D., Waterman M. R. (2007) Conformational dynamics in the F/G segment of CYP51 from Mycobacterium tuberculosis monitored by FRET. Arch. Biochem. Biophys. 464, 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andriani G., Amata E., Beatty J., Clements Z., Coffey B. J., Courtemanche G., Devine W., Erath J., Juda C. E., Wawrzak Z., Wood J. T., Lepesheva G. I., Rodriguez A., Pollastri M. P. (2013) Antitrypanosomal lead discovery: identification of a ligand-efficient inhibitor of Trypanosoma cruzi CYP51 and parasite growth. J. Med. Chem. 56, 2556–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tripathi S., Li H., Poulos T. L. (2013) Structural basis for effector control and redox partner recognition in cytochrome P450. Science 340, 1227–1230 [DOI] [PubMed] [Google Scholar]
- 55. Mast N., Zheng W., Stout C. D., Pikuleva I. A. (2013) Antifungal azoles: structural insights into undesired tight binding to cholesterol-metabolizing CYP46A1. Mol. Pharmacol. 84, 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Villalta F., Dobish M. C., Nde P. N., Kleshchenko Y. Y., Hargrove T. Y., Johnson C. A., Waterman M. R., Johnston J. N., Lepesheva G. I. (2013) VNI cures acute and chronic experimental Chagas disease. J. Infect. Dis. 208, 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cherkesova T. S., Hargrove T. Y., Vanrell M. C., Ges I., Usanov S. A., Romano P. S., Lepesheva G. I. (2014) Sequence variation in CYP51A from the Y strain of Trypanosoma cruzi alters its sensitivity to inhibition. FEBS Lett. 588, 3878–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Papadopoulou N. D., Mewies M., McLean K. J., Seward H. E., Svistunenko D. A., Munro A. W., Raven E. L. (2005) Redox and spectroscopic properties of human indoleamine 2,3-dioxygenase and a His303Ala variant: implications for catalysis. Biochemistry 44, 14318–14328 [DOI] [PubMed] [Google Scholar]
- 59. Efimov I., Parkin G., Millett E. S., Glenday J., Chan C. K., Weedon H., Randhawa H., Basran J., Raven E. L. (2014) A simple method for the determination of reduction potentials in heme proteins. FEBS Lett. 588, 701–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang M., Roberts D. L., Paschke R., Shea T. M., Masters B. S. S., Kim J.-J. P. (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. U.S.A. 94, 8411–8416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Strushkevich N., MacKenzie F., Cherkesova T., Grabovec I., Usanov S., Park H.-W. (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U.S.A. 108, 10139–10143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Soeiro Mde N., de Souza E. M., da Silva C. F., Batista Dda G., Batista M. M., Pavão B. P., Araújo J. S., Aiub C. A., da Silva P. B., Lionel J., Britto C., Kim K., Sulikowski G., Hargrove T. Y., Waterman M. R., Lepesheva G. I. (2013) In vitro and in vivo studies of the antiparasitic activity of sterol 14α-demethylase (CYP51) inhibitor VNI against drug-resistant strains of Trypanosoma cruzi. Antimicrob. Agents Chemother. 57, 4151–4163 [DOI] [PMC free article] [PubMed] [Google Scholar]