

Abstract
Nanostructured lipid carriers (NLCs) have been one of the systems of choice for improving the oral bioavailability of drugs with poor water solubility. In the present study, lovastatin (LVT)-loaded NLCs (LVT-NLCs) were successfully prepared by hot high-pressure homogenization method with high entrapment efficiency, drug loading, and satisfactory particle size distribution. The particles had almost spherical and uniform shapes and were well dispersed with a particle size of <50 nm (23.5±1.6 nm) and a low polydispersity index (0.17±0.05 mV). The result of stability showed that the LVT-NLCs dispersion maintained excellent stability without exhibiting any aggregation, precipitation, or phase separation at 4°C for 6 months of storage. The LVT release data from all developed solid lipid nanoparticles (SLNs) and NLCs were best fitted to a Ritger–Peppas kinetic model (0.9832 and 0.9783 for NLCs and SLNs, respectively). This indicated that the release of LVT from the SLNs and NLCs was due to a combination of drug diffusion and erosion from the lipid matrix. The pharmacokinetic and pharmacodynamic results show that LVT-NLCs were better compared to free drug, which could be attributed to an increase in bioavailability.
Keywords: nanostructured lipid carriers, lovastatin, in vitro release, pharmacokinetic, pharmacodynamic
Introduction
Lovastatin (LVT), a highly lipophilic drug (log P=4.3) isolated from a fungus of Aspergillus terreus, belongs to the class of cholesterol-lowering drugs and is the first clinically used statin.1 It is a prodrug which lowers the cholesterol level through reversible competitive inhibition of 3-hydroxy-3-methyl-glutaryl coenzyme A reductase, an enzyme involved in biosynthesis of cholesterol. However, LVT exhibits poor oral bioavailability (<5%) because of its poor water solubility (0.4×10−3 mg/mL) and short half-life (1–2 hours).2 In addition, it undergoes extensive first-pass metabolism; hepatic extraction leads to low and variable availability of the drug to the general circulation.3–5
Previous researchers have made attempts to improve the aqueous solubility of LVT by preparing nanocrystals,6 nanomatrix-supported lipid bilayers,7 microspheres,8 self-nanoemulsifying drug delivery systems,9,10 methylated beta-cyclodextrin,11 and solid lipid nanoparticles (SLNs).12 In a series of investigations carried out in our lab, LVT-loaded SLN (LVT-SLN) formulations obviously provided sustained release of LVT with potential for improved therapeutic efficacy. However, SLNs suffer from two major limitations: limited drug loading and expulsion of drug during storage. There is a necessity to develop a formulation that would offer rapid dissolution of LVT and improve its bioavailability and finally therapeutic efficacy.
Nanostructured lipid carriers (NLCs), regarded as the second-generation lipid nanoparticles, are attracting foremost consideration as alternative colloidal drug carriers.13,14 NLCs have evolved from SLNs and are composed of a mixture of spatially different lipid molecules; that is, solid lipid is blended with liquid lipid to overcome the limitations of SLNs. This allows accommodation of more drug molecules, minimization of drug expulsion during storage, and modulation of the drug release profile by varying the lipid matrix.15–17 In this study, we have made efforts to investigate the feasibility of improving oral bioavailability of LVT through NLCs.
The objectives of this study were to develop LVT-loaded NLCs (LVT-NLCs) delivery system. First, both SLNs and NLCs containing LVT were formulated. Then, the LVT-NLCs were screened, and the physicochemical characteristics, in vitro drug release properties, pharmacokinetics, and biodistribution were investigated in detail. All the parameters of NLCs were compared with the SLNs. Finally, the possible absorption mechanism of NLC formulation is discussed.
Materials and methods
Materials
LVT was obtained from Meilun Pharm (Dalian, People’s Republic of China). Labrasol and Cremophor ELP were donated by Degussa Chemical Co., Ltd. (Hamburg, Germany). Precirol ATO5 and soybean lecithin were purchased from Sinopharm (Shanghai, People’s Republic of China). All reagents were of high-performance liquid chromatography (HPLC) (Sigma-Aldrich, Co., St Louis, MO, USA) grade. Other reagents were of analytical grade. Purified water from a Milli-Q system (EMD Millipore, Billerica, MA, USA) was used throughout the experiment.
Preparation of LVT-NLCs
LVT-NLCs were prepared by hot high-pressure homogenization method which was based on a preliminary study to optimize the drug incorporation into NLCs.18,19 Briefly, solid lipid (Precirol ATO5 4.65% and soybean lecithin 1.25%), liquid lipid (Labrasol 0.82%), and surfactant (Cremophor ELP 3.2%) were heated at 70°C with continuous stirring to homogeneously mix the lipids. The drug (LVT 0.1%) was dissolved in the melted mixture. Then, distilled water heated to 70°C was poured into the mixture and homogeneously dispersed using a homogenizer (IKA1, RCT, Kassel, Germany) at 2500× g for 10 minutes while maintaining the temperature at 70°C. This pre-emulsion was further homogenized for seven cycles at 850 bar. The prepared LVT-NLC nanosuspensions were immediately placed in an ice bath with gentle stirring for 10 minutes and then cooled down to room temperature. The LVT-NLCs were stored at 4°C before use.
Measurement of size and zeta potential
The particle size and polydispersity index (PDI) of LVT-NLCs were measured with a dynamic light-scattering technique using a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK) at 25°C and an angle of 90°. Zeta potential was measured with the same instrument. Before measurement, the preparations were diluted with deionized water to achieve a suitable scattering intensity. Each sample was analyzed in triplicate.
Morphology
The morphology of NLC formulations was observed by transmission electron microscopy (TEM; JEOL Ltd., Tokyo, Japan) at 160 kV. The samples were placed on air grids and stained with 2% (w/v) phosphotungstic acid for 10 minutes. Any excess fluid was then removed, and the grid surface was air-dried at room temperature.
Encapsulation efficiency and loading capacity
Drug-loading capacity (DL%) and entrapment efficiency (EE%) were calculated as described earlier.20 First, LVT was extracted from the NLCs with water and methanol (24:76, v/v), and then, the extracted solution was properly diluted prior to HPLC analysis. The content of LVT in the NLCs was determined by the HPLC method. Then, DL% and EE% were calculated according to Equations 1 and 2.
| (1) |
| (2) |
where WP is the weight of initial feeding polymer, WM is the weight of drug incorporated in NLCs, and WF is the weight of initial feeding drug.
The amount of the drug entrapped in the NLCs and the total drug were determined by HPLC (Shimadzu LC-10A, Kyoto, Japan) equipped with an ultraviolet detector operated at 238 nm and a Shim-pack VP-ODS (150 mm ×4.6 mm) column. The mobile phase was methanol/water (76:24), the flow rate was kept at 1 mL/min, and the column temperature was 30°C±0.2°C. Drug and excipients were validated to have no interference with each other.
Stability study
The physical stability of the formulations was investigated. Three batches of LVT-NLCs were stored at 4°C in the dark for 6 months. Their appearance, particle size, and zeta potential against storage time were evaluated.
In vitro release
In vitro drug release study of LVT-NLCs was performed by dialysis bag diffusion technique.21,22 LVT-NLC suspensions equivalent to 10 mg LVT were filled in dialysis bag and immersed in a receptor compartment containing 100 mL of phosphate buffer solution (pH 7.4), stirred at 250× g, and maintained at a temperature of 37°C±0.5°C. The receptor compartment was covered to prevent evaporation of the medium. Five milliliters of the release medium was removed at predetermined time intervals (0.5 hour, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 10 hours, 14 hours, 18 hours, 24 hours, 36 hours, 48 hours, and 60 hours), replaced with same volume of fresh dissolution medium, and diluted appropriately, and concentration was measured by HPLC at 238 nm. The experiments were performed in triplicate. Free LVT and LVT-SLNs were the control group (~10 mg LVT).
Animal
The experiments were performed on male Wistar rats weighing between 220±10 g. The animals were kept in cages in a room at a temperature of 25°C±2°C and with a 12:12 hour light–dark cycle. Food and water were freely available. All experiments were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the China National Institutes of Health. Ethical approval was obtained from the institutional review board of the Zhongshan Hospital, Fudan University, People’s Republic of China before initiation of the study.
Pharmacokinetic studies
Eighteen rats were used to investigate the effect of NLCs formulation on the pharmacokinetics of LVT after oral administration. Rats were divided into three groups at random and given a single dose of 10 mg/kg of LVT (free drug, SLNs, and NLCs) by oral administration. All related formulations were prepared freshly the day before the treatments. All rats were fasted for 20–24 hours prior to the administration of the formulations and were fed again 4 hours after administration. Blood samples (0.1 mL) were obtained by individual vein puncture from the caudal vein at designated time points (predose and 0.5 hour, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 16 hours, and 24 hours after administration) and were transferred into tubes. The plasma was separated by centrifugation at 1,500× g (10 minutes) and stored at −20°C until further analysis. In vitro and vivo analysis method was modified according to the previous reports.23,24
Pharmacodynamic studies
The anticholesterolemic effects of different LVT formulations were compared by poloxamer-induced hyperlipidemia model. The animals were divided into four groups of six animals, each group receiving different formulations. Group I received plain water which served as control group, group II received LVT suspension, group III received LVT-SLNs formulation, and group IV received LVT-NLCs formulation. The drug suspension was prepared by suspending proper LVT in water containing 0.5% hydroxypropyl methylcellulose. The rats were fasted overnight prior to study with free access to water. Hyperlipidemia was induced by intraperitoneal injection of 1.0 g/kg of poloxamer F-127 solution (20%, w/v). The rats were orally administered with drug and formulations once after 12 hours of injection of poloxamer (dose =20 mg/kg). This procedure was repeated for 7 days. The blood was withdrawn after 1 day, 2 days, 3 days, 4 days, 5 days, 6 days, and at the end of 7 days of induction of hyperlipidemia. The serum was separated and analyzed for total cholesterol (TC) and high-density lipoproteins (HDLs) using in vitro diagnostic kit (from the Department of Pathology of our hospital).
Statistical analysis
Results were presented as the mean ± standard deviation. Analysis of variance was used to identify the statistical significance of differences among groups. Statistical significance was evaluated using Student’s t-test for the single or multiple comparisons of experimental groups.
Results and discussion
Characterization of LVT-NLC
Usually, there are three methods to prepare NLCs: micro-emulsion, solvent evaporation or diffusion, and high-pressure homogenization. The advantage of microemulsion method is that no special equipment is needed, but the preparation process is relatively complicated; during the solvent diffusion or evaporation method, it is necessary to add a certain amount of organic solvent, which causes inconvenience for the quality evaluation later. Therefore, this article used high-pressure homogenization to successfully make LVT-NLCs. The morphology of NLCs determined by TEM is shown in Figure 1. The particles had almost spherical and uniform shapes and were well dispersed. LVT-NLCs had a particle size of <50 nm (23.5±1.6 nm) with a low PDI (0.17±0.05 mV). In the preparations of NLCs, Precirol ATO5 and soybean lecithin were used as solid lipid of NLCs, and formed its core. The liquid lipid of Labrasol was reported to be well entrapped within solid lipid matrix of Precirol ATO5.25 The magnitude of zeta potential is an indication of the repulsive force that is present in nanoparticles and is a key factor in predicting the long-term stability of colloidal dispersion system.26 In general, particles can be dispersed stably when the absolute value of zeta potential is >30 mV due to the electric repulsion between particles.27,28 In this study, the average zeta potential of LVT-NLCs preparation was approximately −31.2±0.4 mV, indicating that the NLCs obtained were a relatively dynamic stable system. An important issue with respect to the use of nanoparticles as drug carriers is that their capacity for drug loading and drug entrapment efficiency must be investigated. The entrapment efficiency and drug-loading capacity of LVT-NLCs were 94.2%±4.3% and 10.7%±2.1%, respectively.
Figure 1.

Transmission electron microscope photograph of LVT-NLCs: ×50,000.
Abbreviation: LVT-NLCs, lovastatin-loaded nanostructured lipid carriers.
Stability study
The LVT-NLCs dispersion maintained excellent stability without exhibiting any aggregation, precipitation, or phase separation at 4°C for 6 months of storage. No significant changes in appearance, PDI, particle size, or zeta potential were found over the storage period. The zeta potential is a key factor that can predict the stability of a colloidal dispersion. The zeta potential of the NLC formulations remained in the high negative values (>30 mV) during the storage period, which could be considered to be stable dispersion due to the electric repulsion between the particles.27,29
In vitro drug release
The in vitro LVT release profiles of free drug, SLNs, and NLCs are shown in Figure 2. The in vitro release profiles of the NLCs and SLNs were different from the reference formulation (LVT free drug suspensions). In the control group, suspensions showed quick release of LVT, with a cumulative release of more than 90% within only 5 hours. On the other hand, LVT from the SLNs and NLCs formulations exhibited a sustained release up to 60 hours in the release medium. In contrast, only 20%–30% of LVT was released from SLNs or NLCs in the first 5 hours. LVT was gradually released from the nanoparticles as time lapsed (~50% released in 10 hours, ~60% released in 35 hours, and ~70% released in 60 hours), suggesting that LVT release profile was prominently prolonged by the lipid nanoparticles encapsulation. However, no significant changes were observed in terms of release characteristics between SLNs and NLCs. In order to develop a prolonged-release system, it is vital to understand the release mechanism and kinetics. The LVT release data from all developed SLNs and NLCs were best fitted to a Ritger–Peppas kinetic model (0.9832 and 0.9783 for NLCs and SLNs, respectively). This indicated that the release of LVT from the SLNs and NLCs was due to a combination of drug diffusion and erosion from the lipid matrix.
Figure 2.
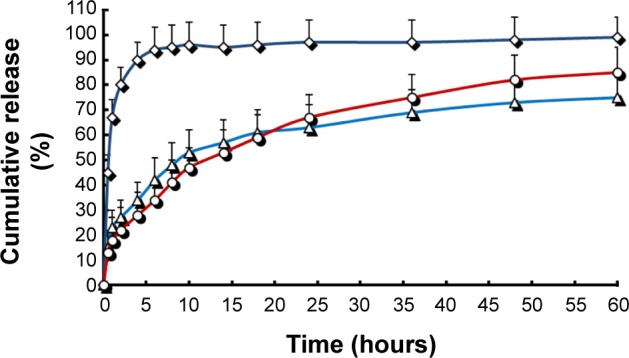
In vitro release profiles of different LVT formulations.
Notes: Release experiments were carried out in phosphate buffer solution (pH 7.4) at 37°C±0.5°C. Each point represents the mean value of three different experiments ± SD. ⋄: free LVT; Δ: LVT-SLNs; ○: LVT-NLCs.
Abbreviations: LVT, lovastatin; SD, standard deviation; LVT-SLNs, LVT-loaded solid lipid nanoparticles; LVT-NLCs, LVT-loaded nanostructured lipid carriers.
Pharmacokinetic studies
The pharmacokinetic parameters in rats after oral administration of LVT in either the aqueous suspension or SLNs or NLCs at same dose are summarized in Table 1. Plasma concentrations vs time profiles are shown in Figure 3. As shown in Table 1, the half-life of LVT suspensions (1.82 hours) was shorter than that of LVT-SLNs (3.65 hours) and LVT-NLCs (7.21 hours), suggesting that LVT suspensions were taken up by other tissues more rapidly than LVT lipid nanoparticles. In comparison to the suspension, the Cmax of LVT administered as SLNs or NLCs was significantly increased. A greater AUC0–t (area under curve) was obtained from the NLC (4.73-fold) and the SLN (three-fold), than from the suspensions group, and indicated that there was a significantly improved systemic exposure to LVT. The clearance (CL) of LVT-SLNs and LVT-NLCs was 0.43 L/h and 0.26 L/h, respectively, smaller than that (0.88 L/h) of the LVT suspensions.
Table 1.
Pharmacokinetic parameters of the three formulations (n=6)
| Parameter | Formulations
|
||
|---|---|---|---|
| LVT suspensions | LVT-SLNs | LVT-NLCs | |
| t1/2 (h) | 1.82±0.21 | 3.65±0.87* | 7.21±2.4*,** |
| Cmax (ng/mL) | 112.3±8.7 | 421.3±82.2* | 216.3±14.5** |
| AUC0–t (ng⋅h/mL) | 712.5±123.3 | 2,141.5±246.5* | 3,370.8±289.2* |
| AUC0–∞ (ng⋅h/mL) | 925.1±129.7 | 2,541.3±267.1* | 3,722.7±296.3* |
| MRT (h) | 3.3±0.8 | 4.9±1.2* | 10.1±4.3*,** |
| CL (L/h) | 0.88±0.09 | 0.43±0.06* | 0.26±0.07* |
Notes:
P<0.05, vs LVT suspensions.
P<0.05, vs LVT-SLNs. Data are represented as mean ± SD.
Abbreviations: LVT, lovastatin; LVT-SLNs, LVT-loaded solid lipid nanoparticles; LVT-NLCs, LVT-loaded nanostructured lipid carriers; h, hours; AUC, area under curve; MRT, mean residence time; CL, clearance; SD, standard deviation; Cmax, peak plasma concentration; t1/2, biological half-life.
Figure 3.
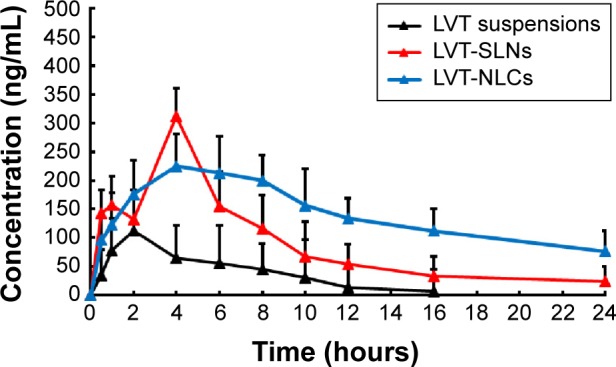
Mean plasma concentration–time profiles of LVT after oral administration of a single 10 mg/kg dose of suspensions, SLNs, and NLCs to rats.
Note: Each point represents the mean ± SD of six rats.
Abbreviations: LVT, lovastatin; SLNs, solid lipid nanoparticles; NLCs, nanostructured lipid carriers; SD, standard deviation; LVT-SLNs, LVT-loaded SLNs; LVT-NLCs, LVT-loaded NLCs.
The low oral bioavailability of LVT could be attributed to a number of reasons. In addition to its poor water solubility (0.4×10−3 mg/mL), it has weakly polarity and short half-life (1–2 hours). Also, it undergoes extensive first-pass metabolism; hepatic extraction leads to low and variable availability of the drug to the general circulation. So in this study, we have made efforts to investigate the feasibility of improving oral bioavailability of LVT through NLCs. Mainly, the significantly smaller particle sizes of the NLCs that occupied a larger surface area than larger particles (eg, SLN), a higher dispersibility, and prolonged residence time provided more amounts of and longer time for the drug to adhere at the absorptive site of the intestinal epithelium.30 Apart from the effects of physical characteristics and the oil used in the NLCs, the lipid used in the formulations was structurally similar to the fat in daily food. It could entrap the LVT in the particles and stimulate bile secretion, and thus enhance the uptake of intact particles by the gut wall and facilitate its draining into the lymphatic system. This is called an absorptive promotion effect of the NLCs.31
Pharmacodynamic studies
From the results of pharmacodynamic studies (Figure 4A and B), it was observed that after 24 hours of poloxamer injection, there was a fivefold increase in the TC level of the control group (1.5 mg/mL). Similar rise was observed in case of LVT treatment groups, which showed around fivefold increase in the TC levels. LVT suspension produced a drop in TC (from 1.48 mg/mL to 1.28 mg/mL) and rise in HDLs (from 0.16 mg/mL to 0.27 mg/mL), whereas LVT-SLN and LVT-NLC formulations produced a significant decrease in serum cholesterol (from 1.51 mg/mL to 1.19 mg/mL and 1.48 mg/mL to 1.08 mg/mL), and a marked increase in HDLs level (from 0.16 mg/mL to 0.36 mg/mL and 0.16 mg/mL to 0.48 mg/mL). The anticholesterolemic activity of lipid nanoparticles of LVT was significantly higher (P<0.05) than that of LVT suspensions. Thus, the higher lipid-lowering activity of the NLCs formulation can be explained by the fact that the NLCs formulation resulted in complete dissolution of LVT, which could have increased absorption and thereby led to higher plasma drug concentration (higher bioavailability).
Figure 4.
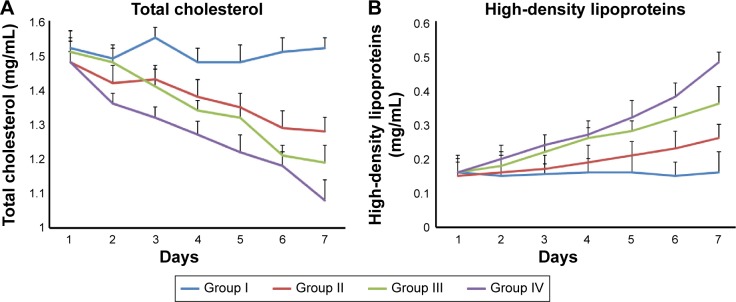
Changes in biochemical index in rats after given LVT suspensions and other LVT lipid nanoparticles for 7 days.
Notes: (A) Total cholesterol. (B) High-density lipoproteins. Group I, control group; group II, LVT suspension; group III, LVT-SLNs formulation; group IV, LVT-NLCs formulation.
Abbreviations: LVT, lovastatin; NLCs, nanostructured lipid carriers; SLNs, solid lipid nanoparticles.
Conclusion
NLCs have been one of the systems of choice for improving the oral bioavailability of drugs with poor water solubility. In the present study, LVT-NLCs were successfully prepared by hot high-pressure homogenization method with high EE%, drug loading, and satisfactory particle size distribution. The particles had almost spherical and uniform shapes and were well dispersed with a particle size of <50 nm (23.5±1.6 nm) and a low PDI (0.17±0.05 mV). The result of stability showed that the LVT-NLCs dispersion maintained excellent stability without exhibiting any aggregation, precipitation, or phase separation at 4°C for 6 months of storage. The LVT release data from all developed SLNs and NLCs were best fitted to a Ritger–Peppas kinetic model (0.9832 and 0.9783 for NLCs and SLNs, respectively). This indicated that the release of LVT from the SLNs and NLCs was due to a combination of drug diffusion and erosion from the lipid matrix. The pharmacokinetic and pharmacodynamic results showed that LVT-NLCs were better compared to free drug which could be attributed to an increase in bioavailability.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Kuzma-Kuzniarska M, Cornell HR, Moneke MC, Carr AJ, Hulley PA. Lovastatin-mediated changes in human tendon cells. J Cell Physiol. 2015;230(10):2543–2551. doi: 10.1002/jcp.25010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alberts AW. Lovastatin and simvastatin – inhibitors of HMG CoA reductase and cholesterol biosynthesis. Cardiology. 1990;77(suppl 4):14–21. doi: 10.1159/000174688. [DOI] [PubMed] [Google Scholar]
- 3.Michalik M, Soczek E, Kosińska M, et al. Lovastatin-induced decrease of intracellular cholesterol level attenuates fibroblast-to-myofibroblast transition in bronchial fibroblasts derived from asthmatic patients. Eur J Pharmacol. 2013;704(1–3):23–32. doi: 10.1016/j.ejphar.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 4.Mailman T, Hariharan M, Karten B. Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J Neurochem. 2011;119(5):1002–1015. doi: 10.1111/j.1471-4159.2011.07474.x. [DOI] [PubMed] [Google Scholar]
- 5.Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101(2):207–213. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- 6.Guo M, Fu Q, Wu C, et al. Rod shaped nanocrystals exhibit superior in vitro dissolution and in vivo bioavailability over spherical like nanocrystals: a case study of lovastatin. Colloids Surf B Biointerfaces. 2015;128:410–418. doi: 10.1016/j.colsurfb.2015.02.039. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Zhang H, Che E, et al. Development of novel mesoporous nanomatrix-supported lipid bilayers for oral sustained delivery of the water-insoluble drug, lovastatin. Colloids Surf B Biointerfaces. 2015;128:77–85. doi: 10.1016/j.colsurfb.2015.02.021. [DOI] [PubMed] [Google Scholar]
- 8.Guan Q, Chen W, Hu X. Development of lovastatin-loaded poly(lactic acid) microspheres for sustained oral delivery: in vitro and ex vivo evaluation. Drug Des Devel Ther. 2015;9:791–798. doi: 10.2147/DDDT.S76676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao S, Tan A, Boyd BJ, Prestidge CA. Synergistic role of self-emulsifying lipids and nanostructured porous silica particles in optimizing the oral delivery of lovastatin. Nanomedicine (Lond) 2014;9(18):2745–2759. doi: 10.2217/nnm.14.37. [DOI] [PubMed] [Google Scholar]
- 10.Beg S, Sandhu PS, Batra RS, Khurana RK, Singh B. QbD-based systematic development of novel optimized solid self-nanoemulsifying drug delivery systems (SNEDDS) of lovastatin with enhanced biopharmaceutical performance. Drug Deliv. 2014:1–20. doi: 10.3109/10717544.2014.900154. [DOI] [PubMed] [Google Scholar]
- 11.Sule A, Szente L, Csempesz F. Enhancement of drug solubility in supramolecular and colloidal systems. J Pharm Sci. 2009;98(2):484–494. doi: 10.1002/jps.21437. [DOI] [PubMed] [Google Scholar]
- 12.Suresh G, Manjunath K, Venkateswarlu V, Satyanarayana V. Preparation, characterization and in vitro and in vivo evaluation of lovastatin solid lipid nanoparticles. AAPS Pharm Sci Tech. 2007;8(1):24. doi: 10.1208/pt0801024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm. 2002;242(1–2):121–128. doi: 10.1016/s0378-5173(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 14.Paliwal R, Rai S, Vaidya B, et al. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 2009;5(2):184–191. doi: 10.1016/j.nano.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Radtke M, Souro EB, Muller RH. Nanostructured lipid carriers: a novel generation of solid lipid drug carriers. Pharm Technol Eur. 2005;17:45–50. [Google Scholar]
- 16.Chen Z, Lai X, Song S, Zhu X, Zhu J. Nanostructured lipid carriers based temozolomide and gene co-encapsulated nanomedicine for gliomatosis cerebri combination therapy. Drug Deliv. 2015:1–5. doi: 10.3109/10717544.2015.1038857. [DOI] [PubMed] [Google Scholar]
- 17.Khan S, Baboota S, Ali J, Narang RS, Narang JK. Chlorogenic acid stabilized nanostructured lipid carriers (NLC) of atorvastatin: formulation, design and in vivo evaluation. Drug Dev Ind Pharm. 2015:1–12. doi: 10.3109/03639045.2015.1040414. [DOI] [PubMed] [Google Scholar]
- 18.Lim WM, Rajinikanth PS, Mallikarjun C, Kang YB. Formulation and delivery of itraconazole to the brain using a nanolipid carrier system. Int J Nanomedicine. 2014;9:2117–2126. doi: 10.2147/IJN.S57565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nikolić S, Keck CM, Anselmi C, Müller RH. Skin photoprotection improvement: synergistic interaction between lipid nanoparticles and organic UV filters. Int J Pharm. 2011;414(1–2):276–284. doi: 10.1016/j.ijpharm.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 20.Lu QZ, Yu AH, Xi YW. Development and evaluation of penciclovir-loaded solid lipid nanoparticles for topical delivery. Int J Pharm. 2009;372(1–2):191–198. doi: 10.1016/j.ijpharm.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 21.de Andrade DF, Zuglianello C, Pohlmann AR, Guterres SS, Beck RC. Assessing the in vitro drug release from lipid-core nanocapsules: a new strategy combining dialysis sac and a continuous-flow system. AAPS Pharm Sci Tech. 2015 doi: 10.1208/s12249-015-0330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan F, Li B, Shen F, Fu Q. Formulation and characterization of albumin microspheres containing norcantharidate for liver tumor targeting. Drug Deliv. 2014 doi: 10.3109/10717544.2014.898715. [DOI] [PubMed] [Google Scholar]
- 23.Silva TD, Oliveira MA, de Oliveira RB, Vianna-Soares CD. Development and validation of a simple and fast HPLC method for determination of lovastatin, pravastatin and simvastatin. J Chromatogr Sci. 2012;50(9):831–838. doi: 10.1093/chromsci/bms079. [DOI] [PubMed] [Google Scholar]
- 24.Hamidi M, Zarei N, Shahbazi MA. A simple and sensitive HPLC-UV method for quantitation of lovastatin in human plasma: application to a bioequivalence study. Biol Pharm Bull. 2009;32(9):1600–1603. doi: 10.1248/bpb.32.1600. [DOI] [PubMed] [Google Scholar]
- 25.Doktorovová S, Araújo J, Garcia ML, Rakovský E, Souto EB. Formulating fluticasone propionate in novel PEG-containing nanostructured lipid carriers (PEG-NLC) Colloids Surf B Biointerfaces. 2010;75(2):538–542. doi: 10.1016/j.colsurfb.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 26.Komatsu H, Kitajima A, Okada S. Pharmaceutical characterization of commercially available intravenous fat emulsions: estimation of average particle size, size distribution and surface potential using photon correlation spectroscopy. Chem Pharm Bull. 1995;43(8):1412–1415. doi: 10.1248/cpb.43.1412. [DOI] [PubMed] [Google Scholar]
- 27.Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. doi: 10.1016/s0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 28.Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Rev. 2001;47(1):3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 29.Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv. 2001;47(2–3):165–196. doi: 10.1016/s0169-409x(01)00105-3. [DOI] [PubMed] [Google Scholar]
- 30.Karn-Orachai K, Smith SM, Phunpee S, et al. The effect of surfactant composition on the chemical and structural properties of nanostructured lipid carriers. J Microencapsul. 2014;31(6):609–618. doi: 10.3109/02652048.2014.911374. [DOI] [PubMed] [Google Scholar]
- 31.Tiwari R, Pathak K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int J Pharm. 2011;415(1–2):232–243. doi: 10.1016/j.ijpharm.2011.05.044. [DOI] [PubMed] [Google Scholar]