

Abstract
A shortage of available organ donors has created a need for engineered tissues. In this context, polymer-based hydrogels that break down inside the body are often used as constructs for growth factors and cells. Herein, we report imine cross-linked gels where degradation is controllable by the introduction of mixed imine cross-links. Specifically, hydrazide-functionalized poly(ethylene glycol) (PEG) reacts with aldehyde-functionalized PEG (PEG-CHO) to form hydrazone linked hydrogels that degrade quickly in media. The time to degradation can be controlled by changing the structure of the hydrazide group or by introducing hydroxylamines to form non-reversible oxime linkages. Hydrogels containing adipohydrazide-functionalized PEG (PEG-ADH) and PEG-CHO were found to degrade more rapidly than gels formed from carbodihydrazide-functionalized PEG (PEG-CDH). Incorporating oxime linkages via aminooxy-functionalized PEG (PEG-AO) into the hydrazone cross-linked gels further stabilized the hydrogels. This imine crosslinking approach should be useful for modulating the degradation characteristics of 3D cell culture supports for controlled cell release.
Keywords: Hydrogel, poly(ethylene glycol), hydrazone, oxime, imine, tissue engineering, degradable, cell release, cell delivery
INTRODUCTION
Hydrogels are a common scaffold for tissue engineering due to their biocompatibility and tunable mechanical properties.1,2 Scaffolds are commonly prepared from synthetic materials as they allow for more precise control over gel properties and circumvent the problems associated with natural materials such as immune response and batch-to-batch variability.3 Many types of hydrogels have been designed to mimic the extracellular matrix (ECM), which provides a complex environment for cells.4–8 The ECM contains various biofactors that serve as cellular cues and is comprised of glycans and fibrous proteins that provide dynamic structural support.9,10 Controlling gel stiffness, elasticity and degradability are therefore important factors in designing synthetic dynamic scaffolds. Cell spreading and mobility, as observed in the ECM, can be achieved in synthetic scaffolds by incorporating integrin-binding peptide sequences, such as RGD, and incorporating hydrolytically degradable cross-links into the hydrogel.3,11 Many current hydrogel systems provide cells with a rigid environment that may not allow for cell migration and proper interaction with cellular and biophysical cues.12 By introducing reversible cross-links that allow for self-healing and selective degradation, synthetic hydrogels can become more dynamic in order to more closely resemble the complex structure of the ECM.
Click chemistry is one of the most widely used methods to easily and rapidly form hydrogels. While numerous click chemistries have been applied to hydrogel formation such as Michael addition, focusing on biocompatible reactions that occur readily at physiological conditions eliminates risks such as cell toxicity and undesirable side products.13–15 Oxime and hydrazone formation are two examples of reversible, biocompatible click reactions. Both reactions are ideal for tissue engineering applications since they occur readily at physiological pH and produce water as the only byproduct.
It has been shown by our group and others that oxime bonds form stable, biocompatible hydrogels in vitro.16,17 We have previously demonstrated that oxime chemistry can be successfully employed to create stable, bioactive hydrogels using 8-arm aminooxy poly(ethylene glycol) (PEG) and glutaraldehyde. Oxime bond formation was pH dependent, which therefore allowed for control of the time to hydrogel formation by adjusting pH. While oximes are reported to degrade via hydrolysis at decreased pH values, this degradation does not occur readily at pH values compatible with cell culture. Degradability could be incorporated through the use of enzyme-sensitive peptide cross-linkers or polymers, although this can add synthetic complexities, or through the introduction of less stable imine cross-links, such as hydrazone bonds.18,19 Imine cross-linked hydrogels are known to exhibit stimuli-responsive and self-healing properties due to bond reversibility,20,21 which make them ideal systems for a number of potential clinical applications, such as drug delivery and tissue engineering.22–25
Anseth and coworkers recently reported rapid formation of PEG-based hydrogels via hydrazone bonds for cell culture.12,26,27 Hydrazone chemistry has also been applied to other hydrogel systems, such as poly(N-isopropylacrylamide) hydrogels. Patenaude and coworkers determined that hydrazone cross-links were the weakest points in these gels, indicating that this is where degradation would occur most readily.28–30 The reversibility of hydrazone bonds in hydrogels has also been probed by Deng and coworkers, who functionalized poly(ethylene oxide) with a hydrazide group to form hydrazone hydrogels. They observed that gelation can be reversed below pH 4.31 In different systems, it has been reported that cells are able to degrade, and even prevent formation of hydrazone-linked hyaluronic acid and alginate-based hydrogels.32
Oxime and hydrazone bonds form from the reaction between carbonyl groups and an aminooxy or hydrazide group, respectively,33,34 and we hypothesized that the mutual reactivity towards carbonyl groups allows these reactions to be used concomitantly to tune degradability in hydrogels. In this paper, we describe a PEG-based hydrogel system that combines oxime and hydrazone chemistries to form hydrogels with tunable degradability and mechanical properties. PEG was strategically used as the polymer scaffold due to its biocompatibility and ease of functionalization.35 PEG was functionalized to contain two different hydrazide groups, adipohydrazide and carbodihydrazide, capable of reacting with aldehyde-functionalized PEG in order to generate hydrazone hydrogels with different degradation rates. Because of the potential of hydrazone bonds to reverse, we chose to use an aldehyde functionalized PEG instead of glutaraldehyde, which is known to be cytotoxic at low millimolar concentrations.36 In addition, PEG was modified with an aminooxy group to incorporate oxime chemistry into the hydrogel to further control gel degradation rates for in vitro applications.
EXPERIMENTAL
Materials
Preparation of all of the PEG precursors and characterization is described in the Supporting Information.
Hydrogel Formation
Equal parts PEG-CHO and PEG-hydrazide/PEG-AO were combined in phosphate buffer for a total polymer concentration of 3.5 or 5.0 wt.%. The polymer solution was vortexed before being pipetted onto a hydrophobic glass slide with 1 mm spacers and sandwiched using a second hydrophobic glass slide. Hydrophobic glass slides were prepared by coating glass slides with a silanization reagent for glass (Sigmacote®) by dipping clean glass slides into the reagent solution for 5–10 minutes. Glass slides were then heated in an oven for 24 hours to allow hydrophobic coating to set before rinsing slides with water.
Rheological Characterization
40 μL gels containing ratios of PEG-CDH/PEG-ADH, PEG-AO and PEG-CHO were made by adding pH 5.5 phosphate buffer and 20 wt.% PEG-CDH/PEG-ADH/PEG-AO solutions and mixing thoroughly. Then 20 wt.% PEG-CHO was added and the solution was mixed for ten seconds. Gel solutions were sandwiched between two hydrophobic glass slides separated by 1 mm spacers. The newly formed gels were added to buffer or media 10 minutes after gelation. Gels were swollen for 18 hours and liquid was refreshed once before taking measurements. Each gel condition was made and tested in triplicate. The gels were measured on a plate-to-plate Anton Paar rheometer (Physica MCR 301, Anton Paar, Ashland, VA) using an 8 mm plate with an angular frequency range of 0.1 to 10 s−1 under a constant strain of 1% at 37°C.
Swelling Studies
Gels were swollen in water for three days before measuring the mass of the swollen hydrogels (ms). The water was refreshed four times before the measurements were taken. The gels were lyophilized to remove water and weighed again to determine the dry mass (md). Gels were made in triplicate for each condition. The degree of swelling was calculated using where ρP is the density of the polymer solution (1.04 g/mL) and ρS is the density of the solution, in this case water (1.00 g/mL).
Degradation Studies
5 wt.% gels were swollen in phosphate buffer (pH 5.6) or Dulbecco’s modified eagle’s medium (DMEM) with or without fetal bovine serum (FBS) or mMSC conditioned DMEM. Buffer and medium were replaced daily during the course of the experiments. Gels were weighed daily over the course of six days. Gels for each condition were prepared in triplicate. Gels containing PEG-ADH degraded in complete DMEM before the six days were over and could therefore not be measured for the full extent of the experiment.
mMSC Encapsulation
AO-RGD (0.1, 0.5 or 1 mM final concentration) and PEG-CHO were dissolved in phosphate buffer. The two solutions were mixed together at the calculated ratios and allowed to react at 37 °C for 3 hours prior to setting up cell experiments. mMSCs in complete DMEM (3,500 or 5,000 cells/μL final concentration) were added to the AO-RGD/PEG-CHO solution and vortexed gently. The final components of the gel solution (5 total wt.% PEG-CDH and/or PEG-AO) were added to the AO-RGD/PEG-CHO/cell solution. 5 μL gels were pipetted onto a hydrophobic glass slide with 1 mm spacers and sandwiched using a second hydrophobic slide. The gels were incubated at 37 °C for 15 minutes to allow for gelation. The gels were then added into the wells of a 96-well plate containing 200 μL complete DMEM.
Cell viability and spreading
mMSC viability was studied with a LIVE/DEAD® viability/cytotoxicity kit (Molecular Probes, Eugene, OR). Briefly, 1 μl of ethidium homodimer-1 and 0.25 μl of calcein AM from the kit were diluted with 500 μl DMEM to make the staining solution. Each gel was stained with 150 μl of staining solution for 30 min at 37 °C in the dark before imaging. To better analyze cell spreading, gels were fixed for 5 min at RT using 4% PFA, rinsed with PBS, treated with 0.1% triton-X for 10 min and stained for 90 min in the dark with DAPI for cell nuclei (1:500 dilution from 5 mg/mL stock, Invitrogen, Grand Island, NY) and Alexa Fluor 488-phalloidin (1:200 dilution, Invitrogen, Grand Island, NY)) in 1% sterile filtered bovine serum albumin solution. The samples were then washed with 0.05% tween-20. For cell viability, an inverted Observer Z1 Zeiss fluorescent microscope was used to visualize samples. To better visualize the distribution throughout the hydrogel, multiple z-stacks 1.9–2.3 μm thick were taken for each image, deconvolved to minimize background, and presented as orthogonal projections. For cell spreading, a Nikon C2 confocal microscope was utilized to visualize samples. For 20X images, z-stacks 160 μm thick were imaged at 1.8 μm intervals. For 40X images, z-stacks 110 μm thick were imaged at 1 μm intervals with a water immersion lens. All confocal images were presented as maximum intensity projections.
RESULTS AND DISCUSSION
Both the aldehyde and amine containing components necessary for imine hydrogel formation were prepared from 8-arm PEG (MW 20,000 Da). Two different hydrazide polymers were synthesized via EDC-coupling of 8-arm PEG-COOH with either adipohydrazide and carbodihydrazide.29 These reactions yielded PEG-adipohydrazide (PEG-ADH) and PEG-carbodihydrazide (PEG-CDH) with 99% and 80% end group conversion, respectively (Figures S1–S2). The 8-arm PEG-aldehyde (PEG-CHO), was made via a Williamson ether synthesis using 8-arm PEG-OH and 2-bromo-1,1-diethoxyethane to yield a protected aldehyde with average end group conversion of 90% in 50% yield (Figure S3). The acetal was cleaved by stirring the polymer solution in pH 2 phosphate buffer at 60°C for 18 hours in 60% yield with 85% end group conversion (Figure S4).37 The stability of PEG-acetal allowed for prolonged storage of the polymer prior deprotection to PEG-aldehyde. Several direct oxidations were attempted as alternative synthesis routes, including PCC and Moffatt oxidation. These approaches resulted in low conversion rates and resulting aldehydes were more difficult to store. Aminooxy-functionalized PEG (PEG-AO) was prepared via a Mitsunobu reaction with N-hydroxyphthalimide, followed by reaction with hydrazine to yield PEG-AO with 94% end-group conversion in 58% overall isolated yield (Figures S5–S6).16
We planned to form hydrogels by mixing equal parts hydrazide and/or aminooxy-functionalized PEG in phosphate buffer to form a hydrogel with hydrazone and/or oxime cross-links (Figure 1). Because we had previously explored hydrogels formed by oxime chemistry,16 initial experiments focused on the hydrazide only gels. Hydrogels consisting of PEG-ADH and PEG-CHO components (1:1 ratio) were studied to determine the optimal pH and wt.%. We carried out hydrazone gel formation experiments at increasing pH levels to determine effects on gelation time. Gel components (5.0 wt.% PEG-ADH and PEG-CHO) were mixed together in phosphate buffer ranging from pH 5–7, and it was observed that all gels formed within five minutes. Since oxime formation slows down significantly around pH 7.0,16 we chose to continue our experiments with phosphate buffer pH 5.5 to allow for efficient oxime and hydrazone bond formation.38 We tested several different polymer concentrations and chose 3.5 wt.% and 5 wt.% PEG, which had optimal gelation rates and physical properties. At these concentrations, gels formed within 60 seconds at pH 5.5, which allowed for easy pipetting of gel solutions. Rheological characterization of PEG-ADH gels revealed that decreasing the total polymer concentration or changing the ratio of hydrazide to aldehyde groups lowers the storage modulus (Figure S7).
Figure 1.
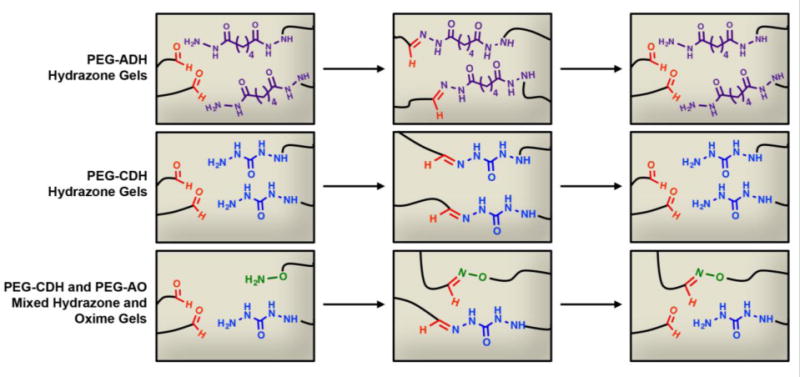
PEG-CHO can react with PEG-ADH and PEG-CDH to form degradable hydrogels via reversible hydrazone cross-links and can also react with PEG-AO to form nonreversible oxime bonds to stabilize the hydrazone gels.
To confirm that the hydrogels formed through hydrazone bonds and not as a result of electrostatic interactions, we performed a model study to analyze bond formation by NMR. Adipohydrazide and propionaldehyde were mixed in water, and the resulting product was analyzed by 1H and 13C NMR to monitor hydrazone bond formation (Figure S8). Propionaldehyde, which has the same structure as the end group of PEG-CHO, can react with adipohydrazide without cross-linking, allowing for straightforward monitoring of bond formation. The appearance of the hydrazone peaks between 7.43–7.28 ppm (1H NMR) and 151.4–148.1 ppm (13C NMR) indicated hydrazone bond formation. The mechanism is expected to be the same for the PEG-based hydrogel system.
An advantage of utilizing hydrazone chemistry is that the bonds are reversible and can break, then re-form again.39 To test this in our system, two different hydrogels containing different colored dyes were first physically cut in half, and the halves of the two gels were swapped and placed together. Within ten minutes the dangling hydrazide and aldehyde groups reacted to re-form a single, uniform gel (Figure 2). After the gel re-formed, the blue dye took on a green color, indicating the two dyes were able to come into contact and mix throughout the gel, further indicating the two gel halves re-formed into one gel. A higher concentration of yellow dye than blue dye was added to the initial gel solutions in order to create a vivid color, which most likely accounts for the observed color change from blue to a teal in the re-healed gels. Since hydrazone bonds are the weakest link in the gel, it is expected that these bonds are broken most easily upon physical cutting and this provides evidence that the bonds can re-form.39 It should be noted that gels containing oxime bonds did not show the same re-healing, likely because these bonds are not readily reversible.
Figure 2.

5.0 wt.% PEG hydrazone hydrogels (40 μL, approximately 1 cm in diameter and 1 mm in height) dyed with food coloring shown during self-healing. PEG-ADH hydrogels formed within one minute (A) and were then cut in half (B). The opposite halves were placed next to each other (C), and after ten minutes, bonds re-formed (D). After fifteen minutes, the two pieces formed one complete gel that can readily be handled (E).
Next, hydrogel swelling in various conditions (e.g. buffer, culture medium) was investigated to analyze the differences between PEG-ADH and PEG-CDH gels (Figure 3). Both PEG-ADH and PEG-CDH containing gels were stable up to at least six days in pH 5.5 phosphate buffer, indicating that both hydrogels had reached equilibrium and did not swell further in aqueous conditions. In DMEM lacking fetal bovine serum (DMEM (−) FBS), PEG-ADH gels exhibited a significant increase in swelling and fully degraded after five days. However, in DMEM with 10% FBS, PEG-ADH based gels swelled rapidly over two days before dissolving completely (Figure 3A). Enzymes present in fetal bovine serum or amines from proteins or peptides could be responsible for the degradation observed for PEG-ADH gels. PEG-CDH based gels, on the other hand, remained intact and exhibited minimal swelling whether in buffer, DMEM (−) FBS, or (+) FBS, indicative of greater bond stability (Figure 3B). This is expected since it has been shown that changing the structure of the dihydrazide group used as a cross-linker can affect hydrazone bond stability by increasing resonance stabilization by shortening the carbon chain between hydrazide groups.40
Figure 3.
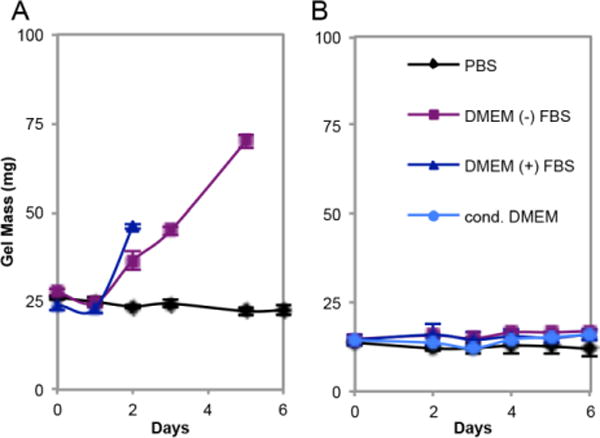
Swelling of 5 wt.% PEG-ADH (A) and 5 wt.% PEG-CDH (B) based hydrogels. 40 μL gels were placed in respective solutions, and PEG-ADH gels degraded after five days in DMEM (−) FBS and after two days in DMEM (+) FBS. PEG-CDH gels retained constant masses throughout the experiment for all conditions, including in cell conditioned DMEM.
To further analyze the difference in time of degradation of our PEG-ADH and PEG-CDH hydrogels, an NMR study was designed to determine the extent to which the two different hydrazone bonds can be reversed in the presence of a nucleophile (Figure 4). CDH and ADH hydrazones were formed by reacting each hydrazide group with one equivalent of propionaldehyde (Figures S9–11). Hydroxylamine was chosen as the nucleophile in this case to match our mixed hydrazone/oxime gel system. After the addition of hydroxylamine to CDH hydrazone, approximately 60% of hydrazone bonds were replaced by oxime bonds after 30 minutes (Figure 4A), which can be seen by the appearance of two new imine peaks at 7.37 and 6.70 ppm corresponding to propionaldehyde oxime (E and Z),41 and a decrease in peak height corresponding to the hydrazone proton. In comparison, 90% of hydrazone bonds were replaced upon the addition of hydroxylamine to ADH hydrazone (Figure 4B) after 30 minutes. In this case, the hydrazone protons at 7.42 and 7.28 ppm are replaced almost entirely by oxime protons at 7.35 and 6.69 ppm. The same ratios of hydrazone to oxime remained constant up to at least 12 hours for both CDH and ADH compounds, suggesting equilibrium is reached. CDH hydrazone was displaced to a lesser extent by hydroxylamine than ADH hydrazone, further indicating that CDH hydrazone bonds are more stable than ADH hydrazones and can help explain why PEG-ADH gels may have degraded in the presence of DMEM, due to amines or other nucleophilic compounds, whereas PEG-CDH gels remained stable.
Figure 4.
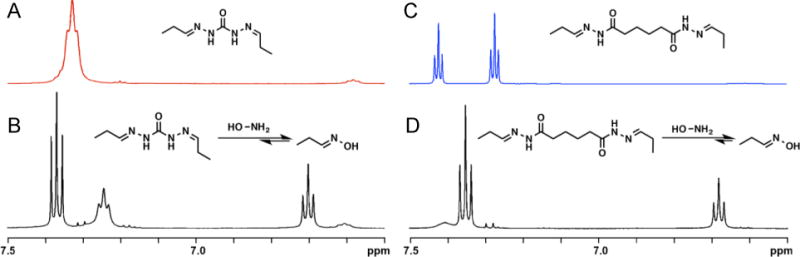
1H NMR of CDH hydrazone (A) after adding one equivalent hydroxylamine amine per hydrazone bond (B), and 1H NMR of ADH hydrazone (C) after adding one equivalent hydroxylamine per hydrazone bond (D).
Next, the stability of the gels was tested in the presence of mouse mesenchymal stem cells (mMSCs). To anchor cells inside the gel, a commonly used integrin-binding peptide sequence, arginine-glycine-aspartic acid (RGD), was synthesized using standard fmoc solid phase synthesis (Figure S12).42–44 We chose to incorporate a synthetic peptide instead of a natural material, such as collagen, to retain a simple and fully synthetic system. A hydroxylamine, (Boc-aminooxy) acetic acid, was incorporated into the peptide sequence (AO-RGD) at the N-terminus to allow for coupling to PEG-CHO via stable oxime bonds. Covalent attachment of the peptide to PEG-CHO was assessed using rheology to analyze the difference in storage modulus of PEG-CDH gels containing 0, 0.1, or 1 mM AO-RGD (Figure S13), which are well within the RGD concentration range (25 μM – 3.5 mM) reported to support cell spreading in PEG-based hydrogels.45–48 For gels containing 1.0 mM RGD, the ratio of aldehyde functional groups to AO-RGD is 9.96:1. Indeed, it was observed that gels prepared with AO-RGD exhibited lower storage moduli than gels containing no RGD, indicating cross-linking sites of PEG-CHO were taken up by the peptide. We chose to continue with gels containing no more than 1 mM RGD to ensure PEG-CHO would retain enough free aldehyde groups to cross-link after reacting with the peptide and that the resulting hydrogels would be stiff enough to handle. To ensure oxime bond formation between AO-RGD (0.5 mM) and PEG-CHO, the peptide and polymer were incubated at 37 °C for 3 hours prior to use in cell encapsulation studies. Gels (5 μL total volume) were prepared by mixing PEG components and cells together in sterile pH 5.5 phosphate buffer. The resulting gels were then placed in 100 μL media in a 96-well plate and incubated at 37 °C. The wells were inspected via light microscope daily. Upon degradation, gels were no longer observed in the wells and previously encapsulated cells were found spread on the bottom of the well. PEG-ADH gels were observed to degrade more rapidly than without cells, within 24 hours, whether 3,500 or 5,000 cells/μL initial cells were added. Interestingly, PEG-CDH gels, which were stable in DMEM in the absence of cells, degraded between 4–5 days at 5,000 cells/μL density, which led us to hypothesize that cells may play a role in the degradation process (Figure 4 A, B).
To further understand the degradation mechanism, PEG-CDH gels were incubated in cell (mMSC) conditioned DMEM without cells; in this case, the gels were completely stable up to at least 6 days (Figure 3B). We believe there are several possible explanations for the observed increased degradation rate in the presence of cells, including that the cells mechanically break the hydrazone bonds or that the mesh size of the gels is altered in the presence of the cells, allowing for more rapid hydrolysis. Because cell-secreted enzyme concentrations are much lower in solution than at the cell-substrate interface,49 we cannot entirely rule out that secreted proteins or other molecules do not cause degradation from this experiment, especially since it has been reported that enzymes released from the cells can affect hydrazone bond stability.50
In order to tune the degradation rate of our hydrazone gels further, we incorporated oxime chemistry since these bonds do not reverse even when cell are present.16 Because PEG-ADH gels degraded so quickly, the rest of the experiments were conducted with the more stable PEG-CDH partner only. Gel solutions containing 3:1:4, 2:2:4, and 1:3:4 parts PEG-CDH: PEG-AO: PEG-CHO were mixed and rapidly formed 3.5 wt.% overall PEG hydrogels. When the same solutions were mixed without PEG-CDH (so the gel solutions contained 1:4, 2:4 and 3:4 parts PEG-AO: PEG-CHO) only the 3:4 gel solution resulted in a gel. This indicates that PEG-CDH is participating in cross-linking in the mixed gels. Rheology was used to assess the stiffness of the mixed gels at 3.5 wt.% and 5 wt.% PEG gels (Figure 5A). For each condition, the combined amount of PEG-AO and PEG-CDH was equal to the total amount of PEG-CHO, and gels containing only hydrazone bonds, only oxime bonds, or a 1:1 mixture of hydrazone and oxime bonds were formed. All 5 wt.% PEG gels exhibited higher storage moduli than the 3.5 wt.% gels, corresponding to the increase in number of cross-links with increasing polymer concentration.
Figure 5.
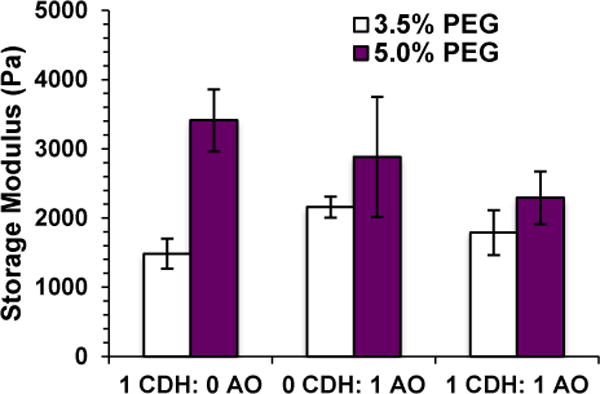
Rheological characterization of hydrazone and oxime hydrogels. Storage modulus can be controlled by changing total polymer concentration and amounts of hydrazone and oxime cross-links. Data is displayed as the average and standard deviation of three independent hydrogels for each condition.
To explore the stabilizing effects of increasing amounts of oxime cross-links in the presence of cells, gels containing 100:0, 75:25, 50:50 and 0:100 hydrazone: oxime bonds were prepared, placed in wells containing media, and examined under a light microscope daily (Figure S14). While we had observed that 100:0 hydrazone: oxime gels degrade fully within 5 days, leaving cells at the bottom of the well, 75:25 hydrazone: oxime gels remained intact at day 6 although a change in gel structure is apparent (Figure S14D). The gel edges were no longer as defined as they were on day 1, and cells are starting to be released from the gel, allowing them to spread on the well surface (Figure S14C–D). Gels containing 50:50 and 0:100 hydrazone: oxime bonds did not show any degradation and cells remained encapsulated (Figure S14E–H).
To study the viability and spreading of cells in the presence of cells, mMSCs were encapsulated in gels containing PEG-CDH and PEG-AO at various ratios. Hydrogels for cell encapsulation studies were formed by adding mMSCs (in DMEM (+) FBS) to the PEG-CHO/AO-RGD (0.1 mM RGD) solution before the addition of PEG-CDH and/or PEG-AO to result in 3.5 and 5 wt.% PEG gels. Live/dead staining of encapsulated cells at days 1 and 7 showed high viability (> 90%) of cells in all gel conditions, indicating that the imine hydrogels at all ratios are biocompatible with cells (Figure 6). Cytotoxicity studies of individual PEG components were also carried out to ensure the polymers are not damaging to the cells upon gel degradation (Figure S15). All polymers (PEG-AO, PEG-CHO, PEG-CDH, and PEG-ADH) were noncytotoxic to mMSCs up to at least 10 mg/mL, which corresponds to twice the amount of each PEG used per gel in the cell studies.
Figure 6.
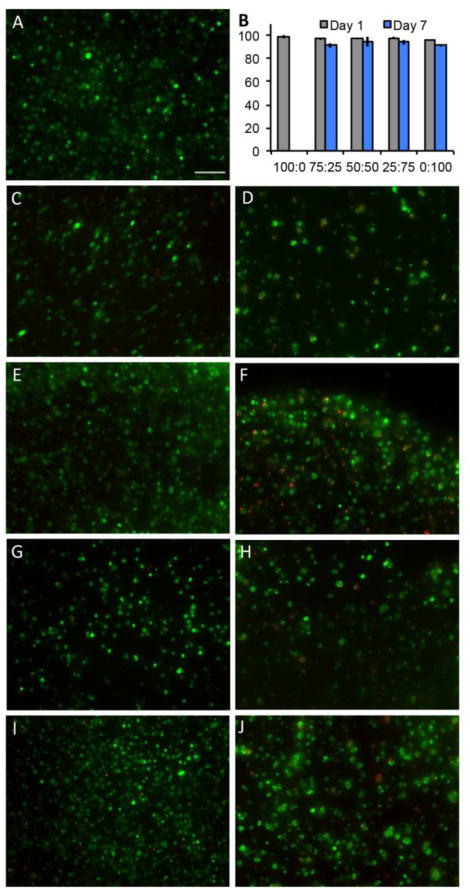
Live/dead staining of encapsulated mMSCs. 100:0 hydrazone: oxime gels showed good viability at day 1 (A) but degraded before imaging at day 7. 75:25 hydrazone: oxime gels at day 1 (C) and day 7 (D); 50:50 gels at day 1 (E) and day 7 (F); 25:75 gels at day 1 (G) and day 7 (H); 0:100 gels at day 1 (I) and day 7 (J). Scale bar = 100 μm. Cell viability of all gel conditions at day 1 and day 7 (B) represented as % live cells per gel (y axis).
Further, the morphology of encapsulated cells in the presence of AO-RGD was investigated. Gels containing 0.1 mM AO-RGD were stained using DAPI/Phalloidin for f-actin after fixing with paraformaldehyde at day 5 (Figure S16). mMSCs exhibited rounded morphologies at all hydrazone: oxime ratios, although 100:0 hydrazone: oxime gels were not imaged due to degradation (Figure 7A–C). The experiment was repeated using human dermal fibroblasts (HDFs), and again, cells were round with little spreading (Figure S16 D–F). In order to image 100:0 hydrazone: oxime gels before degradation, the experiment was repeated and gels were fixed and stained at day 2 (Figure 7). This time, AO-RGD concentration was increased to 0.5 mM to determine if increasing RGD affects cell spreading in our gel system. Gels containing 1 mM RGD were also examined (Figure S17). Cells maintained similar, rounded morphologies for all tested RGD concentrations. This finding is in contrast to a similar hydrazone cross-linked PEG hydrogel system, but the use of 4-arm PEG instead of 8-arm PEG and hydrazine instead of hydrazide end groups introduce several differences that make cell morphologies in the two systems difficult to compare.27
Figure 7.
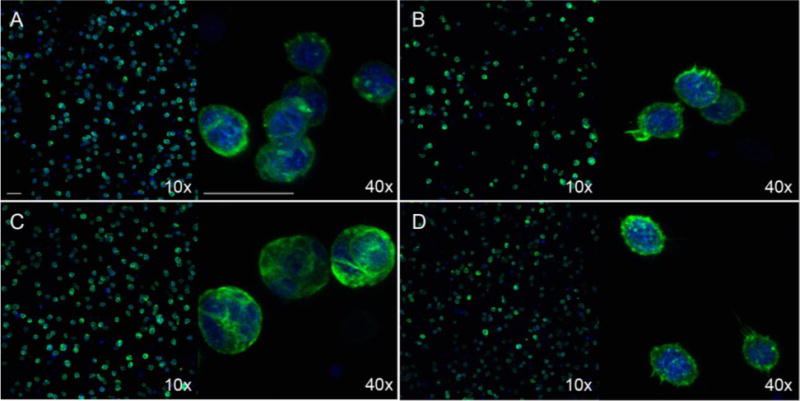
DAPI/Phalloidin staining for cell spreading at day 2. mMSCs were encapsulated in 5 wt.% 100:0 hydrazone: oxime (A), 75:25 hydrazone: oxime (B), 50:50 hydrazone: oxime (C), and 0:100 hydrazone: oxime (D) gels containing 0.5 mM AO-RGD. Scale bars = 50 μm for both 10x and 40x magnification.
With the versatility and tunable design of our imine hydrogels it can be envisioned that the amine component or aldehyde component could be additionally altered to further change the time to degradation in these types of gels. For example, glyoxylamide type carbonyl moieties should degrade more quickly than aldehyde and various hydrazones could be utilized. These gels could then be used in situations where cells or other agents need to be selectively released or, because of the self-healing properties, a mixture of both. In addition, this system could be potentially used not only to encapsulate and deliver cells, but could be used to deliver growth factors or other therapeutic agents. This could be accomplished through simple encapsulation inside the gel or by covalent attachment to the cross-linked PEG network. Due to the possibility of incorporating such biological compounds and the wide range in stiffness of the hydrogels from 1000–3500 Pa, our current system could potentially be used for vascular or endothelial engineering applications.51 Since we have shown that increasing the amount of PEG per gel significantly increases gel stiffness, our gel system could be tuned to match the conditions of a wide variety of stiffer tissues, such as muscle, while retaining the ability to tune degradability of the system as needed.
CONCLUSIONS
In this report, a PEG-based imine hydrogel platform that combines the biocompatibility and stability of oxime bonds with the reversibility of hydrazone bonds to create hydrogels with tunable degradation is reported. The ease with which PEG can be functionalized allows for further modification of these gels through modification of hydrazide groups and incorporation of hydroxylamine groups. It was demonstrated that by combining oxime and hydrazone chemistries, the stability of hydrogels could be tuned from less than 24 hours to greater than 7 days. High cell viability in the presence of a covalently bound RGD peptide was observed. The ability to tune the mechanical properties and degradation of these hydrogels as well as to encapsulate cells will allow for this system to be used for research and clinical applications, such as for cell delivery and tissue engineering.
Supplementary Material
Acknowledgments
H.D.M. thanks the NIH NIBIB (R01 EB 136774-01A1) for funding. N.B. thanks the NIH Funded Biotechnology Training Grant (5T32GM067555-09) for a predoctoral fellowship. E.B. thanks the Netherlands Organization for Scientific Research and Marie Curie Cofund Action for the financial support (Rubicon Grant 680–50–1101).
Footnotes
SUPPORTING INFORMATION AVAILABLE
Experimental procedures, model study, and additional rheology data. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.MacArthur BD, Oreffo ROC. Nature. 2005;433:19–19. doi: 10.1038/433019a. [DOI] [PubMed] [Google Scholar]
- 2.Lenas P, Luyten FP. Ind Eng Chem Res. 2011;50:482–522. [Google Scholar]
- 3.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 4.DeLong SA, Moon JJ, West JL. Biomaterials. 2005;26:3227–3234. doi: 10.1016/j.biomaterials.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 5.Bryant SJ, Anseth KS. J Biomed Mater Res A. 2003;64A:70–79. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 6.DeForest CA, Anseth KS. Nat Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeForest CA, Sims EA, Anseth KS. Chem Mater. 2010;22:4783–4790. doi: 10.1021/cm101391y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeForest CA, Polizzotti BD, Anseth KS. Nat Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tibbitt MW, Anseth KS. Biotechnol Bioeng. 2009;103:655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker EL, Bonnecaze RT, Zamao MH. Biophys J. 2009;97:1013–1021. doi: 10.1016/j.bpj.2009.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.West JL, Hubbell JA. Macromolecules. 1999;32:241–244. [Google Scholar]
- 12.McKinnon DD, Domaille DW, Brown TE, Kyburz KA, Kiyotake E, Cha JN, Anseth KS. Soft Matter. 2014;10:9230–9236. doi: 10.1039/c4sm01365d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Nimmo CM, Shoichet MS. Bioconjugate Chem. 2011;22:2199–2209. doi: 10.1021/bc200281k. [DOI] [PubMed] [Google Scholar]
- 15.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 16.Grover GN, Lam J, Nguyen TH, Segura T, Maynard HD. Biomacromolecules. 2012;13:3013–3017. doi: 10.1021/bm301346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin F, Yu J, Tang W, Zheng J, Defante A, Guo K, Wesdemiotis C, Becker M. L. Biomacromolecules. 2013;14:3749–3758. doi: 10.1021/bm401133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson SB, Lin CC, Kuntzler DV, Anseth KS. Biomaterials. 2011;32:3564–3574. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei YG, Gojgini S, Lam J, Segura T. Biomaterials. 2011;32:39–47. doi: 10.1016/j.biomaterials.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang B, Zhang YL, Zhang XY, Tao L, Li SX, Wei Y. Polym Chem. 2012;3:3235–3238. [Google Scholar]
- 21.Zhang YL, Tao L, Li SX, Wei Y. Biomacromolecules. 2011;12:2894–2901. doi: 10.1021/bm200423f. [DOI] [PubMed] [Google Scholar]
- 22.Lee KY, Alsberg E, Mooney DJ. J Biomed Mater Res. 2001;56:228–233. doi: 10.1002/1097-4636(200108)56:2<228::aid-jbm1089>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 23.Varghese OP, Sun WL, Hilborn J, Ossipov DA. J Am Chem Soc. 2009;131:8781–8783. doi: 10.1021/ja902857b. [DOI] [PubMed] [Google Scholar]
- 24.Hudson SP, Langer R, Fink GR, Kohane DS. Biomaterials. 2010;31:1444–1452. doi: 10.1016/j.biomaterials.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding CX, Zhao LL, Liu FY, Cheng J, Gu JX, Shan-Dan, Liu CY, Qu XZ, Yang ZZ. Biomacromolecules. 2010;11:1043–1051. doi: 10.1021/bm1000179. [DOI] [PubMed] [Google Scholar]
- 26.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Chem Mater. 2014;26:2382–2387. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Adv Mater. 2014;26:865–872. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patenaude M, Hoare T. Biomacromolecules. 2012;13:369–378. doi: 10.1021/bm2013982. [DOI] [PubMed] [Google Scholar]
- 29.Patenaude M, Hoare T. ACS Macro Lett. 2012;1:409–413. doi: 10.1021/mz200121k. [DOI] [PubMed] [Google Scholar]
- 30.Alves MH, Young CJ, Bozzetto K, Poole-Warren LA, Martens PJ. Biomed Mater. 2012;7 doi: 10.1088/1748-6041/7/2/024106. [DOI] [PubMed] [Google Scholar]
- 31.Deng G, Tang C, Li F, Jiang H, Chen Y. Macromolecules. 2010;43:1191–1194. [Google Scholar]
- 32.Dahlmann J, Krause A, Moller L, Kensah G, Mowes M, Diekmann A, Martin U, Kirschning A, Gruh I, Drager G. Biomaterials. 2013;34:940–951. doi: 10.1016/j.biomaterials.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 33.Kool ET, Park DH, Crisalli P. J Am Chem Soc. 2013;135:17663–17666. doi: 10.1021/ja407407h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crisalli P, Kool ET. J Org Chem. 2013;78:1184–1189. doi: 10.1021/jo302746p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 36.Sun HW, Feigal RJ, Messer HH. Pediatr Dent. 1990;12:303–307. [PubMed] [Google Scholar]
- 37.Zhao YJ, Zhai YQ, Ma GH, Su ZG. J Appl Polym Sci. 2009;111:1638–1643. [Google Scholar]
- 38.Dirksen A, Hackeng TM, Dawson PE. Angew Chem Int Ed. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 39.Deng GH, Tang CM, Li FY, Jiang HF, Chen YM. Macromolecules. 2010;43:1191–1194. [Google Scholar]
- 40.Oommen OP, Wang SJ, Kisiel M, Sloff M, Hilborn J, Varghese OP. Adv Funct Mater. 2013;23:1273–1280. [Google Scholar]
- 41.Krylov IB, Terent’ev AO, Timofeev VP, Shelimov BN, Novikov RA, Merkulova VM, Nikishin GI. Adv Synth Catal. 2014;356:2266–2280. [Google Scholar]
- 42.Ruoslahti E, Pierschbacher MD. Cell. 1986;44:517–518. doi: 10.1016/0092-8674(86)90259-x. [DOI] [PubMed] [Google Scholar]
- 43.Ruoslahti E. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 44.Merrifield RB. J Am Chem Soc. 1963;85:2149. –&. [Google Scholar]
- 45.Moon JJ, Saik JE, Poche RA, Leslie-Barbick JE, Lee SH, Smith AA, Dickinson ME, West JL. Biomaterials. 2010;31:3840–3847. doi: 10.1016/j.biomaterials.2010.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, Garcia AJ. Adv Mater. 2012;24:64–70. 62. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bott K, Upton Z, Schrobback K, Ehrbar M, Hubbell JA, Lutolf MP, Rizzi SC. Biomaterials. 2010;31:8454–8464. doi: 10.1016/j.biomaterials.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 48.Adeloew C, Segura T, Hubbell JA, Frey P. Biomaterials. 2008;29:314–326. doi: 10.1016/j.biomaterials.2007.09.036. [DOI] [PubMed] [Google Scholar]
- 49.Bat E, van Kooten TG, Feijen J, Grijpma DW. Biomaterials. 2009;30:3652–3661. doi: 10.1016/j.biomaterials.2009.03.033. [DOI] [PubMed] [Google Scholar]
- 50.Wei Z, Yang JH, Liu ZQ, Xu F, Zhou JX, Zrínyi M, Osada Y, Chen YM. Adv Funct Mater. 2015;25:1352–1359. [Google Scholar]
- 51.Levental I, Georges PC, Janmey PA. Soft Matter. 2007;3:299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.