

Abstract

Transient biomolecular interactions are the cornerstones of the cellular machinery. The identification of the binding sites for low affinity molecular encounters is essential for the development of high affinity pharmaceuticals from weakly binding leads but is hindered by the lack of robust methodologies for characterization of weakly binding complexes. We introduce a paramagnetic ligand tagging approach that enables localization of low affinity protein–ligand binding clefts by detection and analysis of intermolecular protein NMR pseudocontact shifts, which are invoked by the covalent attachment of a paramagnetic lanthanoid chelating tag to the ligand of interest. The methodology is corroborated by identification of the low millimolar volatile anesthetic interaction site of the calcium sensor protein calmodulin. It presents an efficient route to binding site localization for low affinity complexes and is applicable to rapid screening of protein–ligand systems with varying binding affinity.
Introduction
Molecular recognition is essential to biological processes. The determination of the binding cleft, and preferably the binding mode, of lead compounds has become a fundamental step of structure-based drug design. Protein binding site locations for medium-to-high affinity ligands can be pinpointed by crystallization methods, cryo-electron microscopy (cryo-EM),1−3 photoaffinity labeling,4 or by nuclear magnetic resonance (NMR) chemical shift mapping and intermolecular cross-relaxation experiments.5−7 For weakly interacting systems, with affinity constants in the millimolar range, cocrystallization is challenging, and cryo-EM densities, which are based on averaging, become too weak to identify the position of the ligand. The NMR chemical shift response is also population-averaged, with the inherent small molar ratio of the bound state making the majority of the signal content originate from the uncomplexed molecules. Although low-affinity interactions can be confirmed using a variety of techniques, e.g., isothermal titration calorimetry,8 surface plasmon resonance,9 or ligand-detected NMR techniques such as water-LOGSY10 and saturation transfer difference experiments,5 the spatial localization of protein interaction sites for low-affinity ligands is still cumbersome. It is, thereto, commonly impeded by limiting molecular solubility and concomitant unspecific binding events at high ligand concentrations.
We propose the applicability of ligand-transferred paramagnetic NMR restraints for binding site identification of low-affinity drugs and drug candidates. The interaction of the unpaired electrons of a ligand-contained paramagnetic radical or lanthanoid ion with the nuclear spins of a nearby protein results in extensive paramagnetic alterations of conventional biomolecular NMR spectra already at a low ligand concentration. The foremost manifestations are paramagnetic relaxation enhancement (PRE), residual dipolar couplings (RDC), and pseudocontact shifts (δPCS), which can all be obtained from the spectroscopic analysis of the differences in spin relaxation rates, J-couplings, and chemical shifts, respectively, for nuclei interacting with a paramagnetic ion or a diamagnetic reference ion.11 The presented methodology utilizes paramagnetic ligand tagging to identify protein binding sites by the covalent attachment of a paramagnetic lanthanoid ion complexing chelate to the ligand of interest to induce pseudocontact shifts (δPCS) on the interacting protein (Figure 1).
Figure 1.

Schematic illustration of the sensitivity gained when using the presented methodology. The arbitrary chemical shift of a protein nucleus, shown in the 1D NMR trace (in gray), is altered when in fast equilibrium with either a native or diamagnetically tagged ligand (dark gray, δM), or with a paramagnetically tagged ligand (red, δPCS). For weak affinity ligands, the protein nucleus chemical shift change caused by a diamagnetic ligand (δM) may be very small, whereas the pseudocontact shift induced by a paramagnetic ligand (δPCS) is reliably measurable.
The location of the paramagnetic ion and hence the area for the binding site is obtained by fitting the position, relative rotation, and size of the paramagnetic susceptibility tensor to the δPCS and the protein structural coordinates, according to eq 1,
| 1 |
where δPCS is the measured pseudocontact shift for any protein nuclear spin, ΔXax and ΔXrh are the axial and rhombic components of the magnetic susceptibility tensor, whereas the angles θ and φ describe its orientation with respect to the protein coordination frame, and r is the lanthanoid–nuclear spin distance.12 Contact shift contributions can be neglected for protein nuclei due to the noncovalent nature of the interaction.12
Lanthanoid metals have found long use as relaxation agents and chemical shift reagents in both small molecule and biomolecular systems.13,14 Ions from the lanthanoid series (Ln3+) have comparable ionic radii and strongly coordinate with metal binding sites in proteins or with chelating ligands. Except La3+ and Lu3+, which are the commonly used diamagnetic references, all lanthanoid ions are paramagnetic. The methods available for incorporation of lanthanoid ions to invoke protein paramagnetic NMR restraints follow three principles: (i) the lanthanoid ion locates directly to a pre-existing protein metal binding site, (ii) a lanthanoid ion binding tag is covalently engineered to a protein terminus or side chain, or (iii) a lanthanoid ion complexing soluble ligand interacts specifically or nonspecifically with a protein.15,16 Direct metal ion–lanthanoid ion replacement and high affinity lanthanoid ion complexing peptides17−19 or chelating agents16,20 covalently anchored to the protein will, in addition to influencing the NMR signals of the tagged protein itself, induce paramagnetic effects on noncovalent interaction partners. This can be used to convey binding site information on protein–protein21−23 or protein–ligand24−27 interactions. A covalently protein-attached tag based on an EDTA derivative was designed early on.28 Tags with one protein anchor point29−31 were then succeeded by more rigid tags18,32,33 and tags with dual anchoring points in order to restrict tag movements.34−39 Spin labels not containing lanthanoids, such as TEMPO, were also applied.40,41 Although paramagnetic protein tagging may be used to convey information on protein–ligand complexation, the opposite approach, paramagnetic tagging of the ligand, provides a valuable complement. Hitherto it has almost exclusively been applied to measurements of ligand induced protein PRE effects to probe protein solvent accessible areas15,42,43 or used in MRI applications with Gd3+ chelates,44 with the exception of sparse examples of ligand induced protein δPCS used to resolve resonance overlap,45 to facilitate resonance assigning46 and to improve structure determination.47,48 Canales et al. have also elegantly demonstrated the use of a lanthanoid tagged sugar moiety to confirm the previously reported μM affinity galectin-3–lactose interaction.49 Efficient paramagnetic tagging of small molecules was also previously reported.50−54 However, the full potential of ligand lanthanoid tagging as a route to study protein–ligand binding of varying affinity has not yet been explored.
Herein, we demonstrate the applicability of paramagnetic labeling of a weakly binding pharmacon for identification of its binding site. The interaction of the anesthetic agent sevoflurane (Figure 2) and the calcium signaling protein calmodulin was selected for validation of the methodology. This interaction is specific and localized to two hydrophobic binding surfaces, one in each of the two calmodulin lobe domains.55,56 Calmodulin has been found to associate with over 300 intracellular targets, including several ion channels that have been shown to be targeted by anesthetics. For example, calmodulin associates with the ryanodine receptor with nanomolar affinity,57 and in doing so represses its activity.58 Calmodulin is also known to affect inactivation in voltage-gated sodium and calcium channels.59,60 Mutations in the ryanodine receptor gene in combination with sevoflurane may lead to the potentially lethal condition of malignant hyperthermia. Volatile anesthetics, such as sevoflurane have also been shown to perturb the activity of the ligand-gated ion channels (GABA)A and that of glycine receptors, but the low affinities of these molecular recognition phenomena have so far hindered their in-depth mechanistic investigations. Consequently, the mode of anesthetic actions remains unresolved and a subject for further studies, despite its direct clinical relevance.
Figure 2.

1H NMR spectrum at 800 MHz of the Dy3+ complexed sevoflurane analogue showing an unusually wide range of chemical shift dispersion, ca. 800 ppm. The inset shows the structure of sevoflurane.
The calmodulin–sevoflurane interaction was very recently described.55,56 Its binding constants are on the limit of the applicability of conventional NMR techniques, i.e., Kd = 9–18 mM, and the low aqueous solubility and high volatility of sevoflurane renders the detection of its protein binding especially challenging, posing it as a particularly suitable test system for validation of this methodology. For paramagnetic tagging, sevoflurane was attached to DOTA, 1,4,7,10-tetraazacyclododecane-N′-N″-N‴-N′′′′-tetraacetic acid, which has high affinity toward lanthanoid ions.61 In order to cause the smallest possible electronic and structural perturbation, a terminal fluorine of sevoflurane was substituted with oxygen (Figure 2, Scheme S2), the atom most similar in electronic properties to fluorine. This modification retains the hydrogen bond accepting ability and the electron-withdrawing character and simultaneously allows the attachment of DOTA. To exclude a direct DOTA–calmodulin interaction, a truncated analogue in which sevoflurane was replaced by a methyl functionality (Scheme S1) was applied as a negative control. Individual samples of the DOTA-bound sevoflurane were complexed separately to each of the lanthanoid ions La3+, Eu3+, Yb3+, or Dy3+. Intramolecular ligand δPCS of up to 550 ppm were measured after complexation to Dy3+ (Figure 2). We therefore anticipated that binding of the paramagnetically labeled drug to its protein binding site would induce chemical shift changes larger in magnitude than those caused by the magnetic susceptibility of the drug itself, due to the pseudocontact shift phenomenon, see Figure 1. In addition to the increased sensitivity of detection, the known δPCS dependence on the distance to and on the spatial localization of the paramagnetic lanthanoid is expected to enable direct location of the binding site area.
Results
The lanthanoid chelates (Dy3+, Yb3+, Eu3+, or La3+, the latter used as the diamagnetic reference) of the DOTA-bound sevoflurane (Scheme S2) as well as of the control ligand in which a methyl group replaces the sevoflurane adduct (Scheme S1) are highly water-soluble. Tight lanthanoid complexation is evident from the 1H NMR spectrum of the Dy3+ complexed sevoflurane analogue covering an impressive 850 ppm spectral width (Figure 2) despite macrocycle conformational exchange, as expected from previous studies, visible as additional line broadening in the 1H NMR spectrum of the La3+ complexed sevoflurane analogue.61,62
For analysis of each calmodulin binding site independently, the calmodulin N-lobe (residues 1–78) and C-lobe (79–148) were expressed and purified separately. It is well-established that the individual calmodulin lobes retain the same properties as in the full length protein.63 Intact structural integrities of the lobes were sustained after comparison of the lobes’ 1HN–15N chemical shifts to those of native calmodulin under identical conditions. Slight chemical shift deviations as a result of the short 3/6 residue N-terminal sequence expansion (for both lobes, see Materials and Methods) were found for the first N-terminal residues and additionally for residues sequentially located to the flexible interlobe hinge region in native calmodulin, expected from the sequence division being executed at residue 78. As the further analysis is highly dependent on the correct structural coordinates, δPCS for the terminal residues (1–3 and 75–86) were excluded. Chemical shift comparisons of free calmodulin and calmodulin with the La3+ chelated sevoflurane analogue resulted in negligible differences (Figure 3) and ensured that the diamagnetic ligand interaction did not invoke large overall changes to the protein structural coordinates and that the available calmodulin coordinates could be used to fit the magnetic susceptibility tensor and location of the paramagnetic center to both lobes, respectively. Amide 1HN and 15N and methyl 1HMe and 13C δPCS were obtained from pairwise comparison of chemical shifts from 1H–15N/13C-HSQC spectra of 0.1 mM U–13C,15N labeled calmodulin lobes, respectively, titrated with either the La3+ or the Dy3+ complexed sevoflurane analogue at 0.05, 0.1, and 0.2 mM; see Figures 3 and 4. Resonances displaying the most extensive δPCS broadened beyond detection at 0.2 mM ligand concentration due to PRE and ligand induced chemical exchange line broadening.25,64 The optimal trade-off between the number of observable resonances and size of the δPCS were determined to be at a 0.1 mM ligand concentration, resulting in a maximum δPCS of −0.24 ppm for Met109ε 1HMe and 13C. The expected magnitude of the induced protein δPCS can be estimated from the δPCS of the free ligand and the affinity constant. For low millimolar affinity ligands with the current sample conditions, the expected protein occupancy is thus around 1%. The 1H δPCS obtained for the Dy3+ complex of the sevoflurane analogue are −8.9 ppm and −6.5 ppm, for its (CH2) and CH, respectively. Consequently, protein nuclei in the calmodulin–ligand complex at near equidistance of the sevoflurane protons to the paramagnetic center are expected to exhibit δPCS on the order of 0.1 ppm depending on the respective relative orientations of the magnetic susceptibility tensor, the ligand, and the protein. The experimentally observed δPCS, as illustrated for 1HN nuclei in Figure 4, agree well with this assumption. The Dy3+ complexed sevoflurane analogue did not produce measurable protein 1JCH3 RDCs, due to the very small degree of alignment produced by the weak binding of a flexible system and combined RDC dynamic averaging. Ligands chelating to Eu3+ and Yb3+ produced considerably smaller induced protein δPCS than the Dy3+ complex, which was alone found to be sufficient for the study. Identical experiments comprising the control ligand, with the DOTA-attached sevoflurane being replaced by a methyl group, were run to exclude the possibility of the transferred paramagnetic restraints originating from specific interaction of the lanthanoid chelating cage with either of the calmodulin lobes. The protein 1HN δPCS recorded with the Dy3+ (and La3+) complexed control ligand were negligible (< ±0.015 ppm) for both lobes, see Figure 4. The few minor but significant δPCS seen for the control ligand are over an order of magnitude smaller than the δPCS observed for the DOTA-attached sevoflurane analogue ensuring that the parameters fitted in Table 1 truly originate from specific sevoflurane–calmodulin interactions.
Figure 3.
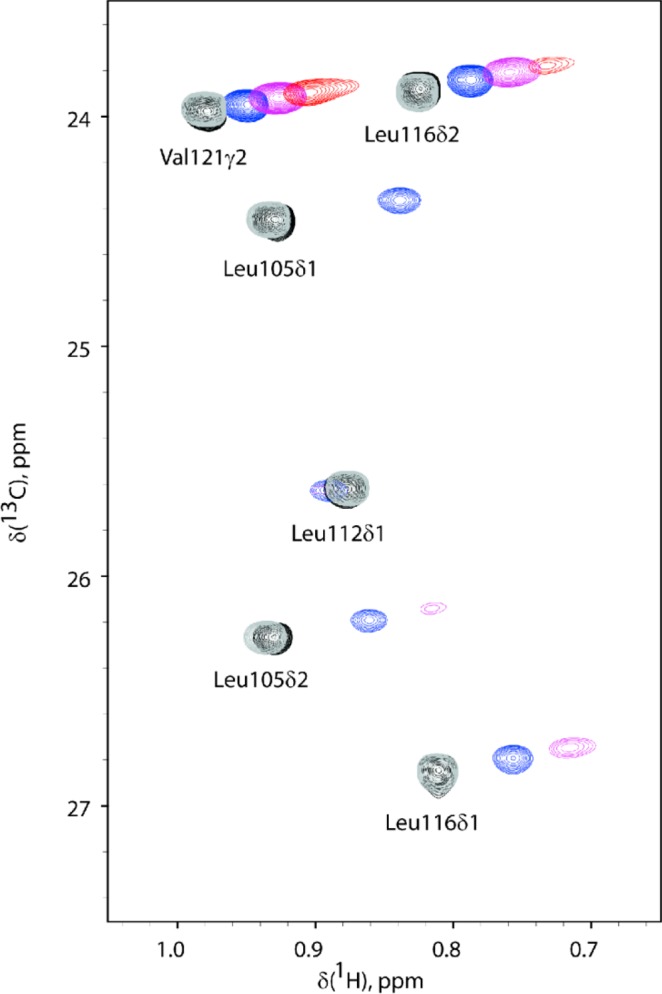
Overlay of 2D NMR 1H–13C HSQC spectra of calmodulin C-lobe titrated with the lanthanoid La3+/Dy3+ complexed sevoflurane analogues. 1H–13C HSQC spectra of 0.1 mM calmodulin C-lobe (black) titrated with 0.05 mM (blue), 0.1 mM (purple), and 0.2 mM (red) Dy3+ complexed sevoflurane analogue are shown. Resonances transverse along approximately parallel lines with the addition of the paramagnetic ligand. Some peaks are broadened beyond detection at the higher concentration of the Dy3+ complexed sevoflurane analogue. The true δPCS are calculated with respect to the chemical shifts detected upon titration with 0.1 mM La3+ complexed sevoflurane analogue (gray). The sensitivity of the methodology is illustrated by the size of the induced pseudocontact shift (chemical shift differences for calmodulin in the presence of 0.1 mM Dy3+ or La3+ complexed sevoflurane analogue, spectra in magenta and gray, respectively) as compared to the size of the chemical shift changes induced by the nonparamagnetic ligand (chemical shift difference for calmodulin in the absence or presence of 0.1 mM La3+ complexed sevoflurane analogue, spectra in black and gray, respectively).
Figure 4.

Calmodulin lobe δPCS induced by Dy3+ labeled sevoflurane and control ligands. Amide proton δPCS in ppm versus sequence for 0.1 mM calmodulin lobes, consecutively plotted, in the presence of 0.1 mM of either the sevoflurane ligand (black, filled circles) or control compound (grey crosses) complexed to Dy3+.
Table 1. Parameters Describing the Individual Lobe Magnetic Susceptibility Tensors Obtained from δPCS Using the Dy3+ Labeled Sevoflurane Analogue and the La3+ Labeled Diamagnetic Referencea.
| N-lobe | std | C-lobe | std | |
|---|---|---|---|---|
| ΔXax (10–32 m3) | –0.208 | 0.034 | 0.839 | 0.065 |
| ΔXrh (10–32 m3) | –0.045 | 0.025 | 0.266 | 0.046 |
| x (Å) | 17.3 | 0.4 | 16.6 | 0.2 |
| y (Å) | 12.3 | 0.4 | 25.8 | 0.4 |
| z (Å) | –5.4 | 0.5 | –25.7 | 0.4 |
| α (deg) | 118 | 7 | 177 | 54 |
| β (deg) | 101 | 4 | 52 | 24 |
| γ (deg) | 36 | 12 | 170 | 51 |
Values are reported for equimolar 0.1 mM calmodulin lobe and ligand concentrations. The axial and rhombic components of the magnetic susceptibility tensor are subject to scaling with the protein occupancy. Parameters were fitted with the Numbat software69 using the experimental δPCS and the calmodulin structure with PDB ID 1X02.68 Errors estimated are reported as one standard deviation (std) based on 1000 times repeated fitting with random removal of 10% of the δPCS in each run.
The affinity constants for the tagged sevoflurane analogue and the control ligand for the respective calmodulin lobes were measured using ITC. Given the limited solubility of sevoflurane, previous measurements of the affinity were conducted using competition experiments,55 prone to larger errors, but the increased ligand solubility upon tagging enabled high ligand concentrations and thus the use of direct ITC measurements. Titrations of tagged sevoflurane analogue into either the calmodulin N-lobe or C-lobe yielded heats significantly higher than those observed for the tagged control ligand (Figure S11). The tagged sevoflurane analogue was observed to have comparable affinity to the two lobes of calmodulin as sevoflurane itself; thus a Kd of ∼4.0 mM was detected for the N-lobe and ∼1.8 mM for the C-lobe. The residual heats for the tagged control ligand were featureless and did not indicate any detectable binding to either lobe.
Measurements of δPCS were also done by ligand titrations to full-length calmodulin (Figure S10). The signs and magnitudes of the δPCS correlated well with those obtained for the individual lobes; however, the concomitant binding of two molecules of paramagnetically labeled sevoflurane to the protein prevented extraction of δPCS induced by each sevoflurane interaction independently. The δPCS obtained for full-length calmodulin were used to verify the interaction, but physical lobe separation was necessary for a single magnetic susceptibility tensor fit and quantitative evaluation. It should, however, be noted that, although the δPCS detected on full-length calmodulin were not quantitatively evaluated, they qualitatively indicated ligand binding at two distinct clefts.
The location of the ligand-attached lanthanoid in the protein–ligand complex can thus be derived from the experimental fit of the induced protein δPCS to one magnetic susceptibility tensor. A single magnetic susceptibility tensor is found for a paramagnetically tagged ligand weakly interacting (fast exchange) with a protein even in the event of ligand diastereomers (as has been shown for the DOTA derivatives) as the induced protein δPCS are motionally averaged.62,65,66 The reliability of the magnetic susceptibility tensor determination from mobile paramagnetic tags has been thoroughly investigated.67 A sizable number of δPCS were extracted for each of the calmodulin lobes, 190 δPCS for the N-lobe and 162 δPCS for the C-lobe. For each lobe, the lanthanoid position and the parameters of the magnetic susceptibility tensor were fitted simultaneously to the structural coordinates of calmodulin (PDB ID 1X02(68)) using Numbat,69 and the values are reported in Table 1.
The previously reported higher calmodulin C-lobe sevoflurane affinity is preserved as reflected in the larger magnitude δPCS and fitted tensor ΔXax and ΔXrh for the C-lobe as compared with the N-lobe. For both lobes, the fitted lanthanoid position is in close proximity to one face of the lobe, see Figure 5, and allowed for the spatial localization of one single binding area in each calmodulin lobe. The sevoflurane binding sites both locate to the exposed, methionine-rich surface areas previously shown to bind free sevoflurane. Although the relative rotation of the sevoflurane adduct with respect to the location of the lanthanoid ion remains undisclosed, the true protein–ligand binding interface is restricted to lay within a radius equal to the maximum distance from the lanthanoid ion to the sevoflurane adduct. This distance, around 14 Å, essentially sweeps out a spherical sector of possible binding sites on the calmodulin surface area. Indeed, the distance from the lanthanoid ion to the methionine Hε protons previously showing intermolecular HOESY cross peaks with free sevoflurane is 10–13 Å.
Figure 5.
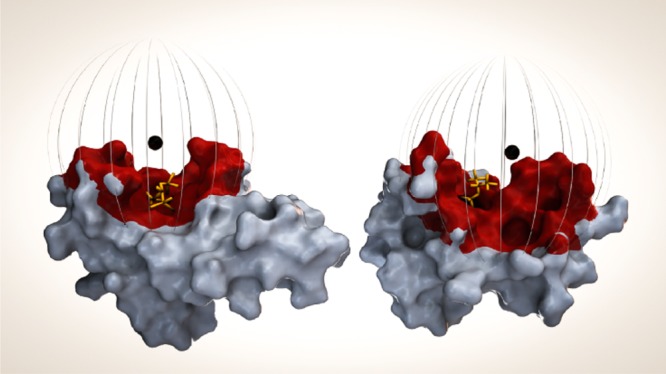
Sevoflurane binding surface as determined by the presented methodology. Calmodulin, N-lobe (left) and C-lobe (right), surface representation with the lanthanoid position represented by a black sphere. Sevoflurane, as positioned previously from conventional NMR restraint methods,55 is indicated in yellow. Atoms within 14 Å of the lanthanoid position are color coded red and represent the determined binding surface. The 14 Å radius is also illustrated by a striped sphere.
Discussion
The presented methodology allows for protein binding site localization at low ligand concentration even for very weakly interacting compounds, in the event that the overall protein structure is preserved upon complexation. Paramagnetic tagging of sevoflurane resulted in sizable induced protein δPCS, adequate for determination of magnetic susceptibility tensors in complexes with either calmodulin lobe, despite the dynamic nature of the low affinity sevoflurane–calmodulin lobe interaction. For both lobes, the determination of the lanthanoid coordinates positions the anesthetic part of the ligand to a calmodulin surface area surrounding the previously determined binding site for free sevoflurane, with the higher C-lobe to N-lobe sevoflurane affinity preserved, illustrating that the synthetic modification did not significantly alter the molecular recognition. ITC affinity measurements further confirmed comparable binding coefficients for the tagged sevoflurane analogue and free sevoflurane, as well as qualitatively supported the higher calmodulin C-lobe to N-lobe binding affinity.
The sensitivity of the method, as compared to conventional chemical shift mapping, is illustrated by the similar calmodulin amide and methyl chemical shift alterations obtained with 10 mM sevoflurane and with 0.1 mM of the Dy3+ complexed sevoflurane analogue, at 0.1–0.2 mM protein concentrations.55 The negative control ligand with the sevoflurane moiety replaced by a methyl group did not induce substantial δPCS, indicating that DOTA itself does not interact with the protein. The δPCS obtained for the paramagnetic-tagged sevoflurane are indeed the result of the sevoflurane–calmodulin interaction and increase the sensitivity of detection by a factor of one hundred, as compared to conventional chemical shift titration. It should be noted that 1H δPCS induced by paramagnetically tagged bioactive compounds are also manifested in 1H NMR spectra of proteins devoid of isotopic enrichment and may therefore be used for rapid screening.
The absolute values of the δPCS can be increased by titration of more ligand to the protein sample, although to some extent this is concomitant with increased line broadening. The magnitude of the δPCS could also be tuned by alternation of the lanthanoid metal ion due to their relative difference in magnetic susceptibility anisotropy. This provides an opportunity for increased accuracy by combined fitting of δPCS from a series of alternating lanthanoid ions even in the event of only a few recordable or assigned δPCS. For the current study, an examination of the data obtained using the paramagnetic lanthanoid Dy3+, with La3+ as reference at equimolar 0.1 mM protein and ligand concentrations, gave sufficient δPCS to corroborate the analysis. Ligands complexed to either Eu3+ or Yb3+ resulted in smaller protein δPCS. Due to the sensitivity of the methodology δPCS alterations were reliably measured at this low, ∼1%, binding site occupancy and physiologically relevant, low sevoflurane analogue concentrations, comparable to that applied in a previous investigation,55 could be used. Attachment of sevoflurane to the lanthanoid binding tag has also increased its solubility, permitting the use of direct ITC measurement to determine its binding constants; however, this did not substantially influence the affinities.
The elucidation of the unknown binding site of a bioactive compound by the current tagging approach requires synthetic modification, i.e., attachment of a paramagnetic tag. This modification is chemically straightforward, as shown here for sevoflurane, but may risk affecting, or in the worst case even prohibiting, the binding in case a functional group involved in the protein recognition is modified. It should be underlined that this, and the fact that the paramagnetic tag is comparable in size to the ligand itself, by no means present risks greater than those taken upon fluorescent labeling, which to date is carried out on a routine basis. However, attachment of the paramagnetic label at several different functionalities of the ligand may be necessary along with control experiments, analogous to those in this study, to ensure that the paramagnetic tag does not bind with high affinity to the protein. A paramagnetic lanthanoid complexing unit,70 such as DOTA,34 DOTA-M8,32 EDTA,50,51,71−73 TAHA,74 DTPA,37,75 4MTDA76 4MMDPA,33 3MDPA,77 or 4MDPA,78 may be easily attached to virtually any pharmacon or pharmaceutical lead compound by an ether linkage, as shown here for sevoflurane, or by simple modification of the phenolic anchoring point of the linker to another suitable functional group and its subsequent attachment using standard organic synthetic transformations. Hence, connection of the tag may be achieved via amidation or esterification, via click chemistry type transformations79 such as the [3 + 2] (Husigen reaction of alkynes and azides),80 [4 + 1] (reaction of isonitriles and tetrazines)81 and Diels–Alder cycloadditions,82,83 via nucleophilic substitution,84 via olefin metathesis85 or via the thiolyne reaction,86 for example. This makes the technique applicable to a very broad range of substances with the greatest advantage of its use being expected for investigation of leads having low affinity for their cellular targets, such as, e.g., the epothilone anticancer natural product.87 A main limitation of the technique is the requirement of compatibility of the functionalities of the small molecule ligand with the conditions of lanthanoid complexation (pH 5–6, 60 °C). Moreover, attachment of a polar metal binding site may introduce additional interactions to the protein that are not available for the drug or lead compound itself, even though dehydration of a metal complexed DOTA tag may pose a significant energetic barrier and therefore is less likely. However, such a scenario can easily be detected by using a control ligand, as in this study. The length and flexibility of the linker connecting the pharmacon/lead and the paramagnetic ion complexing unit defines the accuracy of binding site localization. By careful attention to the length of the linker, the magnitude of the induced δPCS can be modulated. Thus, a shorter linkage, e.g., via omission of the 1,4-substituted benzene linker applied in this study, is expected to increase the δPCS, whereas by its extension, e.g., by incorporation of a biphenyl unit,50 the line broadening due to PRE can be reduced. The highest accuracy is foreseen for a short, rigid linker. The obtained δPCS provide useful experimental input for computational docking into the experimentally established, secluded binding area or as a basis for mutagenesis experiments to provide further insight into the pharmacon binding modes.
The presented tagging approach is best suited for systems with a 1:1 binding stoichiometry as quantitative analysis of protein–ligand complexes of higher stoichiometries would require software packages allowing for simultaneous fits of more than one magnetic susceptibility tensor. However, ligand tagging may allow rapid qualitative screening for multiple protein binding sites, as shown for titration of full-length calmodulin with the Dy3+ labeled sevoflurane analogue clearly indicated binding to both lobes with comparably higher C-lobe than N-lobe affinity (Figure S10).
Paramagnetic ligand tagging to identify protein binding sites provides an attractive alternative to the opposite approach of binding site detection based on analysis of the NMR signals of the ligand11,31,70 interacting with a paramagnetically labeled protein, with both techniques having their strengths and limitations. Using the latter, specular approach, the attachment of the paramagnetic tag, due to the sheer size of the protein, is less likely to influence the binding, and selective attachment of the lanthanoid probe to different side chain positions may improve the accuracy of the location of the binding cleft. This, on the other hand, commonly necessitates protein site-directed mutagenesis. Furthermore, detection of the binding of low affinity ligands may be cumbersome as the observed paramagnetic effect is small due to the low molar fraction of bound ligand. The complementary approach of ligand tagging presented here allows straightforward screening of a bioactive substance against a large number of protein targets and does not necessitate protein engineering. Complexation of a series of different lanthanoids (Dy3+, Yb3+, Tb3+, Er3+, etc.) to the DOTA of the tagged-ligand can provide several sets of δPCS data for the same protein–ligand complex,50 with the combination of these data expectably allowing determination of the binding site with a higher precision. The main advantage of ligand tagging is doubtlessly the great sensitivity enhancement of detection as compared to conventional chemical shift mapping, which reached approximately a factor of hundred in this first study. On the other hand, obviously, modification of the structure of the ligand may affect its binding, and therefore tag attachment at several positions of the ligand may be necessary. Even if the attachment is expected to typically follow standard synthetic transformations, it may require adjustment for each studied ligand. For a highly accurate determination of the binding cleft development of a more rigid linker than that used in this investigation will be necessary. Overall, further exploration of paramagnetic ligand tagging for identification of protein binding sites, as a complementary tool to the alternative of protein tagging, should be expected.
Conclusions
Paramagnetic labeling by a DOTA tag increases the solubility of hydrophobic substances, whose binding site identification using conventional techniques is often seriously limited by their low aqueous solubility. The sustained paramagnetic sevoflurane analogue binding site affinities and binding site locations for both calmodulin lobes indicate that the tagging did not alter the molecular recognition. With the combination of increased substance solubility and higher sensitivity, the described ligand tagging approach offers binding cleft identification at lower ligand concentrations, thereby minimizing the risk of unspecific protein binding. This approach should be particularly attractive for the pharmaceutical industry for rapid screening of protein targets, especially for weakly binding lead compounds.
Materials and Methods
Synthesis
The method for preparation of the paramagnetic control ligand is outlined in Supplementary Scheme S1, whereas that of the paramagnetically (Dy3+, Eu3+, Yb3+) and diamagnetically (La3+) labeled sevoflurane is in Supplementary Scheme S2.
Protein Preparation
Native, full-length human calmodulin and the individual calmodulin lobes, N-lobe: residues 1–78, and C-lobe: residues 79–148, were expressed and purified as hexahistidine, maltose binding protein-Tev protease cleavage site constructs as described previously,55 with the alteration that N-terminal GHWGGM- and GHM- adducts to the native N-lobe and C-lobe sequences, respectively, remained after cleavage. Protein concentrations were determined using the calculated extinction coefficient at 280 nm in the presence of 6 M guanidine.88 The lobe chemical shifts obtained from 1H–15N/13C HSQC spectra were almost identical to full-length calmodulin except for the N-lobe terminal residues (1–3, 75–78) and C-lobe N-terminal residues 79–86. The latter constitutes the flexible hinge linkage between the two lobes in the full-length sequence, and the chemical shifts were expected to change upon cleavage.
NMR Spectroscopy
The lobe assignments were corroborated using HNCA, HNCA+, HNCO, and HNCO+ sequential walks.89 Samples containing either lobe at 0.1 mM in 100 mM KCl, 0.22 mM NaN3, 0.1 mM 2,2-dimethyl-2-silapentane-5-sulfonic acid, and 65 mM CaCl2 were run with 5 μL additions of 2.2 mM sevoflurane analogue ligand or control ligand coordinating to either La3+ or Dy3+. The assignment and ligand titration 1H–15N HSQC and 1H–13C CT-HSQC spectra were performed at 25 °C with a TCI probe on a Bruker Avance III HD spectrometer operating at 800 MHz 1H frequency. Spectral widths of 12820 Hz (1H), 3621 Hz (13C), and 2351 Hz (15N) run with 1024, 200, and 170 complex points, respectively, were employed for the titration experiments. All NMR data were processed using NMRPipe,90 and the spectral analysis was performed using Sparky (T. D. Goddard, D. G. Kneller, UCSF). The 10 μL ligand additions, resulting in 0.1 mM ligand sample concentration, presented the best signal-to-noise ratio for all peaks combined and were used to extract the δPCS. The δPCS were fitted to the calmodulin structure, PDB ID 1X02(68) using the Numbat software,69 in which the location of the paramagnetic ion and the parameters of the magnetic susceptibility tensor were fitted simultaneously.69 For the calmodulin N-lobe and C-lobe, 190 and 162 δPCS, respectively, were used. The data were fitted with and without the built-in RACS correction; the maximum RACS correction were 0.001 and 0.004 ppm for the N-lobe and C-lobe, respectively. Errors estimated were based on 1000 times repeated fitting with random removal of 10% of the δPCS in each run.
ITC Measurements
The calmodulin lobes were dialyzed overnight against 150 mM KCl, 10 mM Na-Hepes pH 7.4, 2 mM CaCl2. The Eu3+ complexed sevoflurane analogue and control ligand were dissolved into the same buffer at a final concentration of 10 mM. Titrations consisted of 20 injections of 2 μL ligand at 10 mM into the cell containing 1 mM calmodulin lobe. The background heats from dilution of the ligands were determined by titrating them into buffer. Experiments were performed at 25 °C and a stirring speed of 750 rpm on an ITC200 instrument (GE Healthcare). The data were processed using Origin 7.0 and fit to a single-site fitting model after background buffer subtraction.
Acknowledgments
We thank the Swedish Research Council (2012:3819) and the Swedish Foundation for International Cooperation in Research and Higher Education (STINT, YR 2010-7045) for financial support. The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 259638. We thank Sven Arenz for helpful discussions in an early phase of the project and Kelvin Lau for calmodulin sample preparations. NMR data was collected at the SWEDSTRUCT research infrastructure.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06220.
Details on synthesis and characterization, spectra of calmodulin lobes in the presence of either dia- or paramagnetically labeled sevoflurane analogue, plots of ITC measurements, calculated δPCS, and back-calculated versus experimental δPCS (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kuhlbrandt W. eLife 2014, 3, e03678. 10.7554/eLife.01412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca P. C.; Morris E. P. Nat. Commun. 2015, 6, 7573. 10.1038/ncomms8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A.; Merk A.; Banerjee S.; Matthies D.; Wu X.; Milne J. L.; Subramaniam S. Science 2015, 348, 1147. 10.1126/science.aab1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda S.; Tomohiro T.; Yamaguchi S.; Morimoto S.; Hatanaka Y. Bioorg. Med. Chem. Lett. 2015, 25, 1675. 10.1016/j.bmcl.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Meyer B.; Peters T. Angew. Chem., Int. Ed. 2003, 42, 864. 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- Roberts G. C. K. Drug Discovery Today 2000, 5, 230. 10.1016/S1359-6446(00)01479-3. [DOI] [PubMed] [Google Scholar]
- Fernandez C.; Jahnke W. Drug Discovery Today: Technol. 2004, 1, 277. 10.1016/j.ddtec.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Leavitt S.; Freire E. Curr. Opin. Struct. Biol. 2001, 11, 560. 10.1016/S0959-440X(00)00248-7. [DOI] [PubMed] [Google Scholar]
- Mann D. A.; Kanai M.; Maly D. J.; Kiessling L. L. J. Am. Chem. Soc. 1998, 120, 10575. 10.1021/ja9818506. [DOI] [Google Scholar]
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. J. Biomol. NMR 2001, 21, 349. 10.1023/A:1013302231549. [DOI] [PubMed] [Google Scholar]
- Otting G. Annu. Rev. Biophys. 2010, 39, 387. 10.1146/annurev.biophys.093008.131321. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 249. 10.1016/S0079-6565(02)00002-X. [DOI] [Google Scholar]
- Dwek R. A.; Richards R. E.; Morallee K. G.; Nieboer E.; Williams R. J. P.; Xavier A. V. Eur. J. Biochem. 1971, 21, 204. 10.1111/j.1432-1033.1971.tb01457.x. [DOI] [PubMed] [Google Scholar]
- Reuben J.; Leigh J. S. J. Am. Chem. Soc. 1972, 94, 2789. 10.1021/ja00763a042. [DOI] [Google Scholar]
- Yagi H.; Loscha K. V.; Su X.-C.; Stanton-Cook M.; Huber T.; Otting G. J. Biomol. NMR 2010, 47, 143. 10.1007/s10858-010-9416-x. [DOI] [PubMed] [Google Scholar]
- Liu W.-M.; Overhand M.; Ubbink M. Coord. Chem. Rev. 2014, 273, 2. 10.1016/j.ccr.2013.10.018. [DOI] [Google Scholar]
- Ma C.; Opella S. J. J. Magn. Reson. 2000, 146, 381. 10.1006/jmre.2000.2172. [DOI] [PubMed] [Google Scholar]
- Su X.-C.; Huber T.; Dixon N. E.; Otting G. ChemBioChem 2006, 7, 1599. 10.1002/cbic.200600142. [DOI] [PubMed] [Google Scholar]
- Wöhnert J.; Franz K. J.; Nitz M.; Imperiali B.; Schwalbe H. J. Am. Chem. Soc. 2003, 125, 13338. 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
- Dvoretsky A.; Gaponenko V.; Rosevear P. R. FEBS Lett. 2002, 528, 189. 10.1016/S0014-5793(02)03297-0. [DOI] [PubMed] [Google Scholar]
- Hass M. A. S.; Ubbink M. Curr. Opin. Struct. Biol. 2014, 24, 45. 10.1016/j.sbi.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; Park A. Y.; Keniry M. A.; Dixon N. E.; Otting G. J. Am. Chem. Soc. 2006, 128, 3696. 10.1021/ja057008z. [DOI] [PubMed] [Google Scholar]
- Saio T.; Yokochi M.; Kumeta H.; Inagaki F. J. Biomol. NMR 2010, 46, 271. 10.1007/s10858-010-9401-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J.-Y.; Keizers P. H. J.; Liu W.-M.; Loehr F.; Skinner S. P.; Heeneman E. A.; Schwalbe H.; Ubbink M.; Siegal G. J. Am. Chem. Soc. 2013, 135, 5859. 10.1021/ja401323m. [DOI] [PubMed] [Google Scholar]
- John M.; Pintacuda G.; Park A. Y.; Dixon N. E.; Otting G. J. Am. Chem. Soc. 2006, 128, 12910. 10.1021/ja063584z. [DOI] [PubMed] [Google Scholar]
- Zhuang T.; Lee H.-S.; Imperiali B.; Prestegard J. H. Protein Sci. 2008, 17, 1220. 10.1110/ps.034561.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Zarco A. R.; Munari F.; Wegstroth M.; Liu W.-M.; Ubbink M.; Becker S.; Zweckstetter M. Angew. Chem., Int. Ed. 2015, 54, 336. 10.1002/anie.201408615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoretsky A.; Gaponenko V.; Rosevear P. R. FEBS Lett. 2002, 528, 189. 10.1016/S0014-5793(02)03297-0. [DOI] [PubMed] [Google Scholar]
- Saio T.; Ogura K.; Shimizu K.; Yokochi M.; Burke T. R. Jr.; Inagaki F. J. Biomol. NMR 2011, 51, 395. 10.1007/s10858-011-9566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting B.; Strauss A.; Fendrich G.; Manley P. W.; Jahnke W. J. Biomol. NMR 2004, 30, 205. 10.1023/B:JNMR.0000048947.28598.ea. [DOI] [PubMed] [Google Scholar]
- Graham B.; Loh C. T.; Swarbrick J. D.; Ung P.; Shin J.; Yagi H.; Jia X.; Chhabra S.; Barlow N.; Pintacuda G.; Huber T.; Otting G. Bioconjugate Chem. 2011, 22, 2118. 10.1021/bc200353c. [DOI] [PubMed] [Google Scholar]
- Haussinger D.; Huang J. R.; Grzesiek S. J. Am. Chem. Soc. 2009, 131, 14761. 10.1021/ja903233w. [DOI] [PubMed] [Google Scholar]
- Su X. C.; Man B.; Beeren S.; Liang H.; Simonsen S.; Schmitz C.; Huber T.; Messerle B. A.; Otting G. J. Am. Chem. Soc. 2008, 130, 10486. 10.1021/ja803741f. [DOI] [PubMed] [Google Scholar]
- Keizers P. H.; Desreux J. F.; Overhand M.; Ubbink M. J. Am. Chem. Soc. 2007, 129, 9292. 10.1021/ja0725201. [DOI] [PubMed] [Google Scholar]
- Keizers P. H.; Saragliadis A.; Hiruma Y.; Overhand M.; Ubbink M. J. Am. Chem. Soc. 2008, 130, 14802. 10.1021/ja8054832. [DOI] [PubMed] [Google Scholar]
- Liu W. M.; Skinner S. P.; Timmer M.; Blok A.; Hass M. A.; Filippov D. V.; Overhand M.; Ubbink M. Chem. - Eur. J. 2014, 20, 6256. 10.1002/chem.201400257. [DOI] [PubMed] [Google Scholar]
- Prudencio M.; Rohovec J.; Peters J. A.; Tocheva E.; Boulanger M. J.; Murphy M. E.; Hupkes H. J.; Kosters W.; Impagliazzo A.; Ubbink M. Chem. - Eur. J. 2004, 10, 3252. 10.1002/chem.200306019. [DOI] [PubMed] [Google Scholar]
- Swarbrick J. D.; Ung P.; Su X. C.; Maleckis A.; Chhabra S.; Huber T.; Otting G.; Graham B. Chem. Commun. 2011, 47, 7368. 10.1039/c1cc11893e. [DOI] [PubMed] [Google Scholar]
- Vlasie M. D.; Comuzzi C.; van den Nieuwendijk A. M.; Prudencio M.; Overhand M.; Ubbink M. Chem. - Eur. J. 2007, 13, 1715. 10.1002/chem.200600916. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Rudisser S.; Zurini M. J. Am. Chem. Soc. 2001, 123, 3149. 10.1021/ja005836g. [DOI] [PubMed] [Google Scholar]
- Jahnke W. ChemBioChem 2002, 3, 167.. [DOI] [PubMed] [Google Scholar]
- Hocking H. G.; Zangger K.; Madl T. ChemPhysChem 2013, 14, 3082. 10.1002/cphc.201300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assfalg M.; Gianolio E.; Zanzoni S.; Tomaselli S.; Lo Russo V.; Cabella C.; Ragona L.; Aime S.; Molinari H. J. Med. Chem. 2007, 50, 5257. 10.1021/jm070397i. [DOI] [PubMed] [Google Scholar]
- Viswanathan S.; Kovacs Z.; Green K. N.; Ratnakar S. J.; Sherry A. D. Chem. Rev. 2010, 110, 2960. 10.1021/cr900284a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M.; Fesik S. W. J. Am. Chem. Soc. 1997, 119, 7885. 10.1021/ja971356m. [DOI] [Google Scholar]
- Dick L. R.; Geraldes C.; Sherry A. D.; Gray C. W.; Gray D. M. Biochemistry 1989, 28, 7896. 10.1021/bi00445a052. [DOI] [PubMed] [Google Scholar]
- Su X.-C.; Liang H.; Loscha K. V.; Otting G. J. Am. Chem. Soc. 2009, 131, 10352. 10.1021/ja9034957. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Yang Y.; Li Q.-F.; Huang F.; Zuo H.-H.; Su X.-C. Chem. - Eur. J. 2013, 19, 5758. 10.1002/chem.201204152. [DOI] [PubMed] [Google Scholar]
- Canales Á.; Mallagaray Á.; Berbís M. Á.; Navarro-Vázquez A.; Domínguez G.; Cañada F. J.; André S.; Gabius H.-J.; Pérez-Castells J.; Jiménez-Barbero J. J. Am. Chem. Soc. 2014, 136, 8011. 10.1021/ja502406x. [DOI] [PubMed] [Google Scholar]
- Erdelyi M.; d'Auvergne E.; Navarro-Vazquez A.; Leonov A.; Griesinger C. Chem. - Eur. J. 2011, 17, 9368. 10.1002/chem.201100854. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Yamaguchi T.; Erdelyi M.; Griesinger C.; Kato K. Chem. - Eur. J. 2011, 17, 9280. 10.1002/chem.201100856. [DOI] [PubMed] [Google Scholar]
- Mallagaray A.; Canales A.; Dominguez G.; Jimenez-Barbero J.; Perez-Castells J. Chem. Commun. 2011, 47, 7179. 10.1039/c1cc11860a. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Zhang Y.; Yamaguchi T.; Kameda T.; Kato K. Chem. Commun. 2012, 48, 4752. 10.1039/c2cc30353a. [DOI] [PubMed] [Google Scholar]
- Leonov A.; Voigt B.; Rodriguez-Castaneda F.; Sakhaii P.; Griesinger C. Chem. - Eur. J. 2005, 11, 3342. 10.1002/chem.200400907. [DOI] [PubMed] [Google Scholar]
- Brath U.; Lau K.; Van Petegem F.; Erdélyi M. Pharmacol. Res. Perspect. 2014, 2, e00025. 10.1002/prp2.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juranic N. O.; Jones K. A.; Penheiter A. R.; Hock T. J.; Streiff J. H. J. Serb. Chem. Soc. 2013, 78, 1655. 10.2298/JSC130523090J. [DOI] [Google Scholar]
- Lau K.; Chan M. M.; Van Petegem F. Biochemistry 2014, 53, 932. 10.1021/bi401502x. [DOI] [PubMed] [Google Scholar]
- Van Petegem F. J. Mol. Biol. 2015, 427, 31. 10.1016/j.jmb.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Ben-Johny M.; Yue D. T. J. Gen. Physiol. 2014, 143, 679. 10.1085/jgp.201311153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F.; Lobo P. A.; Ahern C. A. Biophys. J. 2012, 103, 2243. 10.1016/j.bpj.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desreux J. F. Inorg. Chem. 1980, 19, 1319. 10.1021/ic50207a042. [DOI] [Google Scholar]
- Marques M. P. M.; Geraldes C.; Sherry A. D.; Merbach A. E.; Powell H.; Pubanz D.; Aime S.; Botta M. J. Alloys Compd. 1995, 225, 303. 10.1016/0925-8388(94)07083-0. [DOI] [Google Scholar]
- Dalgarno D. C.; Klevit R. E.; Levine B. A.; Williams R. J. P.; Dobrowolski Z.; Drabikowski W. Eur. J. Biochem. 1984, 138, 281. 10.1111/j.1432-1033.1984.tb07913.x. [DOI] [PubMed] [Google Scholar]
- Iwahara J.; Clore G. M. Nature 2006, 440, 1227. 10.1038/nature04673. [DOI] [PubMed] [Google Scholar]
- Aime S.; Botta M.; Fasano M.; Marques M. P. M.; Geraldes C.; Pubanz D.; Merbach A. E. Inorg. Chem. 1997, 36, 2059. 10.1021/ic961364o. [DOI] [PubMed] [Google Scholar]
- Babailov S. P.; Dubovskii P. V.; Zapolotsky E. N. Polyhedron 2014, 79, 277. 10.1016/j.poly.2014.04.067. [DOI] [Google Scholar]
- Shishmarev D.; Otting G. J. Biomol. NMR 2013, 56, 203. 10.1007/s10858-013-9738-6. [DOI] [PubMed] [Google Scholar]
- Kainosho M.; Torizawa T.; Iwashita Y.; Terauchi T.; Ono A. M.; Güntert P. Nature 2006, 440, 52. 10.1038/nature04525. [DOI] [PubMed] [Google Scholar]
- Schmitz C.; Stanton-Cook M. J.; Su X.-C.; Otting G.; Huber T. J. Biomol. NMR 2008, 41, 179. 10.1007/s10858-008-9249-z. [DOI] [PubMed] [Google Scholar]
- Liu W. M.; Overhand M.; Ubbink M. Coord. Chem. Rev. 2014, 273, 2. 10.1016/j.ccr.2013.10.018. [DOI] [Google Scholar]
- Gaponenko V.; Altieri A. S.; Li J.; Byrd R. A. J. Biomol. NMR 2002, 24, 143. 10.1023/A:1020948529076. [DOI] [PubMed] [Google Scholar]
- Ikegami T.; Verdier L.; Sakhaii P.; Grimme S.; Pescatore B.; Saxena K.; Fiebig K. M.; Griesinger C. J. Biomol. NMR 2004, 29, 339. 10.1023/B:JNMR.0000032611.72827.de. [DOI] [PubMed] [Google Scholar]
- Haberz P.; Rodriguez-Castaneda F.; Junker J.; Becker S.; Leonov A.; Griesinger C. Org. Lett. 2006, 8, 1275. 10.1021/ol053049o. [DOI] [PubMed] [Google Scholar]
- Peters F.; Maestre-Martinez M.; Leonov A.; Kovacic L.; Becker S.; Boelens R.; Griesinger C. J. Biomol. NMR 2011, 51, 329. 10.1007/s10858-011-9560-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Castaneda F.; Haberz P.; Leonov A.; Griesinger C. Magn. Reson. Chem. 2006, 44, S10. 10.1002/mrc.1811. [DOI] [PubMed] [Google Scholar]
- Huang F.; Pei Y. Y.; Zuo H. H.; Chen J. L.; Yang Y.; Su X. C. Chem. - Eur. J. 2013, 19, 17141. 10.1002/chem.201302273. [DOI] [PubMed] [Google Scholar]
- Man B.; Su X. C.; Liang H.; Simonsen S.; Huber T.; Messerle B. A.; Otting G. Chem. - Eur. J. 2010, 16, 3827. 10.1002/chem.200902904. [DOI] [PubMed] [Google Scholar]
- Jia X.; Maleckis A.; Huber T.; Otting G. Chem. - Eur. J. 2011, 17, 6830. 10.1002/chem.201003573. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004.. [DOI] [PubMed] [Google Scholar]
- Spiteri C.; Moses J. E. Angew. Chem., Int. Ed. 2010, 49, 31. 10.1002/anie.200905322. [DOI] [PubMed] [Google Scholar]
- Stockmann H.; Neves A. A.; Stairs S.; Ireland-Zecchini H.; Brindle K. M.; Leeper F. J. Chem. Sci. 2011, 2, 932. 10.1039/c0sc00631a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. J. Am. Chem. Soc. 2008, 130, 13518. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Weissleder R.; Hilderbrand S. A. Bioconjugate Chem. 2008, 19, 2297. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashemirov B. A.; Bala J. L.; Chen X.; Ebetino F. H.; Xia Z.; Russell R. G.; Coxon F. P.; Roelofs A. J.; Rogers M. J.; McKenna C. E. Bioconjugate Chem. 2008, 19, 2308. 10.1021/bc800369c. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum K.; Arora P. S. Nat. Chem. Biol. 2008, 4, 527. 10.1038/nchembio0908-527. [DOI] [PubMed] [Google Scholar]
- Lowe A. B. Polymer 2014, 55, 5517. 10.1016/j.polymer.2014.08.015. [DOI] [Google Scholar]
- Buey R. M.; Diaz J. F.; Andreu J. M.; O'Brate A.; Giannakakou P.; Nicolaou K. C.; Sasmal P. K.; Ritzen A.; Namoto K. Chem. Biol. 2004, 11, 225. 10.1016/S1074-5521(04)00025-0. [DOI] [PubMed] [Google Scholar]
- Edelhoch H. Biochemistry 1967, 6, 1948. 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- Gil-Caballero S.; Favier A.; Brutscher B. J. Biomol. NMR 2014, 60, 1. 10.1007/s10858-014-9847-x. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. J. Biomol. NMR 1995, 6, 277. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.