

Background: The regulation of gap junction protein connexin 43 (Cx43), involved in natural killer (NK) cell-mediated tumor killing, is still elusive.
Results: Hypoxia-induced autophagy selectively degrades gap junctional Cx43 from immune synapses and impairs NK-mediated melanoma cell lysis.
Conclusion: A hypoxic microenvironment induces melanoma resistance to NK cells via modulation of Cx43 channels.
Significance: Targeting autophagy prevents gap-junctional Cx43 degradation and potentiates NK-based tumor immunotherapies.
Keywords: autophagy, gap junction, hypoxia, melanoma, natural killer (NK) cells, tumor immunology, connexin 43
Abstract
Although natural killer (NK) cells play an important role in the control of melanoma, hypoxic stress in the tumor microenvironment may impair NK-mediated tumor cell killing by mechanisms that are not fully understood. In this study, we investigated the effect of hypoxia on the expression and channel activity of connexin 43 (Cx43) in melanoma cells and its impact on their susceptibility to NK cell-mediated lysis. Our results demonstrated that hypoxic stress increases Cx43 expression in melanoma cells via hypoxia-inducible factor-1α (HIF-1α) transcriptional activity. Hypoxic cells displaying increased Cx43 expression were less susceptible to NK cell-mediated lysis compared with normoxic cells expressing a moderate level of Cx43. Conversely, when overexpressed in normoxic tumor cells, Cx43 improves their susceptibility to N cell-mediated killing. We show that the NK cell immune synapse formed with normoxic melanoma cells is more stable and contains a high level of gap-junctional Cx43 whereas that formed with hypoxic cells is less stable and contains a significant lower level of gap-junctional Cx43. We provide evidence that the activation of autophagy in hypoxic melanoma cells selectively degrades gap-junctional Cx43, leading to the destabilization of the immune synapse and the impairment of NK cell-mediated killing. Inhibition of autophagy by genetic or pharmacological approaches as well as expression of the non-degradable form of Cx43 significantly restore its accumulation at the immune synapse and improves N cell-mediated lysis of hypoxic melanoma cells. This study provides the first evidence that the hypoxic microenvironment negatively affects the immune surveillance of tumors by NK cells through the modulation of Cx43-mediated intercellular communications.
Introduction
Natural killer (NK)3 cells are innate lymphocytes endowed with the ability to detect and kill tumor cells. The elimination of target cells by NK cells largely relies on the formation of an immunological synapse at the intercellular interface (1). Given its important role in tumor regression, NK cell-based immunotherapies have emerged recently as promising treatments for hematopoietic malignancies and solid tumors, including melanoma (2).
Gap junctions (GJs) are channels formed of connexin (Cx) subunits that mediate the direct cell-cell exchange of small molecules. GJ-mediated intercellular communication participates in the regulation of a plethora of cell functions, including differentiation, survival, and cell death (3). Non-gap-junctional Cx hemichannels (Cx-HCs) also participate in cell behaviors via the release of small mediator molecules to the extracellular medium (4). Because Cxs have short half-lives, their turnover regulation and trafficking critically impact the control of GJ-mediated intercellular communication (5). Connexin 43 (Cx43), the main GJ protein found in immune cells (6), has been shown recently to accumulate at the immunological synapse and allows GJ-mediated intercellular communication between effector immune and tumor target cells (7–10). These observations suggest that loss of Cx43 expression during tumor progression may represent a mechanism by which cancer cells evade immune control by the local microenvironment (11).
It is well established that the tumor microenvironment supports tumor growth and limits the effectiveness of solid tumor immunotherapies by promoting neoplastic transformation and cell plasticity and by inducing tumor cell resistance to host immunity (12, 13). Accumulating data suggest that hypoxic stress in the tumor microenvironment promotes the acquisition of tumor cell resistance to NK cells (14, 15), and cytotoxic T lymphocytes attack by several mechanisms (16, 17) via the induction of hypoxia-inducible factor 1α (HIF-1α) (18). We have reported recently that hypoxia reduces breast cancer cell susceptibility to NK cell-mediated lysis by a mechanism involving the activation of autophagy and the subsequent degradation of NK cell-derived granzyme B (GzmB) (19).
It has been reported that hypoxia regulates Cx43 expression and channel activity in different cell models (20, 21). However, no data are currently available addressing the role of Cx43 channels in melanoma cells in the context of the hypoxic microenvironment and their putative involvement in tumor cell susceptibility to NK cell-mediated lysis. In this study, we provide evidence that ectopic overexpression of Cx43 under normoxia in melanoma tumor cells increases their susceptibility to NK cell-mediated killing via the stabilization of the cytolytic immune synapse. However, under hypoxic stress, although melanoma cells overexpressed Cx43, they were less susceptible to NK cell-mediated killing. We therefore unraveled that hypoxia-induced autophagy selectively degraded GJ-Cx43, leading to destabilization of the immunological synapse and, subsequently, the acquisition of resistance to NK cell-mediated lysis.
Experimental Procedures
Cell Cultures and Treatments
Melanoma cells were established from biopsies of primary lesions (M4T, T1, and I2) or from metastatic lymph nodes (M4T2 and G1) as described previously (22). The M4T melanoma cells used in this study were generated from melanoma biopsies at the Hospital Clinic, Barcelona, Spain (1991–1993) and defined by clinicopathological characteristics as primary melanoma cells. M4T cells displayed a loss of heterozygosity at the microsatellite marker locus D9S265 of the chromosome 9p13–23, a region that contains genes involved in melanoma tumorigenesis (23). M4T cells express wild-type BRAF. The A753 and SKMel30 cell lines were from the ATCC and Deutsche Sammlung von Mikroorganismen und Zellkulturen, respectively. NK92 and MCF7 cells were obtained from the ATCC. Cells were maintained in culture as described previously (7). NK cells were maintained in RPMI medium supplemented with 300 units/ml recombinant human IL-2.
Hypoxic treatments were conducted in hypoxia chambers as described previously (16). Autophagy was inhibited by addition of 50 μm hydroxychloroquine (HCQ) or 5 mm 3-methyladenine (both from Sigma-Aldrich). Cx43 channels were inhibited using Cx43-mimetic peptides (300 μm, gap27, SRPTEKTIFII, Synprosis) (10, 24). A scrambled peptide (TFEPIRISITK) was used as a control.
Transfections
Transfections were performed as described previously (16). Cells were stably transfected with the following plasmids: hCx43-pUNO1 or control (Invivogen), pcDNA3.1/V5/His-Cx43Y286A (25), ptdTomato-Cx43 or pEGFP-Cx43, ptdTomato-LC3, and psiRNA-hATG5 or control vector (psiRNA-LucGL3) (Invivogen). Knockdowns were performed by transfection with siRNA against HIF-1α (a pool of three independent target-specific siRNAs, as described previously (16, 17)) or p62 (all from Qiagen). Luciferase-specific siRNA (Sigma-Proligo) was used as a negative control. siRNAs sequences are available upon request.
Confocal Microscopy
Melanoma cells were precultured under normoxic or hypoxic conditions and incubated with NK cells at a 3/1 effector/target (E/T) ratio in μ-Slide eight-well chambered coverslips (IBIDI). Time-lapse microscopy was performed as described previously (19). Cells were maintained at 37 °C in a CO2 incubator mounted on the microscope stage. Time-lapse microscopy was performed using an Axiovert 200 M microscope (Carl Zeiss MicroImaging), and images were captured with a ×40 oil objective lens. Confocal microscopy of fixed melanoma/NK cell cocultures was performed as described previously (16). Cells were stained with polyclonal antibodies (anti-Cx43, Sigma-Aldrich), mAb (anti-Tyr(P), Millipore), or rhodamine-phalloidin (Invitrogen). Cx43 and Tyr(P) expression were visualized by using secondary goat anti-rabbit (Alexa Fluor 647-conjugated) and anti-mouse (Alexa Fluor 546-conjugated) Abs, respectively (Invitrogen). Cells were analyzed with a Zeiss LSM-510 Meta laser-scanning confocal microscope (Carl Zeiss), and images were captured with a ×63 oil objective lens. The recruitment of Cx43 to the cell-to-cell contact site (immune synapses) was quantified using ImageJ (National Institutes of Health) software as described previously (10). Cx43-LC3 colocalization was analyzed using Imaris software.
Cytotoxicity Assay
The cytotoxic activity of NK cells was measured by conventional 4-h 51Cr release assays as descried previously (16).
Immunoblots
Cells were lysed in lysis buffer (50 mm Tris (pH 7.4), 150 mm NaCl, and 1% Triton X-100) supplemented with protease and phosphatase inhibitor mixtures (Roche) for 30 min on ice. Western blotting was conducted as described previously (17). Proteins were detected by using Abs to Cx43 (catalog no. C6219, Sigma-Aldrich), HIF-1α (clone 54, BD Transduction Laboratories), HIF-2α (clone D9E3, Cell Signaling Technology), GzmB (catalog no. 4275, Cell Signaling Technology), p62 (clone 3, BD Transduction Laboratories), ATG5 (catalog no. 2630, Cell Signaling Technology), and actin (clone c-11, Santa Cruz Biotechnology). Alternatively, cells were treated for 6 h with 50 mg/ml cycloheximide (Sigma-Aldrich), and Triton X-100 fractionation assays were performed as described previously (5, 26).
ChIP
Normoxic or hypoxic (16 h) M4T cells were harvested and processed with the Simple ChIP enzymatic chromatin IP kit (Cell Signaling Technology) according to the instructions of the manufacturer. For HIF-1α-specific ChIP, chromatin was immunoprecipitated with 5 μg mAb (BD Transduction Laboratories). The relative amounts of chromatin immunoprecipitated by the anti-HIF-1α Abs were determined by SYBR Green quantitative PCR method (Applied Biosystems) using specific primers for hypoxic response elements (HREs) in the GJA1 gene promoter. Primer sequences are available upon request.
Luciferase Reporter Assay
A 2500-bp fragment corresponding to the human GJA1 gene promoter containing HRE1–5 sequences was inserted into the NheI-XhoI sites of the pGL3-Basic vector (Promega). Mutations of HRE3 and/or HRE5 were performed by site-directed mutagenesis and verified by sequencing. M4T cells were cotransfected with 0.2 μg of pGL4-hRluc/SV40 vector (which contains Renilla luciferase sequences downstream of the SV40 promoter) and 1 μg of the pGL3 HRE3/5 WT, pGL3 HRE3 Mut, pGL3 HRE5 Mut, or pGL3 HRE3–5 Mut vectors. After 48 h, the cells were grown under normoxia or hypoxia for an additional 24 h, and firefly and Renilla luciferase activities were measured using the Dual-Luciferase reporter assay (Promega).
Cx43-HC Activity
Cx43-HC activity was determined by EtBr (25 μm) uptake experiments using flow cytometry as described previously (27).
Formation and Stabilization of Cell Conjugate Analysis
Melanoma and NK92 cells were loaded with the red Dil-CM (Invitrogen) or the blue TFL4 (OncoImmunin) cell trackers according to the instructions of the manufacturer and cocultured for 10 min at a 3/1 E/T ratio. The percentages of target cells conjugated with NK cells were immediately analyzed by flow cytometry. To determine stability, cell conjugates were subjected to increasing dissociation forces by 30 s of vortexing (low, 2; medium, 5; high, 9; Heidolph TopMix, 94323-Bioblock Scientific) and analyzed as described previously (28).
Flow Cytometry Analysis
Phycoerythrin (PE)-conjugated anti-CD69 (Immunotech) and Alexa Fluor 488-conjugated anti-CD56 (BD Biosciences) Abs were used for cell staining. Flow cytometry analysis was performed using a BD AccuriTM C6 flow cytometer. Data were processed using BD Accuri software for acquisition, analysis, and calculation of cell counts.
NK Cell-derived GzmB Detection in Target Cells
GzmB activity was measured in TFL4 prestained melanoma target cells with a GranToxiLux kit (OncoImmunin) according to the instructions of the manufacturer, after coculture with NK cells for 1 h at a 1/3 T/E ratio, in the presence of a permeable fluorogenic substrate for GzmB. GzmB activity was evaluated in target cells (TFL4+) by flow cytometry. The level of GzmB in target cells was assessed by Western blot analysis as described previously (19).
Microarray
Gene expression was profiled using an 8 × 60,000 human whole genome expression array (Agilent Technologies) according to the instructions of the manufacturer at the Genomics and Bioinformatics platform of the Gustave Roussy Cancer Campus. Total RNA from four independent clones of M4T-EV and M4T-Cx43 cells was used as samples. Image analyses (quantification and normalization) were performed with Feature Extraction software (Agilent Technologies), and gene expression analysis was performed using Resolver software (Rosetta Inpharmatics). Analysis of genes expressed differentially between M4T-EV and M4T-Cx43 melanoma cells was performed with an absolute -fold change of more than 2 and a p value of less than 10.
Statistical Analysis
Data were analyzed with GraphPadPrism. Statistical analyses were performed using a two-tailed Student's t test or, where appropriate, by analysis of variance. Differences were considered statistically significant at p < 0.05.
Results
Hypoxia Increases the Expression of Cx43 in Melanoma Cells via HIF-1α-dependent Transcriptional Activation
We analyzed the effect of hypoxia on the expression of Cx43 in human melanoma cells. Five human melanoma cells tested (M4T, T1, G1, I2, and M4T2) showed a clear hypoxia time-dependent increase in Cx43 protein levels (Fig. 1A). The increase in the expression of Cx43 is correlated with the induction of HIF-1α because no difference in the expression of HIF-2α was observed in M4T cells cultured under normoxia or hypoxia (Fig. 1B). We next investigated the molecular mechanism by which hypoxia up-regulates the expression of Cx43 using the M4T cell line as a model. We showed that a pool of three independent siRNAs targeting HIF-1α resulted in complete inhibition of Cx43 protein expression in hypoxic M4T cells (Fig. 1C). In silico analysis of the Cx43 promoter region revealed the presence of six putative HREs (Fig. 1D, left panel). The ChIP data indicate that, under hypoxia, HIF-1α directly interacts with HRE-3 and HRE-5 (Fig. 1D, right panel). The functional interaction between HIF-1α and HRE-3 and HRE-5 was further assessed by luciferase reporter assay using vectors encoding either WT or mutated (Mut) HREs at the indicated positions (Fig. 1E, left panel). Our results showed that the activity of the Cx43 promoter containing WT HREs increased upon hypoxic stress and that it was not affected by the mutation of HRE-3. However, it was decreased significantly by the mutation of HRE-5. Because no additional decrease in luciferase activity was observed by the double mutations of HRE-3 and HRE-5, our results indicate that only HRE-5 is the functionally active cis element in the Cx43 promoter (Fig. 1E, right panel).
FIGURE 1.

Hypoxic stress increases the expression of Cx43 by transcriptional activation through HIF-1α. A and B, cells were incubated under normoxia (N) or hypoxia (H) at the indicated times (in B, 24 h). The expression of Cx43, HIF-1α, HIF-2α (in B only), and β-actin in the indicated melanoma cell lines (M4T, T1, G1, I2, and M4T2) were determined by Western blot analysis, and Cx43/Actin ratios were quantified by ImageJ software. C, M4T cells were incubated under normoxia or hypoxia in the absence (−) or presence (+) of a pool of three independent HIF-1α-specific siRNAs. The expression of HIF-1α, Cx43, and Actin was assessed by Western blot analysis, and HIF/Actin and Cx43/Actin ratios were quantified by ImageJ software. D, left panel, schematic of the Cx43 (GJA1) promoter. The positions of six putative HREs are shown. Right panel, ChIP was performed in M4T cells incubated under normoxia or hypoxia using anti-HIF-1α antibodies. The HIF-1α ChIP/input DNA ratios for HRE-3 and HRE-5 are shown. E, left panel, sequence of HRE-5 and HRE-3. The mutation positions in HRE-5 and HRE-3 are indicated by arrowheads. Right panel, M4T cells were transfected with pGL3 vectors encoding WT or mutated HRE-3, HRE-5, or both (3–5) and grown under normoxia or hypoxia. After 24 h, firefly and Renilla luciferase activities were measured. Luciferase activity is reported as -fold change relative to normoxia. ***, p < 0.001.
Ectopic Overexpression of Cx43 Enhances NK-mediated Tumor Cell Killing by Stabilizing the Immune Synapse
It has been reported that the inhibition of Cx43 channels strongly decreased NK-mediated tumor cell lysis (10). In keeping with this, we investigated whether the overexpression of Cx43 in the M4T melanoma cell model influences their susceptibility to NK cell-mediated lysis. Our results confirm that Cx43 overexpressed in M4T melanoma cells (Fig. 2A, left panel) forms functional Cx43-HCs (Fig. 2A, right panel) and that M4T melanoma cells overexpressing Cx43 (M4T-Cx43) were significantly more susceptible to NK cell-mediated lysis compared with M4T cells transfected with control empty vector (M4T-EV) (Fig. 2B). The increased susceptibility of M4T-Cx43 to NK cell killing was no longer observed when the cells were treated with gap27 mimetic peptide (a specific Cx43 inhibitor) (Fig. 2B), suggesting that, in the M4T cell model, NK cell-mediated killing is enhanced by Cx43 expression at the surface of target cells. Similar results were obtained by overexpressing Cx43 in other melanoma cell lines (T1 and M4T2) and in the breast cancer cell line MCF7 (Fig. 2C). In addition, the increased susceptibility of M4T-Cx43 cells to NK cell-mediated lysis was associated with a higher accumulation of Cx43 at the immunological synapse between NK/target, as demonstrated by Tyr(P) staining (Fig. 2D), and this was not associated with an increase in their ability to form conjugates with NK cells because no significant difference in conjugate formation was observed between NK cells and either control or Cx43-overexpressing M4T cells (Fig. 2E). However, Cx43-overexpressing cells formed more stable NK cell/melanoma conjugates compared with control cells (Fig. 2F). Furthermore, expression of the NK cell activation marker CD69 was increased at the surface of NK cells when cocultured with Cx43-overexpressing melanoma cells (Fig. 2G). This result indicates that NK cells become more active when cocultured with target cells displaying higher expression levels of Cx43. In line with this data, we showed that Cx43-positive melanoma cells (Cx43+) displayed higher level of NK cell-derived active GzmB compared with Cx43-negative melanoma cells (Cx43−). The level of active GzmB in target cells seems to be strikingly related to the expression of Cx43 because the Cx43-mimetic peptide gap27 dramatically decreases GzmB activity only in Cx43+ target cells (Fig. 2H). Moreover, it is unlikely that overexpression of Cx43 increases tumor cell susceptibility to NK cell-mediated killing by regulating the global gene expression profile (Fig. 2I) or by regulating NK cell-activating (MHC class I-related chain A/B, ULBP1, ULBP2, and ULBP3) and inhibitory (MHC class I) ligand expression on surface of target cells (data not shown). Together, these results show that overexpression of Cx43 in melanoma cells results in an increase in their susceptibility to NK cell-mediated lysis, most likely because of a mechanism involving the stabilization of the cytolytic immunological synapse.
FIGURE 2.

Ectopic overexpression of Cx43 in melanoma cells improves their susceptibility to NK cell-mediated lysis. A, left panel, M4T melanoma cells were transfected with EV or a vector encoding Cx43. The overexpression of Cx43 was determined by Western blot analysis. Right panel, the uptake of EtBr by M4T-EV or M4T-Cx43 cells was determined in the presence of a control (−) or the Cx43 inhibitor peptide gap27 (+). The percentage of EtBr+ cells is reported. B, the percentage of NK cell-mediated lysis of M4T-EV or M4T-Cx43 cells. Cells were cocultured with NK92 cells at a 10/1 (10) or 20/1 (20) E/T ratio in the presence of a control (Ctrl) or the Cx43 inhibitor peptide gap27. C, top panel, the melanoma cell lines T1 and M4T2 and the breast cancer cell line MCF7 were stably transfected with EV or a vector encoding Cx43. Cx43 and β-actin protein levels were determined by Western blot analysis. Bottom panel, NK cell-mediated cytotoxicity was determined at a 10/1 E/T ratio. D, M4T-EV or M4T-Cx43 cells were cocultured with NK92 cells. NK cell/target (T) conjugates were analyzed by phase-contrast (PC) microscopy. Cx43 and Tyr(P) accumulation at the immune synapse is indicated by white arrows. Scale bar = 5 μm. E and F, M4T-EV or M4T-Cx43 cells were loaded with Dil-CM and cocultured for 10 min with TFL4-loaded NK92 cells at a 3/1 E/T ratio. E, the percentages of target cells conjugated with NK cells were assessed by flow cytometry. F, cell conjugates were subjected to increasing dissociation forces (vortex setting: 1, low; 2, medium; 3, high velocity) for 30 s. The percentages of target cells conjugated with NK cells are reported as the percentage relative to the maximum (no vortex). A.U., arbitrary unit. G, NK92 cells were cocultured for 4 h with M4T-EV or M4T-Cx43 cells. The expression of CD69 on the surface of NK cells was determined by flow cytometry. H, M4T cells were transfected with Tomato fused to Cx43 (Tom-Cx43). Cells were labeled with TFL4 and cocultured with NK92 cells for 1 h at a 3/1 E/T ratio in the presence of a permeable fluorogenic substrate for GzmB (GranToxiLux). GzmB activity was evaluated on TFL4+Tom-Cx43− or TFL4+Tom-Cx43+ M4T cells by flow cytometry. Shown is the percentage of GranToxiLux-positive target cells cocultured with NK92 cells in the presence of control (−) or gap27 peptides (+). I, volcano plots of gene expression (log2 -fold change, FC) and adjusted (adj) p values for M4T cells stably transfected with Cx43 versus the control EV. Data were obtained by analyzing the gene expression profiles of four independent clones per condition. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Hypoxia-dependent Expression of Cx43 Is Associated with a Decrease in Melanoma Cell Susceptibility to NK Cell-mediated Killing
Our results described above predict that hypoxic M4T cells, which express high levels of Cx43, must be more susceptible to NK-mediated killing compared with normoxic M4T cells. Surprisingly, hypoxic melanoma cells were significantly less susceptible to NK-mediated lysis compared with normoxic cells (Fig. 3A). This paradoxical role of Cx43 overexpressed, either ectopically or under hypoxic stress, in NK cell-mediated killing susceptibility raises the important issue of whether Cx43 overexpressed on hypoxic cells forms functional Cx43-HCs. We therefore evaluated the activity of Cx43-HCs in normoxic and hypoxic M4T cells by EtBr uptake. As depicted in Fig. 3B, an increase in EtBr uptake was observed in hypoxic cells, and incubation of these cells with the Cx43-inhibitor mimetic peptide (gap27) significantly decreased EtBr uptake. These results provide evidence that hypoxia-dependent induction of Cx43 expression contributes to the formation of functional Cx43-HCs in hypoxic melanoma cells.
FIGURE 3.
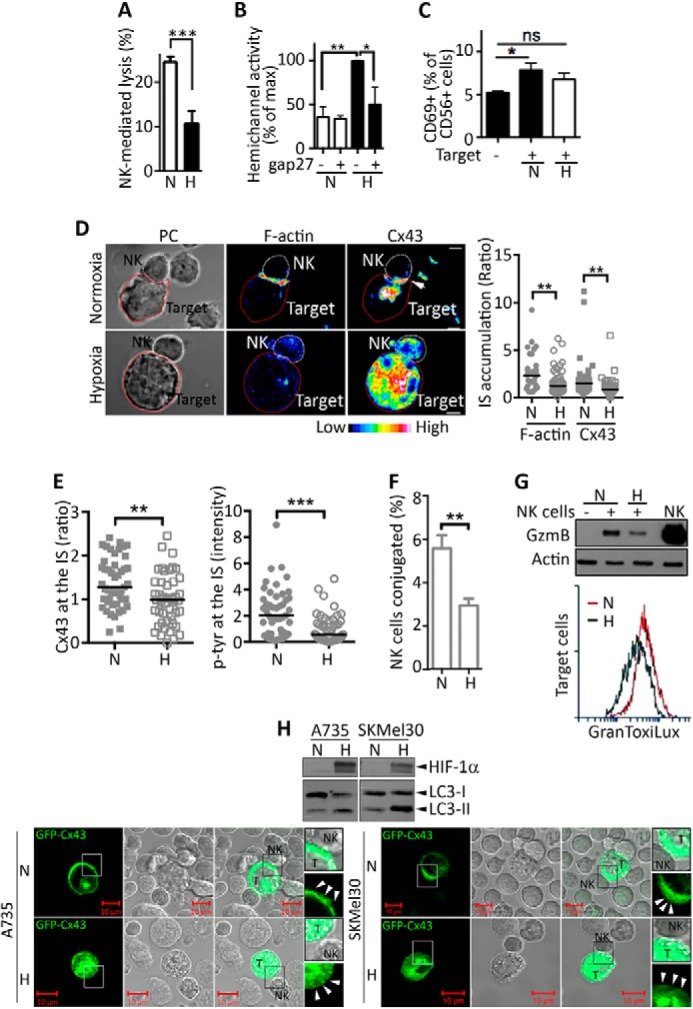
Hypoxia decreases both the accumulation of Cx43 at the immune synapse and the NK cell-mediated lysis of melanoma cells. A, M4T cells were incubated for 24 h under normoxia (N) or hypoxia (H). The percentage of NK92-mediated cytotoxicity was determined at a 10/1 E/T ratio. B, hemichannel activity was evaluated in M4T cells cultured under normoxia or hypoxia during 24 h in the presence of a control (−) or the Cx43 inhibitor peptide gap27 (+). The activity is reported as a percentage of the maximum. C, NK92 cells were cocultured for 4 h with normoxic or hypoxic M4T cells at a 3/1 E/T ratio. CD69 surface expression was determined by flow cytometry. D, left panel, M4T-Cx43 cells were incubated under normoxia or hypoxia for 24 h and cocultured with NK92 cells. Conjugates between NK and target cells were analyzed by phase-contrast (PC) microscopy. F-actin (rhodamine-phalloidin) and Cx43 were stained and analyzed by fluorescence microscopy. The accumulation of Cx43 at the immune synapse (IS) is indicated by an arrow. Scale bar = 5 μm. Right panel, quantification of F-actin and Cx43 accumulation at the IS. E, quantification of Cx43 accumulation (left panel) and Tyr(P) fluorescence intensity (right panel) at the IS was determined by analysis of fluorescence microscopy images. F, M4T-Cx43 cells were loaded with Dil-CM and incubated under normoxia or hypoxia for 24 h. Subsequently, melanoma cells were cocultured for 10 min with TFL4-preloaded NK92 cells at a 3/1 E/T ratio. The percentages of NK cells conjugated with target cells were evaluated by flow cytometry. G, top panel, Normoxic or hypoxic M4T-Cx43 cells were cultured alone (−) or with NK92 cells (+) at a 5/1 E/T ratio for 30 min. Melanoma cells separated from NK92 cells were subjected to immunoblot analysis to evaluate the intracellular GzmB. NK92 cell lysate (NK) was used as a control for GzmB detection. Bottom panel, normoxic or hypoxic M4T-Cx43 cells were labeled with TFL4 and cocultured with NK92 cells for 1 h at a 3/1 E/T ratio in the presence of a permeable fluorogenic substrate for GzmB (GranToxiLux). GzmB activity was evaluated in TFL4+ target cells by flow cytometry. H, A735 and SKMel30 melanoma cells were cultured under normoxia or hypoxia for 24 h. Top panel, the expression of HIF-1α and LC3-I and LC3-II (an autophagy marker) was determined by Western blot analysis. Bottom panel, A735 and SKMel30 melanoma cells expressing GFP-Cx43 were subsequently cocultured with NK92 cells, and the localization of Cx43 at the IS (arrowheads) was analyzed by confocal microscopy. Enlarged (×200) regions (boxes) are shown on the right. T, target cells. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Our results showed that, despite the increased level of Cx43 protein expression and the formation of functional Cx43-HC, the NK-mediated killing of hypoxic cells was impaired compared with that of normoxic M4T cells. This impairment was not related to a decreased expression of NK cell-activating (MHC class I-related chain A/B, ULBP1, ULBP2, ULBP3) and inhibitory (MHC class I) ligands on the surface of M4T-Cx43 cells (data not shown).
We next assessed the expression of CD69 as an NK cell activation marker. We observed a moderate but statistically significant increase in the expression of CD69 at the surface of NK cells cocultured for 4 h with normoxic tumor cells compared with its basal expression level of NK cells cultured alone (Fig. 3C). Surprisingly, the increase in expression of CD69 at the surface of NK cells cocultured with hypoxic tumor cells remained very low, or even not significant, after 4 h of coculture. This result indicates that NK cells become slightly more active when cocultured with normoxic target cells compared with their activity when they are cocultured with hypoxic target cells.
Nevertheless, the induction of CD69 in NK cells remains low. We therefore speculate that this could be related to the time point we used to measure the expression of CD69 rather than to an impairment in the activity of NK cells. Indeed, a recent report by Tran et al. (29) provided evidence that the expression of lymphocytic CD69 is detected after 6 h of stimulation, reached a peak at 24 h, and down-regulated after 48 h.
Hypoxia Decreases the Accumulation of Cx43 at the Immune Synapse Formed between Melanoma and NK Cells
We next investigated whether hypoxic stress affects the localization of Cx43 at the cytolytic immunological synapse between NK and M4T cells, where it is supposed to stabilize the synapse and improve NK cell-mediated lysis. Although Cx43 is primarily detected at the immune synapse formed between normoxic M4T and NK cells, its localization at the synapse formed between hypoxic M4T target and NK cells was decreased dramatically (Fig. 3D). The reduction in Cx43 immune synapse localization correlated with the formation of less stable immune synapses characterized by a decrease in both actin cytoskeleton polarization (Fig. 3D) and Tyr(P) staining (Fig. 3E), fewer conjugates with NK cells (Fig. 3F), and a lower level and less activity of NK cell-derived GzmB (Fig. 3G, top and bottom panels, respectively). The hypoxia-mediated decrease in the immunological synapsis localization of Cx43 was also observed in other melanoma cell lines, such as A735 and SKMel30 cells (Fig. 3H). Taken together, these results strongly argue that the defect in the localization of Cx43 at the immunological synapse between hypoxic target and NK cells could be responsible for the impairment of melanoma cell susceptibility to NK cell-mediated killing.
Hypoxia-induced Autophagy Promotes the Degradation of Gap-junctional Cx43 at the NK Cell/Melanoma Immune Synapse
Recent evidence has demonstrated that autophagy contributes to the turnover of the gap junctional Cx43 (GJ-Cx43) (5, 30). We therefore analyzed whether the activation of autophagy in hypoxic melanoma cells is involved in a selective decrease in GJ-Cx43 expression at the immune synapse. Fig. 4A showed that hypoxic M4T cells displayed an increased number of autophagosomes and decreased expression of p62 (Fig. 4B), indicating that the autophagy flux is activated in M4T cells under hypoxia. This was supported by our data showing that the number of LC3-positive puncta per cell was increased following treatment of hypoxic cells with the autophagy inhibitor HCQ (Fig. 4C).
FIGURE 4.

Hypoxia-induced autophagy impairs Cx43 expression at the immune synapse between hypoxic melanoma cells and NK cells. A, the formation of autophagosomes was analyzed by confocal microscopy analysis in Tomato-LC3-expressing M4T-Cx43 cells cultured for 24 h under normoxia or hypoxia. Scale bar = 10 μm. B, the expression of HIF-1α, p62, and β-actin in normoxic (N) or hypoxic (H) M4T-Cx43 cells was determined by Western blot analysis. C, the number of LC3+ puncta per cell was quantified in M4T-Cx43 cells cultured under normoxia or hypoxia for 24 h in the absence or presence of 50 μm HCQ. D, M4T-GFP-Cx43 cells were cultured under normoxia or hypoxia for 24 h in the absence or presence of 50 μm HCQ. Cells were cocultured with NK92, and the localization of Cx43 was evaluated. Immune synapses between target (T) and NK cells are indicated (arrows). Scale bar = 10 μm. E, left panel, M4T cells expressing Tom-Cx43 (red) were transfected with a GFP control plasmid or a GFP siRNA plasmid targeting ATG5 (siATG5). Cells were precultured for 24 h under hypoxia and cocultured for 15 min with NK92 cells. Cx43 localization at the immune synapse is indicated by an arrowhead. Scale bar = 10 μm. Right panel, ATG5 and p62 expression was assessed by Western blot in normoxic or hypoxic M4T-Tom-Cx43 cells stably transfected with psiRNA-hATG5 (psiATG5) or control (Ctrl) empty vectors. The ATG5/actin and p62/actin ratios were calculated by densitometric quantification of the blot. F–H, M4T-GFP-Cx43 cells were transfected with Tom-LC3 (red) and cultured under normoxia or hypoxia in the absence or presence of HCQ (F–H) or 3-methyladenine (3MA, G and H). F, melanoma cells were then cocultured with NK92 cells, and the colocalization of Cx43 with LC3+ autophagosomes was analyzed by confocal microscopy. Enlarged images of the immune synapses (boxes) are shown below each representative image. Cx43-containing autophagosomes are indicated (arrows). PC, phase-contrast images. Scale bar = 10 μm. G, the percentage of Cx43 colocalized with LC3 was evaluated over the time. H, quantification of the number of GFP-Cx43 positive autophagosomes (LC3+) at the IS proximity (2 μm). I, hypoxia-induced autophagy selectively degrades GJ-Cx43 in hypoxic melanoma cells. Left panel, M4T-Cx43 cells were incubated under normoxic or hypoxic conditions for 24 h and subsequently cocultured with NK92 cells for 30 min. Triton X-100-insoluble (TX100-ins) or soluble (TX100-sol) fractions were analyzed by immunoblot for Cx43 detection. The Triton X-100-soluble fraction (center panel) resolves predominately a faster-migrating isoform (P0) that represents non-junctional Cx43 (which is largely unphosphorylated). The Triton X-100-insoluble fraction (top left panel) resolves at least three distinct Cx43 isoforms: the unphosphorylated P0 and two (P1 and P2) phosphorylated isoforms. Top right panel, M4T-Tom-Cx43 cells transfected with control (C) or siRNA-plasmids targeting ATG5 (siATG5) were incubated under normoxia or hypoxia for 24 h in the absence or presence of HCQ. Cells were treated with the protein synthesis inhibitor cycloheximide for an additional 6 h and subsequently cocultured with NK92 cells for 30 min. Triton X-100-soluble or Triton X-100-insoluble fractions were processed for immunoblot against Cx43 and actin. Bottom right panel, densitometric quantification of the hypoxia-induced Cx43 degradation in Triton X-100-soluble or Triton X-100-insoluble fractions. Values are reported as percent of Cx43 levels relative to normoxic conditions. **, p < 0.01.
We next evaluated the impact of inhibiting autophagy on the localization of melanoma Cx43 at the immune synapse formed between NK and normoxic or hypoxic target cells. Fig. 4D shows a significant decrease in the localization of Cx43 at the immune synapse of hypoxic M4T/NK cells, which was rescued by both HCQ treatment (Fig. 4D) and by targeting ATG5 (Fig. 4E). Indeed, M4T stably expressing siRNA against ATG5 showed 80% of ATG5 protein inhibition, and this was correlated with an increase in the expression of p62, indicating that the autophagy flux was blocked efficiently (Fig. 4E, right panel). Taken together, our results strongly argue that hypoxia-induced autophagy could be responsible for a selective degradation of GJ-Cx43 localized at the immune synapse. Consistent with this, we showed a colocalization of Cx43 and LC3 at the proximity of the immune synapse of hypoxic M4T/NK cells, which was abrogated following HCQ or 3-methyladenine treatment (Fig. 4, F–H).
To further assess the effect of hypoxia-induced autophagy on the selective degradation of GJ-Cx43 localized at the immune synapse, we analyzed the accumulation of GJ-Cx43 isolated from normoxic or hypoxic M4T/NK cell cocultures left untreated or pretreated with cycloheximide. Previous reports have shown that GJ-Cx43 is enriched in Triton X-100 insoluble fraction, whereas non-GJ-Cx43 is enriched in Triton X-100 soluble fraction (26). Our results (Fig. 4I, left panel) showed that, even under hypoxic conditions where Triton X-100 soluble Cx43 was up-regulated, the Triton X-100-insoluble GJ-Cx43 was down-regulated dramatically, most likely in an autophagy-dependent degradation manner. Similarly, in cycloheximide-treated hypoxic cells, a selective decrease in the accumulated GJ-Cx43 was observed, which was rescued by HCQ treatment or by targeting ATG5 (Fig. 4I, right panel). However, no significant difference in non-GJ-Cx43 (intracellular Cx43 and Cx43-HCs) was observed under the same conditions. Overall, these results indicate that hypoxia accelerates the removal of Cx43 at the NK cell/target immune synapse by selective degradation of GJ-Cx43 through an autophagy-dependent mechanism.
Inhibition of Autophagy-mediated GJ-Cx43 Degradation in Hypoxic Melanoma Cells Restores Their Susceptibility to NK Cell-mediated Lysis
We next assessed how autophagy selectively degrades GJ-Cx43. Indeed, autophagy has long been viewed as a random cytoplasmic degradation system. However, emerging new evidence suggests that autophagy could be a selective degradation process (31). The discovery and characterization of the autophagic adapter protein cargos have provided mechanistic insights into this process. The most common of these cargo recognition molecules is p62 (32). p62 is able to act as a cargo receptor for the degradation of ubiquitinated substrates. A direct interaction between the autophagic adapter p62 and the autophagosomal marker protein LC3, mediated by a so-called LC3-interacting region motif, is required for efficient selective autophagy. To investigate whether GJ-Cx43 is selectively degraded by autophagy in a p62-dependent manner, we targeted p62 by siRNA and analyzed the expression of GJ-Cx43 at the immunological synapse of hypoxic melanoma cells. Our results clearly showed that targeting the cargo protein p62 (Fig. 5A, left panel) inhibited the degradation of synaptic GJ-Cx43 in hypoxic M4T melanoma cells cocultured with NK cells (Fig. 5A, right panel). These results indicate that p62 is required for the selective degradation of GJ-Cx43 by autophagy.
FIGURE 5.
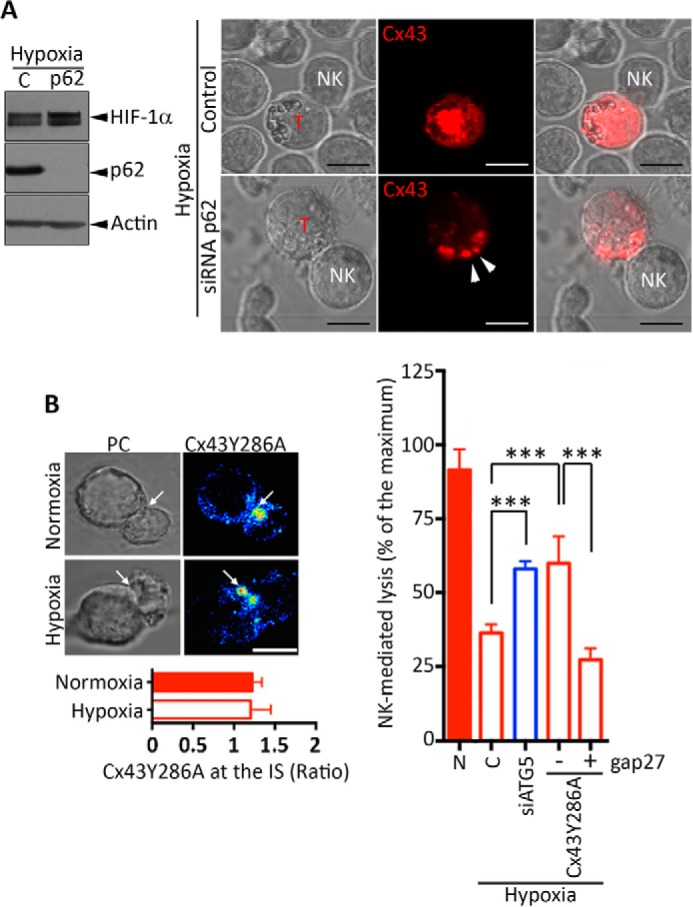
Prevention of autophagy-mediated degradation of Cx43 in hypoxic melanoma cells restores their susceptibility to NK-mediated lysis. A, the role of the cargo protein p62 in the selective degradation of GJ-Cx43 in hypoxic M4T melanoma cells. M4T cells expressing Tomato-Cx43 protein were transfected with control (C) or p62 siRNA (p62) and cultured under hypoxia. The expression of HIF-1α and p62 was assessed by Western blot analysis (left panel). Cells were cocultured with NK92 cells at a 5/1 effector/target ratio, and the accumulation of synaptic Cx43 (arrowheads) was monitored by confocal microscopy. T, target cell. Scale bars = 10 μm. B, left panel, M4T cells were stably transfected with Cx43Y286A. Cells were incubated under normoxia or hypoxia for 24 h and cocultured with NK92 cells for 15 min. Cx43 accumulation at the immune synapse (arrows) was analyzed by confocal microscopy and quantified (bottom left panel). PC, phase-contrast images. Scale bar = 5 μm. Right panel, M4T melanoma cells expressing the wild-type form of Cx43 (columns 1–3) were transfected with a control or a plasmid encoding ATG5 siRNA (siATG5) as well as with Cx43Y286A (columns 4 and 5). Cells were incubated under normoxia (N) or hypoxia for 24 h. NK92-mediated cytotoxicity was determined in the presence of a control (−) or the inhibitory Cx43-mimetic peptide gap27 (+). NK-mediated lysis values are shown as a percentage of the maximum. ***, p < 0.001.
To further support our data showing that hypoxia-induced autophagy is responsible for a selective degradation of synaptic GJ-Cx43, we used an endocytically impaired form of Cx43 (Cx43Y286A) mutated in the tyrosine sorting signal. Cx43Y286A has been described previously to be undegradable by the autophagy machinery (5, 25). We first showed that Cx43Y286A was localized at the immune synapse formed between NK cells and either normoxic or hypoxic M4T cells, where it is supposed to stabilize the synapse and improve NK cell-mediated lysis. Our results (Fig. 5B, left panel) clearly showed that Cx43Y286A variant was still present at the immune synapse of hypoxic cells, indicating that Cx43Y286A resists autophagy-mediated degradation. We next assessed NK-mediated lysis of hypoxic cells expressing Cx43Y286A. Our results provided clear evidence that, similar to targeting ATG5, the expression of Cx43Y286A in hypoxic M4T cells significantly restored their susceptibility to NK cell-mediated lysis. Cx43Y286A was functionally involved in the restoration of NK cell-mediated lysis of hypoxic target cells because such restoration was no longer observed when the Cx43-mimetic peptide gap27 was used (Fig. 5B, right panel). Taken together, our results strongly argue that blocking autophagy in hypoxic M4T cells restores NK-mediated killing mainly by preventing the degradation of GJ-Cx43.
Discussion
Impaired expression and function of Cxs have been observed in different tumor tissues and cell lines, including melanoma (11, 33). Recent data strongly suggest that Cx43 may act as a tumor suppressor gene and predicts clinical outcomes in cancer patients treated with chemotherapy (34). Indeed, several reports have demonstrated that Cx43 expression increased the sensitivity of tumor cells to apoptosis in GJ channel- and HC-dependent and -independent manners (35). Therefore, GJ channels allow the passage of apoptotic signals such as Ca2+ or inositol 1,4,5-trisphosphate (36).
Several reports support a prominent role for Cx43-mediated intercellular communications in different immunological processes, including tumor immunity (6, 37, 38). In line with this, we showed that ectopic overexpression of Cx43 in normoxic melanoma cells stabilizes immune synapse formation and improves NK cell-mediated killing through the formation of GJ-Cx43 between NK and target cells. Under hypoxic stress, Cx43 is overexpressed, but its localization at the immune synapse is impaired dramatically by autophagy. Such an impairment leads to the destabilization of the immune synapse and to tumor escape from NK cell-mediated killing. Given that normoxic melanoma cells formed stable conjugates and that hypoxic cells displaying a defect in GJ-Cx43 formed fewer conjugates with NK cells than with normoxic M4T-Cx43 cells, it is tempting to speculate that Cx43 localized at the immunological synapse may contribute to cell adhesion between NK cells and their targets, as described previously in endothelial cell/B lymphocyte interactions (39).
In response to hypoxia, the transcription factor HIF-1α is rapidly stabilized and translocated to the nucleus, where it binds to the HRE motif (5′-RCGTG-3′) to induce the transcription of many critical genes to sustain cell survival under hypoxic stress (18). Here we identified that HIF-1α induces Cx43 expression in M4T melanoma cells through binding to the promoter of the Cx43 gene (GJA1). In line with this result, we showed that hypoxia induces the expression of Cx43 in M4T and several melanoma cells, including T1, G1, I2, and M4T2 cells. Similar to M4T, there is a hypoxia time-dependent increase in the expression of Cx43 in G1 and I2 cells. However, in T1 cells, the expression of Cx43 was increased gradually after 24 and 48 h and reached a plateau at 72 h of hypoxia. In the case of M4T2 cells, we observed an increase in Cx43 after 24 h of hypoxia, and then its level declined slightly after 48–72 h, although it remained higher than in normoxia. As suggested by our results, it seems that the levels of Cx43 in melanoma cells exposed to chronic hypoxia depend on the balance between its expression by HIF-1α and its degradation through autophagy. Moreover, Cx43 has an intrinsically short half-life (1–3 h) that can vary in different cell lines.
Although the relationship between hypoxic stress and Cxs is not well documented in tumor cells, it has been proposed that the expression of Cx46 may protect breast cancer cells from hypoxia-induced cell death (40). Indeed, the role of Cx43 in hypoxia has been well described in other cell types. For instance, Cx43 is a key mediator of cell protection during ischemia/hypoxia in cardiomyocytes (41).
It has been proposed that human melanoma cells are addicted to functional autophagy for their survival and invasion (42). In this regard, we have reported previously that the activation of autophagy under hypoxic conditions resulted in the degradation of NK cell-derived GzmB in tumor cells, therefore compromising the ability of NK cells to eliminate their targets in vitro and in vivo (19). In this study, we demonstrated that autophagy impairs NK cell-mediated killing of hypoxic melanoma cells through an additional mechanism involving immune synapse destabilization by selective degradation of GJ-Cx43 localized at the NK/melanoma interface.
Recently, several lines of evidence highlighted the role of autophagy in the selective degradation of the pool of Cx43 in plasma membrane GJ plaques (5, 30). Briefly, using Cx43-GFP-expressing HeLa cells, it has been reported that the GJ-Cx43 is internalized in structures called annular GJs, which colocalized with autophagosomes. The inhibition of autophagy resulted in an increase in total levels of exogenously expressed Cx43 and a significant increase in cytoplasmic annular GJs. Using mouse liver tissue and NIH3T3 cells, Bejarano et al. (5) confirmed the degradation of GJ-Cx43 by autophagy and highlighted the role for ubiquitination in the selective targeting of the GJ-Cx43 to starvation-induced autophagosomes. Furthermore, they demonstrated that ubiquitin ligase Nedd4-mediated ubiquitination and the subsequent Eps15-mediated endocytosis were required for Cx43 autophagic degradation. Moreover, it has been shown that a mutation in the PY motif (Tyr-286) of Cx43 blocked its autophagic degradation. Indeed, Cx43 carrying such a mutation fails to interact with Nedd4 (25) and, therefore, prevents the subsequent interaction with Eps15 (5). In this study, we strongly argue that a similar mechanism of autophagy-mediated degradation occurs during NK cell/target interactions under hypoxic conditions. In support of this, we showed that hypoxia did not affect the accumulation of the Cx43-Y286A mutants at the NK/M4T cytolytic immune synapses.
Together, the results reported here provide new insights into the role of Cx43 in regulating melanoma susceptibility to NK-mediated lysis and could, therefore, lead to new therapeutic approaches that involve manipulation of Cx43 expression. Our data also suggest that strategies targeting autophagy in combination with compounds that selectively enhance Cx43 expression in tumor cells (43, 44) would significantly enhance the therapeutic efficacy of NK cell-based melanoma immunotherapies.
Author Contributions
A. T. and B. J. designed the study and performed and analyzed the experiments shown in all figures. B. J. and S. C. wrote the paper. K. V. M. and M. Z. N. performed and analyzed the experiments. S. C. conceived and coordinated the study. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Henrique Girão (University of Coimbra, Portugal) for the Cx43Y286A pcDNA3.1/V5/His vector.
This project was supported by a grant “Equipe labellisée Ligue Contre le Cancer,” European Union Seventh Framework Program Grant FP7/2007–2013 under grant agreement 246556 (European Project RBUCE-UP) and by grants from INSERM and the Université Paris-Sud, by Luxembourg Institute for Health Grant LHCE-2013-1105, and by Fondation Cancer Luxembourg Grant FC/2012/02. The authors declare that they have no conflicts of interest with the contents of this article.
- NK
- natural killer
- GJ
- gap junction
- Cx
- connexin
- HC
- hemichannel
- GzmB
- granzyme B
- HCQ
- hydroxychloroquine
- E/T
- effector/target
- Ab
- antibody
- HRE
- hypoxia-responsive element
- Mut
- mutated
- EV
- empty vector
- IS
- immune synapse.
References
- 1. Mace E. M., Dongre P., Hsu H. T., Sinha P., James A. M., Mann S. S., Forbes L. R., Watkin L. B., Orange J. S. (2014) Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol. Cell Biol. 92, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burke S., Lakshmikanth T., Colucci F., Carbone E. (2010) New views on natural killer cell-based immunotherapy for melanoma treatment. Trends Immunol. 31, 339–345 [DOI] [PubMed] [Google Scholar]
- 3. Nielsen M. S., Axelsen L. N., Sorgen P. L., Verma V., Delmar M., Holstein-Rathlou N. H. (2012) Gap junctions. Compr. Physiol. 2, 1981–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Orellana J. A., Froger N., Ezan P., Jiang J. X., Bennett M. V., Naus C. C., Giaume C., Sáez J. C. (2011) ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 118, 826–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bejarano E., Girao H., Yuste A., Patel B., Marques C., Spray D. C., Pereira P., Cuervo A. M. (2012) Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol. Biol. Cell 23, 2156–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neijssen J., Pang B., Neefjes J. (2007) Gap junction-mediated intercellular communication in the immune system. Progress Biophys. Mol. Biol. 94, 207–218 [DOI] [PubMed] [Google Scholar]
- 7. Benlalam H., Carré T., Jalil A., Noman Z., Caillou B., Vielh P., Tittarelli A., Robert C., Chouaib S. (2013) Regulation of gap junctions in melanoma and their impact on Melan-A/MART-1-specific CD8(+) T lymphocyte emergence. J. Mol. Med. 91, 1207–1220 [DOI] [PubMed] [Google Scholar]
- 8. Elgueta R., Tobar J. A., Shoji K. F., De Calisto J., Kalergis A. M., Bono M. R., Rosemblatt M., Sáez J. C. (2009) Gap junctions at the dendritic cell-T cell interface are key elements for antigen-dependent T cell activation. J. Immunol. 183, 277–284 [DOI] [PubMed] [Google Scholar]
- 9. Mendoza-Naranjo A., Bouma G., Pereda C., Ramírez M., Webb K. F., Tittarelli A., López M. N., Kalergis A. M., Thrasher A. J., Becker D. L., Salazar-Onfray F. (2011) Functional gap junctions accumulate at the immunological synapse and contribute to T cell activation. J. Immunol. 187, 3121–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tittarelli A., Mendoza-Naranjo A., Farías M., Guerrero I., Ihara F., Wennerberg E., Riquelme S., Gleisner A., Kalergis A., Lundqvist A., López M. N., Chambers B. J., Salazar-Onfray F. (2014) Gap junction intercellular communications regulate NK cell activation and modulate NK cytotoxic capacity. J. Immunol. 192, 1313–1319 [DOI] [PubMed] [Google Scholar]
- 11. Naus C. C., Laird D. W. (2010) Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 10, 435–441 [DOI] [PubMed] [Google Scholar]
- 12. Chouaib S., Janji B., Tittarelli A., Eggermont A., Thiery J. P. (2014) Tumor plasticity interferes with anti-tumor immunity. Crit. Rev. Immunol. 34, 91–102 [DOI] [PubMed] [Google Scholar]
- 13. Swartz M. A., Iida N., Roberts E. W., Sangaletti S., Wong M. H., Yull F. E., Coussens L. M., DeClerck Y. A. (2012) Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res. 72, 2473–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sceneay J., Chow M. T., Chen A., Halse H. M., Wong C. S., Andrews D. M., Sloan E. K., Parker B. S., Bowtell D. D., Smyth M. J., Möller A. (2012) Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 72, 3906–3911 [DOI] [PubMed] [Google Scholar]
- 15. Siemens D. R., Hu N., Sheikhi A. K., Chung E., Frederiksen L. J., Pross H., Graham C. H. (2008) Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. 68, 4746–4753 [DOI] [PubMed] [Google Scholar]
- 16. Noman M. Z., Buart S., Van Pelt J., Richon C., Hasmim M., Leleu N., Suchorska W. M., Jalil A., Lecluse Y., El Hage F., Giuliani M., Pichon C., Azzarone B., Mazure N., Romero P., Mami-Chouaib F., Chouaib S. (2009) The cooperative induction of hypoxia-inducible factor-1 α and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J. Immunol. 182, 3510–3521 [DOI] [PubMed] [Google Scholar]
- 17. Noman M. Z., Janji B., Kaminska B., Van Moer K., Pierson S., Przanowski P., Buart S., Berchem G., Romero P., Mami-Chouaib F., Chouaib S. (2011) Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 71, 5976–5986 [DOI] [PubMed] [Google Scholar]
- 18. Semenza G. L. (2010) HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baginska J., Viry E., Berchem G., Poli A., Noman M. Z., van Moer K., Medves S., Zimmer J., Oudin A., Niclou S. P., Bleackley R. C., Goping I. S., Chouaib S., Janji B. (2013) Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. U.S.A. 110, 17450–17455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen J., He L., Dinger B., Stensaas L., Fidone S. (2002) Chronic hypoxia upregulates connexin43 expression in rat carotid body and petrosal ganglion. Journal of applied physiology 92, 1480–1486 [DOI] [PubMed] [Google Scholar]
- 21. Le H. T., Sin W. C., Lozinsky S., Bechberger J., Vega J. L., Guo X. Q., Sáez J. C., Naus C. C. (2014) Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. J. Biol. Chem. 289, 1345–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dufour E., Carcelain G., Gaudin C., Flament C., Avril M. F., Faure F. (1997) Diversity of the cytotoxic melanoma-specific immune response: some CTL clones recognize autologous fresh tumor cells and not tumor cell lines. J. Immunol. 158, 3787–3795 [PubMed] [Google Scholar]
- 23. Puig S., Ruiz A., Lázaro C., Castel T., Lynch M., Palou J., Vilalta A., Weissenbach J., Mascaro J. M., Estivill X. (1995) Chromosome 9p deletions in cutaneous malignant melanoma tumors: the minimal deleted region involves markers outside the p16 (CDKN2) gene. Am. J. Hum. Genet. 57, 395–402 [PMC free article] [PubMed] [Google Scholar]
- 24. Evans W. H., Bultynck G., Leybaert L. (2012) Manipulating connexin communication channels: use of peptidomimetics and the translational outputs. J. Membr. Biol. 245, 437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Catarino S., Ramalho J. S., Marques C., Pereira P., Girão H. (2011) Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem. J. 437, 255–267 [DOI] [PubMed] [Google Scholar]
- 26. Hunter A. W., Barker R. J., Zhu C., Gourdie R. G. (2005) Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 16, 5686–5698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhaskaracharya A., Dao-Ung P., Jalilian I., Spildrejorde M., Skarratt K. K., Fuller S. J., Sluyter R., Stokes L. (2014) Probenecid blocks human P2X7 receptor-induced dye uptake via a pannexin-1 independent mechanism. PloS ONE 9, e93058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Callewaert D. M., Radcliff G., Waite R., LeFevre J., Poulik M. D. (1991) Characterization of effector-target conjugates for cloned human natural killer and human lymphokine activated killer cells by flow cytometry. Cytometry 12, 666–676 [DOI] [PubMed] [Google Scholar]
- 29. Tran K., Clor J., Tyagarajan K. (2013) Kinetics of lymphocyte activation in cytokine expression studies (P6317). J. Immunol. 190, 184–12023225889 [Google Scholar]
- 30. Fong J. T., Kells R. M., Gumpert A. M., Marzillier J. Y., Davidson M. W., Falk M. M. (2012) Internalized gap junctions are degraded by autophagy. Autophagy 8, 794–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rogov V., Dötsch V., Johansen T., Kirkin V. (2014) Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 53, 167–178 [DOI] [PubMed] [Google Scholar]
- 32. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 33. Haass N. K., Smalley K. S., Herlyn M. (2004) The role of altered cell-cell communication in melanoma progression. J. Mol. Histol. 35, 309–318 [DOI] [PubMed] [Google Scholar]
- 34. Sirnes S., Bruun J., Kolberg M., Kjenseth A., Lind G. E., Svindland A., Brech A., Nesbakken A., Lothe R. A., Leithe E., Rivedal E. (2012) Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int. J. Cancer 131, 570–581 [DOI] [PubMed] [Google Scholar]
- 35. Carette D., Gilleron J., Chevallier D., Segretain D., Pointis G. (2014) Connexin a check-point component of cell apoptosis in normal and physiopathological conditions. Biochimie 101, 1–9 [DOI] [PubMed] [Google Scholar]
- 36. Kameritsch P., Khandoga N., Pohl U., Pogoda K. (2013) Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis. 4, e584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mazzini E., Massimiliano L., Penna G., Rescigno M. (2014) Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity 40, 248–261 [DOI] [PubMed] [Google Scholar]
- 38. Saccheri F., Pozzi C., Avogadri F., Barozzi S., Faretta M., Fusi P., Rescigno M. (2010) Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Sci. Transl. Med. 2, 44ra57. [DOI] [PubMed] [Google Scholar]
- 39. Machtaler S., Dang-Lawson M., Choi K., Jang C., Naus C. C., Matsuuchi L. (2011) The gap junction protein Cx43 regulates B-lymphocyte spreading and adhesion. J. Cell Sci. 124, 2611–2621 [DOI] [PubMed] [Google Scholar]
- 40. Banerjee D., Gakhar G., Madgwick D., Hurt A., Takemoto D., Nguyen T. A. (2010) A novel role of gap junction connexin46 protein to protect breast tumors from hypoxia. Int. J. Cancer 127, 839–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waza A. A., Andrabi K., Hussain M. U. (2014) Protein kinase C (PKC) mediated interaction between conexin43 (Cx43) and K(+)(ATP) channel subunit (Kir6.1) in cardiomyocyte mitochondria: implications in cytoprotection against hypoxia induced cell apoptosis. Cell. Signal. 26, 1909–1917 [DOI] [PubMed] [Google Scholar]
- 42. Ma X. H., Piao S., Wang D., McAfee Q. W., Nathanson K. L., Lum J. J., Li L. Z., Amaravadi R. K. (2011) Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. 17, 3478–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Conklin C. M., Bechberger J. F., MacFabe D., Guthrie N., Kurowska E. M., Naus C. C. (2007) Genistein and quercetin increase connexin43 and suppress growth of breast cancer cells. Carcinogenesis 28, 93–100 [DOI] [PubMed] [Google Scholar]
- 44. Liu H., He Z., Simon H. U. (2013) Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin. Cancer Biol. 23, 352–360 [DOI] [PubMed] [Google Scholar]