

Abstract
G protein-coupled receptors (GPCRs) represent ~25% of current drug targets. Ligand binding to these receptors activates G proteins and arrestins, which are involved in differential signaling pathways. Functionally selective or biased ligands activate one of these two pathways and may be superior medications for certain diseases states. The identification of these ligands requires robust drug screening assays for both G protein and arrestin activity. Here we describe in detail the technical aspects of two bioluminescence resonance energy (BRET)-based assays that can be used to monitor arrestin recruitment to GPCRs. One assay requires modification of GPCRs by fusion to a BRET donor or acceptor moiety, whereas the other can detect recruitment of arrestin to unmodified GPCRs.
Keywords: G protein-coupled receptors (GPCRs), arrestin, bioluminescence resonance energy transfer (BRET)
INTRODUCTION
G protein-coupled receptors (GPCRs), which are targeted by ~25% of drugs currently on the market, signal differentially through G proteins and arrestins. In certain disease contexts it can be desirable to selectivity activate a single pathway (Overington et al., 2006; Violin et al., 2014). Therefore, significant efforts were invested into the discovery of compounds that selectively activate either G proteins or arrestins (so called functionally-selective or biased ligands). To screen for bias it is critical to accurately probe the ability of compounds to activate these pathways. High-throughput assays that are currently commercially available for screening of drug dependent-arrestin recruitment to GPCRs (e.g., PathHunter, DiscoveRx; Tango, Life Technologies) require the use of modified receptors and arrestins that are fused to accessory protein fragments that complement upon receptor activation (van der Lee et al., 2009; Zhang and Xie, 2012). These modifications have the potential to alter normal signaling, and the complementation-based assays also may artificially augment the pharmacological activity of the compounds tested (e.g., efficacy, kinetic profile).
Bioluminescence resonance energy transfer (BRET) has been used to dynamically monitor arrestin recruitment to GPCRs. It is widely used to measure protein-protein interactions in living cells (Hamdan et al., 2006; Kocan and Pfleger, 2011; Pfleger et al., 2006; Salahpour et al., 2012). To study protein-protein interactions in living cells using BRET, one protein is genetically fused with a donor protein, typically a variant of Renilla luciferase, and the other is fused to an acceptor protein, typically a variant of green fluorescent protein (GFP). When the donor and acceptor are within ~10 nm of each other the donor non-radioactively transfers energy to and excites the acceptor, which results in emission from the acceptor protein. Here we describe two BRET-based assays that are capable of monitoring subtype-specific arrestin recruitment to a wide variety of GPCRs: the traditional approach in which the receptor is labeled with a donor molecule (referred to here as the receptor-arrestin BRET assay; see Basic Protocol 1), and a novel approach that does not require receptor labeling (referred to here as the arrestin translocation BRET assay; see Basic Protocol 2).
BASIC PROTOCOL 1
Receptor-arrestin BRET assay
A widely used cell-based assay to study ligand induced-recruitment of arrestin to receptors utilizes receptors that are fused to a BRET donor (typically Renilla luciferase - Rluc or Rluc8) and arrestins that are fused to an acceptor (typically variants of GFP, e.g., YFP or Venus)(Hamdan et al., 2005) (Figure 1A). Rluc must be fused to the cytoplasmic C-terminus of receptors, either directly or through linkers (e.g., the amino acids SGGGS) that facilitate proper folding of both proteins and prevent steric hindrance between the receptor and the sensor. Receptors are generally linked to the donor, e.g., Rluc8 (Loening et al., 2006)). N- or C- terminally acceptor fused-arrestins have been described previously (Hamdan et al., 2005; Klewe et al., 2008; Vishnivetskiy et al., 2011).
Figure 1. BRET-based arrestin-GPCR interaction assays.
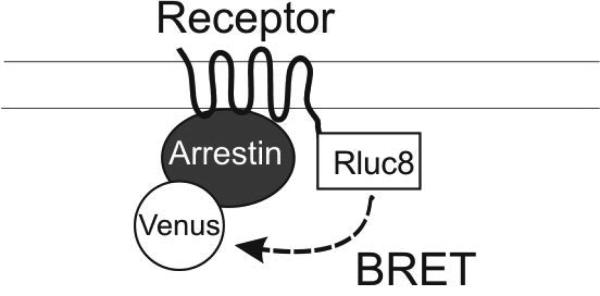



A. Schematic of the receptor-arrestin BRET assay, wherein a receptor is fused to the donor Rluc8 at its C-terminus and arrestin is fused to the acceptor Venus. Receptor activation results in an increase in BRET between the donor and acceptor molecules. B. Representative dose-response curves for the receptor-arrestin BRET assay shown in panel A. D2R-Rluc8 was activated dose dependently with dopamine (agonist mode). Additionally, activation of D2R with 1 μM dopamine was inhibited dose-dependently with the antagonist sulpiride (antagonist mode). C. Schematic of arrestin translocation BRET assay. Arrestin is fused to Rluc8 and an unrelated plasma membrane marker is fused to the acceptor citrine. Activation of the unmodified receptor results in translocation of arrestin to the plasma membrane and an increase in BRET between the donor and acceptor molecules. D. Representative dose-response curves for the arrestin translocation BRET assay shown in panel C. The experiments were performed, as described for panel B.
The receptor-arrestin BRET assay can be used to screen compounds in a medium- to high-throughput manner in immortalized mammalian cell lines including HEK293 and CHO (Hamdan et al., 2005). Receptor and arrestin biosensors can be transiently or stably transfected, and can be co-expressed with G protein receptor kinases (GRKs) to enhance agonist-dependent phosphorylation of the receptor and subsequent arrestin recruitment (Clayton et al., 2014). This protocol can be applied to a wide variety of GPCRs. Here, as an example, we describe the use for recruitment BRET of Venus fused to the N-terminus of arrestin-3 (Venus-arrestin3) to dopamine D2 receptor (D2R) fused with Rluc8 at its C-terminus (D2R-Rluc8) in transiently transfected HEK293T cells (Figure 1A).
Materials
Mammalian expression plasmids (the plasmids that are not commercially available can be obtained from the authors for non-profit use):
Plasmid encoding donor-fused GPCR, e.g., pcDNA3.1-D2R-linker-Rluc8
Plasmid encoding acceptor-fused arrestin, e.g., pIRES-puro-Venus-linker-arrestin-3
Plasmid encoding GRK, e.g., pcDNA3.1-GRK2
Empty vector plasmid, e.g., pcDNA3.1 (Life Technologies)
HEK293T cells (ATCC, CRL-3216)
HEK293T culture medium (see recipe below)
Dulbecco's Phosphate buffered saline, e.g., DPBS; Cellgro (21-031-CV)
DPBS, pH 7.4, 40mg/L sodium bisulfite (sodium bisulfite is used in the assay buffer to reduce the level of dopamine oxidation and is not universally required)
Glucose stock (0.5 M)
DPBS, pH 7.4, 5 mM glucose
Trypsin, e.g., Cellgro, 25-052-Cl
Polyethyleimine, 1 μg/μL stock, e.g., PEI; Linear, MW 25,000, Polysciences (23966-2); see supporting protocols for details on resuspension
DMEM, e.g., Gibco, 11965-092)
Dimethyl Sulfoxide
Dopamine hydrochloride, e.g., Sigma-Aldrich, H8502
S(-)-Sulpiride, e.g., Sigma RBI (S7771)
22 mM or 44 mM stocks of compounds of interest (e.g., dopamine, sulpiride) in appropriate solvent (e.g., distilled water, DMSO), depending on whether screening is planned in agonist or antagonist mode, respectively (see below)
10 cm tissue culture plates, e.g., BD Falcon (353003)
Compound plate, e.g., Greiner 96-well V-bottom plates (651101)
BRET assay plate, e.g., Greiner 96-well white plates, Flat bottom (655075) or PerkinElmer
Black/White 96-well Isoplate (6005030)
Pipette basin, e.g., USA scientific (2320-2620)
12-channel multichannel pipettor, e.g., Labsystems Finnpipette 50-300 μL (Z368989), or 5-50 μL (Z678031)
Repeater pipettor, e.g. Eppendorf Repeater Plus Pipettor (022260201)
5 mM coelenterazine H in absolute ethanol, e.g., Dalton (DC001437) or NanoLight (301); see supporting protocols for details on resuspension
DPBS, pH 7.4, 40 mg/L sodium bisulfite, 50 μM coelenterazine H
Plate reader for luminescence, fluorescence and BRET detection, e.g., Pherastar FS, BMG; Tecan F500
Software data for analysis (Microsoft Excel and GraphPad Prism)
HEK293T Culture Medium
DMEM (Gibco, 11965-092) supplemented with:
10% fetal bovine serum, e.g., FBS, Atlanta Biologics (D13056)
100 U/ml penicillin-streptomycin, e.g., Cellgro (30-002-Cl)
Protocol steps
The following protocol is described for either a single dopamine agonist curve or sulpiride antagonist curve at a single dose of dopamine. However, this procedure can be scaled up, depending on the number of compounds to be screened.
Note: All mammalian tissue culture should be carried out using aseptic technique in a laminar flow hood. Cells should be maintained in an incubator at 37°C at 5% CO2.
Day 1. Cell seeding
The following is described for the use of HEK293T cells, in this case with a pre-prepared, fully confluent 10 cm plate of cells.
Aspirate media. Wash plates with 3 mL sterile DPBS and aspirate.
Add 1 mL trypsin. Incubate at room temperature for 30 seconds to 1 minute.
Detach cells with 5 mL HEK293T culture medium and place in 15 mL conical tube.
Spin for 3 minutes at 600 rcf at room temperature.
Aspirate media. Resuspend in 15 mL HEK293T culture medium.
Count cells using a hemocytometer or cell counter.
Seed 3-4 million HEK293T cells into a 10 cm tissue culture plate in a total volume of 10 mL.
Day 2. Transfection
-
8.
After 24 hours, prepare plasmids for transient transfection. Combine the appropriate amounts of each plasmid, including the donor-fused receptor, the acceptor-fused arrestin, and if desired, the appropriate amount of GRK, in a 1.5 mL microfuge tube. The amount of each plasmid will need to be optimized to obtain the appropriate expression level as indicated by luminescence and fluorescence on the day of the assay. The amounts of DNA will vary, depending on the identity of the receptor-donor construct and the efficiency of the transient transfection. In general, the highest signal-to-noise ratio will be obtained when the acceptor is in stoichiometric excess. Adjust the total amount of plasmid DNA to 20 μg using the empty vector plasmid. As an example, for the D2R-arrestin-3 BRET, use the following amounts of plasmid DNA [D2R-linker-Rluc8 (0.2 μg); Venus-linker-arrestin-3 (8 μg); if desired, GRK2 (5 μg); pCDNA3.1 (6.8 or 11.8 μg the empty vector depending on whether GRK2 is included)].
-
9.
In a tissue culture hood, add non-supplemented DMEM to the tube containing the plasmid DNA so that the final volume is 500 μL.
-
10.
Vortex the 1 μg/μL PEI stock solution.
-
11.
In a 1.5 mL microfuge tube, add non-supplemented DMEM, followed by PEI pipetted directly into the DMEM. Use optimized PEI ratio as determined by PEI optimization (see support protocol). The final volume of the DMEM/PEI solution should be 500 μL.
-
12.
Vortex the DMEM/PEI solution well.
-
13.
Add 500 μL of DMEM/PEI solution to 500 μL of DMEM/DNA solution.
-
14.
Vortex. Incubate at room temperature for 15 minutes.
-
15.
Drip pipette the 1 mL mixture onto the media of the 10 cm plate containing HEK293T cells. Gently rock the 10 cm plate back and forth to mix, and return the plate to the incubator.
Day 3
-
16.
After 24 hours, aspirate the media and replace with 10 mL fresh HEK293T cell culture media.
Day 4. BRET assay
All subsequent steps do not require aseptic technique and can be performed outside of the tissue culture hood.
Compound preparation
Agonist mode
The following is described for screening the activity of an agonist in a dose-dependent manner. In a 96-well, V-bottom compound plate, prepare in triplicate an appropriate dilution series of agonist depending on its expected activity range. In each well of a 96-well BRET assay plate, 45 μL of compound will be added to a final volume of 100 μL. Therefore, each concentration needs to be 2.22-fold higher than the desired final concentration to adjust for this dilution. The preparation of a dilution series of dopamine for a single curve ranging from 10 pM to 100 μM is described below as an example:
-
a)
Dilute 3 μL of the freshly made 22 mM dopamine stock 100-fold into a final volume of 300 μL DPBS with sodium bisulfite for a maximum concentration of 222 μM.
-
b)
From this high concentration, carry out a 10-fold dilution series to a minimum concentration of 22 pM (a total of seven dilutions).
-
c)
Aliquot 60 μL of each individual dilution into three wells of the V-bottom plate, with an additional three wells for the vehicle (DPBS with sodium bisulfite). This dilution series (24 wells in total) can either be aliquoted into three columns or two rows of the V-bottom compound plate 96-well plate.
Antagonist mode
To screen the activity of an antagonist in a dose-dependent manner, a series of appropriate dilutions of the antagonist (to yield final concentrations that range from ineffective to those that completely suppress receptor activity) are prepared in triplicate in a 96-well, V-bottom compound plate, together with a single dose of agonist (typically at its EC80 concentration, i.e., the concentration that elicits 80% of the maximum response). If the inhibitory constant (Ki) of a particular compound at the receptor of interest is known, the dilution series can be adjusted appropriately around this concentration. Otherwise, a broad concentration range of 10 pM to 100 μM should be used initially to ensure that antagonist activity is detected. In each well of a 96-well BRET assay plate, 22.5 μL of antagonist and agonist will be injected into final volume of 100 μL. Therefore, each concentration needs to be set to 4.44-fold higher than the desired final concentration. The preparation of a dilution series of sulpiride for a single curve ranging from 10 pM to 100 μM, as well as a single dose of dopamine at 1 μM is described below as an example:
-
a)
Dilute 1.5 μL of the 44 mM sulpiride stock 100-fold into a final volume of 150 μL DPBS with sodium bisulfite for a maximum concentration of 444 μM.
-
b)
From this high concentration carry out a 10-fold dilution series to a minimum concentration of 44 pM (a total of seven dilutions).
-
c)
Aliquot 30 μL of each individual dilution into three wells of the V-bottom plate, with an additional three wells for the vehicle to serve as a negative control (DPBS with sodium bisulfite). This dilution series (24 wells in total) can either be aliquoted into three columns or two rows of a 96-well plate.
-
d)
Prepare 1 mL of 4.44 μM dopamine in a 1.5 mL microfuge tube. Aliquot 30 μL into 24 wells of a 96-well plate in a similar format as that performed for the sulpiride antagonist dilution series.
Note: For compound preparation in both agonist and antagonist mode, some compounds are dissolved in DMSO or other solvents that may affect the BRET assay, and therefore it is critical to adjust each dilution so that the concentration of solvent is equivalent throughout the entire concentration curves.
Cell preparation
-
17.
Aspirate the media from the transfected 10 cm plate.
-
18.
Wash the plate with 3 mL DPBS. Be careful not to detach the cells.
-
19.
Aspirate the DPBS.
-
20.
Detach by scrapping with a pipettor using a volume of 2.5 mL DPBS with 5 mM glucose and transfer to a 15 mL conical tube.
-
21.
Using a multichannel pipette, aliquot 45 μL of cells into wells from either three columns or two rows of the BRET assay plate, depending on the format chosen for the compound plate.
Perform BRET experiment
-
22.
Dilute 10 μL of 5 mM coelenterazine stock in DPBS 100-fold to a concentration of 50 μM (final volume of 1 mL). Because coelenterazine is light-sensitive, store it in a dark tube.
Agonist mode
-
23.
At time 0 minutes, use a repeater pipettor to inject 10 μL of 50 μM coelenterazine into each well of the 96-well BRET assay plate containing cells. Stagger the injection time for each well to account for the amount of time it will take the plate reader to read one well (~1-2 seconds). Inject in the pattern that the plate reader will read across the plate (e.g., in a serpentine fashion across columns or rows).
-
24.
After 8 minutes, inject 45 μL of agonist one row or column at a time using a multipipettor. Stagger the injection to account for the amount of time it will take the plate reader to read each entire row or column (~10-20 seconds).
-
25.
Read the plate after 2, 10, and 20 minutes from the start of agonist injection using a BRET plate reader with detection filters or monochrometers set for Rluc8 (~485 nm) and Venus (~525nm).
Antagonist mode
Pre-incubate the antagonist for the desired amount of time prior to the addition of agonist (10 minutes for sulpiride) by injecting 22.5 μL of antagonist one row or column at a time using a multichannel pipettor. Stagger the injection into each row or column ~10-20 seconds to account for the amount of time it will take the plate reader to read each well.
Eight minutes prior to the injection of agonist, using a repeater pipettor inject 10 μL of 50 μM coelenterazine into each well of the 96-well BRET assay plate containing cells. Stagger the injection time for each well to account for the amount of time it will take the plate reader to read one well (~1-2 seconds). Inject in the pattern that the plate reader will read across the plate (e.g., in a serpentine fashion across columns or rows).
Inject 22.5 μL of agonist one row or column at a time using a multichannel pipettor. Stagger the injection to account for the amount of time it will take the plate reader to read each row or column (~10-20 seconds).
Read the plate after 2, 10, and 20 minutes from the start of agonist injection using a BRET plate reader with detection filters set for Rluc8 (~485 nm) and Venus (~525nm).
Data Analysis
-
5.
Export the raw data for each filter set. Ensure that the luminescence counts for the 485 nm filter (which corresponds to emission from Rluc8) is not saturated. If it is saturated, reduce the amount of plasmid DNA encoding for the donor-fused protein. Generally these counts should be in the range of ~100,000-1,000,000. However, this will depend greatly on the instrument and exact filter sets used, and will need to be determined empirically.
-
6.
In Microsoft Excel, calculate the BRET ratio by dividing the raw counts from the 525 nm filter set by that of the 485 nm readings.
-
7.
Organize the data with the appropriate compound concentrations.
-
8.
Import into a points-only, XY plot in GraphPad Prism, with three replicate values.
-
9.
Fit the data to a non-linear regression curve. For fitting an agonist curve or antagonist curves, use the log(agonist) vs. response or log(inhibitor) vs. response fits, respectively.
To calculate the compound-induced effect, transform the Y-values using the Y=Y-K function with the K-value being the bottom fit of the non-linear regression. Examples of dopamine agonist and sulpiride antagonist curves are shown in Figure 1B.
ALTERNATE PROTOCOL
Arrestin translocation BRET assay
Like the commercially available PathHunter and Tango assays, fusion of a protein directly to the C-terminus of the receptor in the receptor-arrestin BRET assay may affect receptor function. Therefore, we have developed a novel BRET based-assay to overcome this problem (Clayton et al., 2014). Rather than being directly fused to the receptor, the BRET sensor is instead fused to an unrelated plasma membrane marker. Translocation of arrestin to the plasma membrane by activated receptor increases the proximity of arrestin to this marker, resulting in increased resonance energy transfer (RET). Arrestin translocation to the plasma membrane is dynamically regulated as receptor antagonists can reverse agonist induced-arrestin translocation. Moreover, this assay is capable of characterizing GPCR ligands with varying efficacy, potency, and kinetic profiles, potentially in a high-throughput manner.
Specifically, in immortalized mammalian cells lines, e.g., human embryonic kidney cells (HEK293), unlabeled GPCRs are coexpressed with Rluc8-arrestin-3 labeled with the weak helper peptide Sp1 at its C-terminus (Rluc8-arrestin3-Sp1) and a membrane marker composed of a doubly palmitoylated fragment of GAP43 linked to citrine and the weak helper peptide SH3 through a serine and glycine rich linker (mem-linker-citrine-SH3). Although not strictly necessary for the detection of arrestin translocation to the plasma membrane, the Sp1 and SH3 helper peptides, adapted from the helper-interaction FRET (hiFRET) system described recently (Grunberg et al., 2013), are used to enhance the interaction between arrestin recruited to the plasma membrane by receptor and the membrane marker. Although this modification moderately increases the basal signal between the plasma membrane marker and arrestin, it also increases the dynamic range of the assay. As in the receptor-arrestin BRET assay, G protein receptor kinases (GRKs) can be coexpressed to enhance receptor phosphorylation and arrestin recruitment.
Unlike the commercially available assays and the receptor-arrestin BRET assay, this assay can be used to screen any GPCR by simply co-expressing the unlabeled receptor with the arrestin and plasma membrane biosensors. Therefore, this assay does not require the generation and optimization of a fusion receptor. Here we use translocation of arrestin-3 to the plasma membrane by D2R as an example (Figure 1C).
Materials
Mammalian expression plasmids:
Plasmid encoding for GPCR (e.g., pcDNA3.1-D2R)
Plasmid encoding for Rluc8-Arrestin-3-Sp1
Plasmid encoding for mem-linker-citrine-SH3
All other reagents and protocol steps are identical to that described for the receptor-arrestin BRET assay outlined above, except for plasmid DNA preparation described below.
The procedure for the transient transfection of HEK293T cells for the arrestin translocation assay is identical to the procedure described above except for the plasmids used. In this case, the donor is the Rluc8-Arrestin-3-Sp1 and the acceptor is mem-linker-citrine-SH3. However, unlike in the receptor-arrestin BRET assay, the receptor is not fused with a BRET sensor. The following amounts of plasmid DNA is an example of the amounts used for transfection [Rluc8-Arrestin-3-Sp1 (0.25 μg); mem-linker- citrine-SH3 (5 μg); if desired, GRK2 (5 μg); D2R (2μg); pCDNA3.1 (4.8 or 9.8 μg the empty vector depending on whether GRK2 is included)].
For the rest of the protocol, see direct arrestin-receptor BRET protocol above. Examples of dopamine agonist and sulpiride antagonist curves are shown in Figure 1D.
SUPPORT PROTOCOLS
Support Protocol 1
Polyethylenimine (PEI) preparation and optimization
Different lots of PEI, even if from the same manufacturer, vary in activity (the efficiency of transfection). PEI is also cytotoxic to cells. Therefore, the DNA:PEI ratio must be optimized for each lot to maximize transfection efficiency and reduce cytotoxicity.
PEI Stock (1 μg /μL) stock preparation
Add 25 mg of PEI to 25 mL dH20 and stir continuously with a stir bar.
Add concentrated HCl dropwise to the solution to adjust the pH to less than 2.0.
Stir until the PEI is dissolved (30 minutes to 1 hour). Maintain a pH of less than 2.0 throughout this step.
Add concentrated NaOH dropwise to the solution to adjust the pH to 7.2.
Sterile-filter solution in a tissue culture hood using a 0.2 micron filter.
Aliquot the solution into 1.5 mL microfuge tubes (1 mL/tube).
Store at −20° C.
PEI transfection optimization
To optimize that DNA:PEI ratio, utilize a similar protocol as that performed in the arrestin BRET assays described above (Day 1-3), except with the following modifications:
Day 1. Cell seeding
1. Seed six 10 cm plates with 3-4 million cells (e.g., HEK293T).
Day 2. Transfection
-
2.
Prepare six 1.5 mL microfuge tubes, each containing the 20 μg of plasmid DNA described in the arrestin BRET assays.
-
3.
Prepare mixtures of a 1:1 to 1:6 ratio of DNA:PEI (either 20, 40, 60, 80, 100, or 120 μL PEI to 20 μg of plasmid DNA) and transfect into 10 cm plates as described in the protocol above for the arrestin BRET assays.
Day 3
-
4.
After 24 hours, aspirate the media and replace with 10 mL fresh HEK293T cell culture media.
Day 4. Assessment of transfection efficiency
-
5.
Before preparation of cells, take note of the cell health for each condition. At higher PEI levels, cells may noticeably be unhealthy or detached from the 10 cm plate.
-
6.
Prepare cells as described above and aliquot 45 μL of cells in triplicate from each transfection into a 96-well BRET plate (a total of 18 wells).
-
7.
Adjust the volume in each well to a final volume of 90 μL using 45 μL DPBS with sodium bisulfite.
-
8.
Inject 10 μL of DPBS with sodium bisulfite, 50 μM coelenterazine H.
-
9.
Read the plate after 8 minutes from the start of coelenterazine H injection using a BRET plate reader with detection filters set for Rluc8 (~485 nm) and Venus (~525 nm).
-
10.
Compare the luminescence counts from the 485 nm filter, using these data as a measure of transfection efficiency across the PEI titration range. Select the ratio that results in the highest luminescence with the least cell death for all further experiments using this lot of PEI.
Support Protocol 2
Coelenterazine H preparation
Coelenterazine H is temperature, light and oxygen sensitive and will lose activity over time. Because the amount of coelenterzine H purchased is typically in great excess of the amount required for a single experiment, it must be stored properly to avoid decomposition. Take note of the following upon preparation of the stock:
Dissolve the coelenterzine H in absolute ethanol to the appropriate concentration (5 mM) under low light conditions, and aliquot into 1.5 mL microfuge tubes.
After aliquoting, fill each tube with enough argon or nitrogen gas to cover the solution and displace atmospheric air.
Seal each tube with Parafilm and store at −20° C in the dark. This reagent is typically stable for at least 3 years under these conditions.
COMMENTARY
Background Information
Arrestin and drug development
GPCRs canonically couple to heterotrimeric G proteins, to initiate a variety of downstream signaling processes. These receptors also recruit arrestins, which terminate G protein signaling and facilitate receptor internalization (Lefkowitz, 2013). It has been shown recently that arrestins, once recruited to GPCRs, can signal independently of G proteins by acting as scaffolds for a variety of signaling molecules (Lefkowitz, 2013). Some ligands (so called functionally selective or biased ligands) can selectivity activate either G protein or arrestin, and may be superior medications for diseases in which one pathway downstream of a target receptor is associated with efficacy and the other is associated with side effects (Violin et al., 2014). Indeed, substantial efforts have gone towards the development of these ligands, some of which are currently in clinical trials (TRV027, TRV130; Trevena; clinicaltrials.gov).
There are a number of commercially available assays for screening G protein activity, both at the receptor-G protein level and downstream of the activation of G protein (Zhang and Xie, 2012). However, screening for arrestin activity is more limited due to a paucity of reliable readouts specifically downstream of arrestin. Thus, most available assays for screening arrestin activity (BRET based-assays; PathHunter, DiscoveRx; Tango, Life Technologies) rely on monitoring arrestin recruitment to receptor (Zhang and Xie, 2012). Commercially available assays, including PathHunter and Tango, require the use of modified receptors that are fused to accessory proteins that pseudo-irreversibly complement upon recruitment: e.g., in the PathHunter assay, two inactive fragments of β-galactosidase recombine to form an active enzyme upon arrestin recruitment to the receptor (van der Lee et al., 2009). These accessory proteins in and of themselves have affinity for each other, and thus may artificially enhance the stability the receptor-arrestin complex. Additionally, protein complementation usually is not reversible. Therefore, it is difficult to accurately characterize pharmacological properties such as efficacy, potency, and kinetics relative to reference agonists because these assays are inherently at non-equilibrium conditions.
BRET as a tool to study GPCR pharmacology
Bioluminescence resonance energy transfer (BRET) is a photophysical phenomenon that involves non-radiative transfer of energy from a donor (usually the product of a luciferasecatalyzed oxidation reaction) to a nearby acceptor (most often a fluorescent protein). This is seen in nature where BRET allows marine organisms to emit bright green rather than blue light (Morin and Hastings, 1971; Morise et al., 1974). In pharmacological research, BRET has been widely applied to detect dynamic protein-protein interactions, to monitor conformational changes in individual proteins and within multi-protein complexes, and to report on changes in cellular activity and metabolism (Marullo and Bouvier, 2007). BRET has been especially useful for studying the structure and function of G protein-coupled receptors (GPCRs), as well as their interactions with G proteins and arrestins, and is suitable for medium- to high-throughput drug screening.
Generally, implementation of BRET assays is straightforward, as both donor and acceptor can be genetically encoded as fusion proteins, and because BRET assays are relatively simple to perform and quantify without specialized equipment. One of the advantages of BRET is that this assay is extremely sensitive. Because BRET is based on luminescence, there is no background signal from cellular autofluorescence and photons can be collected for long periods of time without a significant accumulation of noise. Another advantage is that unlike Förster (or fluorescence) resonance energy transfer (FRET), there is no possibility of unintended direct excitation of the acceptor; therefore, error is not introduced by correction procedures. Additionally, BRET is ratiometric and therefore insensitive to variables such as cell number and transfection efficiency. As a consequence, photon emission ratios can be measured with very high precision and reproducibility, allowing detection of very weak BRET efficiency.
Critical Parameters and Troubleshooting
Generating Fusion Proteins
The most widely used BRET based-assay described above (Basic Protocol) requires the fusion of a BRET sensor at the C-terminus of receptors. As in the commercially available PathHunter and Tango assays, this may affect receptor function. BRET requires the fusion of relatively large probes (luciferases and fluorescent proteins) to proteins of interest. The presence of these probes can easily change the behavior of the proteins being studied in an unpredictable manner. Thus, it is necessary to ascertain that luciferase-fused receptors are still active, i.e., can activate G proteins comparably to unfused WT receptors (Gimenez et al., 2014). To achieve this, receptors are often connected to luciferase via a flexible linker (Hamdan et al., 2006).
Poor expression or disruption of the activity of receptor-Rluc fusions requires further optimization, either by extending the linker length, introducing a linker with different physical properties, e.g., a rigid linker composed of one or more repeats of EAAAK, or by exchanging the donor and acceptor on the receptor and arrestin. These modifications may help facilitate the proper folding of the receptor and BRET sensor domains of the fusion protein. However, they can also affect both the proximity and orientation of donor and acceptor moieties relative to one another, which dictate the degree of RET between the receptor and arrestin fusion proteins and, thus, the dynamic range of the assay.
N- or C-terminal acceptor-fused arrestin constructs have been developed previously, which allow for characterization of arrestin recruitment in a subtype-specific manner (Hamdan et al., 2005; Klewe et al., 2008; Vishnivetskiy et al., 2011). Although arrestins with C-terminally fused GFP or Venus are recruited to receptors, caution must be taken given that this region of the protein plays an important role in arrestin response to the receptor-attached phosphates (Celver et al., 2002; Gurevich, 1998; Hirsch et al., 1999; Kovoor et al., 1999; Shukla et al., 2013; Vishnivetskiy et al., 1999). Disruption of this phosphate sensor may increase basal interactions between arrestin and clathrin or AP-2 (Kim and Benovic, 2002; Nobles et al., 2007).
Specific and non-specific BRET
In order for BRET to occur, the energy donor and acceptor must be in close proximity (~10 nm). Therefore, as a general rule BRET signals can be generated in cells when either: 1) donor- and acceptor-fused proteins are specifically associated, either directly or as part of a macromolecular complex, or 2) when both donor and acceptor are associated with the same membrane, even without a stable direct or indirect interaction between the two proteins. BRET signals produced by these two mechanisms are referred to as “specific” and “nonspecific” BRET, respectively, although the latter is also sometimes referred to as “bystander” BRET. Specific and nonspecific signals are not mutually exclusive, and both can be useful for pharmacological assays.
In the case of the receptor-arrestin BRET assay (Basic Protocol), the increase in BRET seen upon activation of the receptor is presumably due to an increase in the direct interaction between donor-fused receptor and acceptor-fused arrestin. However, it is also possible that the increase in BRET is nonspecific, due to activation of endogenous receptors that are activated by the compound of interest. For example, HEK cells express high levels of chemokine receptor CXCR4 (Atwood et al., 2011), and off-target activation of this receptor may increase the nonspecific interaction between arrestin recruited to the plasma membrane by CXCR4 and the fused receptor of interest. Therefore, compounds identified as hits in drug screens must be validated by control experiments using specific antagonists of the fused receptor of interest that fully inhibit agonist signaling via that particular receptor.
The arrestin translocation assay (Alternate Protocol) takes advantage of nonspecific BRET in order to obviate the use of fused receptors. Donor-fused arrestin is recruited to the membrane by an unfused receptor of interest from the cytosol. Due to random proximity, the increased localization of arrestin in this compartment can be sufficient to increase the nonspecific BRET between this sensor and many proteins localized in the plasma membrane (in this case a fragment of GAP43 that is doubly palmitoylated). This is not the case if the two proteins are localized in different micro-compartments of the plasma membrane (e.g., clathrin coated pits).
The magnitude of the nonspecific interaction can be increased in several ways: 1) increasing the expression level of the unfused receptor of interest, which increases the total amount of cytosolic arrestin that can be recruited to the membrane upon receptor activation, 2) overexpression of GRKs, which phosphorylate the receptor and increase the affinity of the receptor for arrestin, 3) increasing the expression level of the acceptor-fused plasma membrane marker, which increases the probability that arrestins recruited to the plasma membrane will be in close proximity to or randomly collide with the membrane marker. This assay is also subject to nonspecific BRET associated with off-target activation of endogenous receptors, and thus it is critical to use an appropriate antagonist control to ensure agonist activity is solely mediated by the receptor of interest.
Negative controls
Regardless of the assay used, it is helpful to include negative controls. Arrestin-KNC mutants that have substitutions of 12 key receptor-binding residues on the surface and thus do not bind GPCRs were developed recently (Vishnivetskiy et al., 2011). The use of these control arrestins N-terminally fused with Venus, which are the same size as wildtype arrestin, allow for the detection with reasonable confidence of “basal” (e.g., agonist-independent) interaction of arrestin-3 with GPCRs, which KNC and some other arrestin-3 mutants do not demonstrate (Gimenez et al., 2014; Gimenez et al., 2012a; Gimenez et al., 2012b). Ideally, to have full set of controls, one would also require receptor mutants that do not bind either G proteins or arrestins. In some cases, such mutants have recently become available (reviewed in (Wess et al., 2013)).
Interpretation of arrestin recruitment
The commercially available PathHunter and Tango assays, as well as both BRET-based arrestin assays, measure the degree of recruitment of arrestin from the cytosol to the plasma membrane. However once recruited to the receptor, arrestin can adopt multiple conformations that may be involved differentially in various arrestin fuctions, i.e., inhibition of G protein-mediated signaling, internalization, and activation of downstream signaling pathways (Gurevich and Gurevich, 2006; Lefkowitz, 2013). These conformations can be differentially stabilized in a ligand-specific manner, and this has been shown using an alternative BRET assay in which arrestin is doubly fused with both a donor and acceptor sensor (Charest et al., 2005). Upon activation of the receptor, arrestin is recruited to the receptor and undergoes a conformational change, which is indicated by a change in BRET. Unfortunately, this assay has a poor dynamic range relative to both BRET-based arrestin assays, and is not suitable for robust drug screening. Additionally, there is a paucity of available screening assays that read out the signaling that is unambiguously downstream of arrestin. For example, phospho-ERK (pERK) assays are commonly used to study arrestin activity, but interpretation of the results of this assay is complicated by the contribution of G protein to this endpoint (Ahn et al., 2004; Coffa et al., 2011; Eishingdrelo and Kongsamut, 2013). Thus, caution must be applied in interpreting arrestin recruitment as a readout of arrestin activity because arrestin-dependent inhibition of G protein signaling, internalization of the receptor, and activation of downstream signaling proteins are not measured.
Anticipated Results
The background BRET ratio seen in the arrestin translocation is generally higher than that in the receptor-arrestin BRET assay, which is indicative of higher basal interaction between arrestin and the plasma membrane assay (~0.5-0.7 and ~0.4-0.5, respectively, depending on the expression level of the biosensors and BRET filter sets specific to each plate reader). This is due the hiBRET peptides (Sp1 and SH3) used in this assay. The maximal drug-induced BRET values (ranging from 0 to ~0.3) depend on the ability of the receptor to recruit arrestin, the expression level of receptor and the other components of the assay, and the presence or absence of overexpressed GRKs. These values are not consistently higher in one arrestin BRET assay versus another, and therefore these assays must be compared empirically.
Time Considerations
For transient transfections, both assays are generally performed over a period of four days and include cell seeding, transfection and expression over a period of 24-48 hours, and processing and assaying. When stable cell lines are available, only cell seeding, processing and assaying are needed, which reduces the overall time by 24-48 hours. Overall, assaying can take anywhere from 10 minutes to two hours depending on the number of samples and plates being run. The time it takes to read a full 96-well plate can take anywhere from one to five minutes depending on the plate reader.
ACKNOWLEDGEMENTS
Supported by NIH grants GM077561, GM109955, EY011500 (VVG), NIMH-R01 MH54137 (PCD, JRQ, JAJ), DA022413 (JAJ), GM078319 (NAL).
LITERATURE CITED
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. The Journal of biological chemistry. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC genomics. 2011;12:14. doi: 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- Charest PG, Terrillon S, Bouvier M. Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO reports. 2005;6:334–340. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton CC, Donthamsetti P, Lambert NA, Javitch JA, Neve KA. Mutation of Three Residues in the Third Intracellular Loop of the Dopamine D2 Receptor Creates an Internalization-Defective Receptor. The Journal of biological chemistry. 2014 doi: 10.1074/jbc.M114.605378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffa S, Breitman M, Spiller BW, Gurevich VV. A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry. 2011;50:6951–6958. doi: 10.1021/bi200745k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eishingdrelo H, Kongsamut S. Minireview: Targeting GPCR Activated ERK Pathways for Drug Discovery. Current chemical genomics and translational medicine. 2013;7:9–15. doi: 10.2174/2213988501307010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Babilon S, Wanka L, Beck-Sickinger AG, Gurevich VV. Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell Signal. 2014;26:1523–1531. doi: 10.1016/j.cellsig.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Kook S, Vishnivetskiy SA, Ahmed MR, Gurevich EV, Gurevich VV. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. The Journal of biological chemistry. 2012a;287:9028–9040. doi: 10.1074/jbc.M111.311803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Vishnivetskiy SA, Baameur F, Gurevich VV. Manipulation of Very Few Receptor Discriminator Residues Greatly Enhances Receptor Specificity of Non-visual Arrestins. The Journal of biological chemistry. 2012b;287:29495–29505. doi: 10.1074/jbc.M112.366674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunberg R, Burnier JV, Ferrar T, Beltran-Sastre V, Stricher F, van der Sloot AM, Garcia-Olivas R, Mallabiabarrena A, Sanjuan X, Zimmermann T, Serrano L. Engineering of weak helper interactions for high-efficiency FRET probes. Nature methods. 2013;10:1021–1027. doi: 10.1038/nmeth.2625. [DOI] [PubMed] [Google Scholar]
- Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. The Journal of biological chemistry. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharm Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Audet M, Garneau P, Pelletier J, Bouvier M. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. Journal of biomolecular screening. 2005;10:463–475. doi: 10.1177/1087057105275344. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Percherancier Y, Breton B, Bouvier M. Monitoring protein-protein interactions in living cells by bioluminescence resonance energy transfer (BRET). Current protocols in neuroscience / editorial board, Jacqueline N. Crawley ... [et al.] 2006 doi: 10.1002/0471142301.ns0523s34. Chapter 5:Unit 5 23. [DOI] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Kim YM, Benovic JL. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. The Journal of biological chemistry. 2002;277:30760–30768. doi: 10.1074/jbc.M204528200. [DOI] [PubMed] [Google Scholar]
- Klewe IV, Nielsen SM, Tarpo L, Urizar E, Dipace C, Javitch JA, Gether U, Egebjerg J, Christensen KV. Recruitment of beta-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology. 2008;54:1215–1222. doi: 10.1016/j.neuropharm.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocan M, Pfleger KD. Study of GPCR-protein interactions by BRET. Methods in molecular biology. 2011;746:357–371. doi: 10.1007/978-1-61779-126-0_20. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. Arrestins come of age: a personal historical perspective. Progress in molecular biology and translational science. 2013;118:3–18. doi: 10.1016/B978-0-12-394440-5.00001-2. [DOI] [PubMed] [Google Scholar]
- Loening AM, Fenn TD, Wu AM, Gambhir SS. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein engineering, design & selection : PEDS. 2006;19:391–400. doi: 10.1093/protein/gzl023. [DOI] [PubMed] [Google Scholar]
- Marullo S, Bouvier M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends in pharmacological sciences. 2007;28:362–365. doi: 10.1016/j.tips.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Morin JG, Hastings JW. Energy transfer in a bioluminescent system. Journal of cellular physiology. 1971;77:313–318. doi: 10.1002/jcp.1040770305. [DOI] [PubMed] [Google Scholar]
- Morise H, Shimomura O, Johnson FH, Winant J. Intermolecular energy transfer in the bioluminescent system of Aequorea. Biochemistry. 1974;13:2656–2662. doi: 10.1021/bi00709a028. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Guan Z, Xiao K, Oas TG, Lefkowitz RJ. The active conformation of beta-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of beta-arrestins1 and -2. The Journal of biological chemistry. 2007;282:21370–21381. doi: 10.1074/jbc.M611483200. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nature reviews. Drug discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Seeber RM, Eidne KA. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nature protocols. 2006;1:337–345. doi: 10.1038/nprot.2006.52. [DOI] [PubMed] [Google Scholar]
- Salahpour A, Espinoza S, Masri B, Lam V, Barak LS, Gainetdinov RR. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Frontiers in endocrinology. 2012;3:105. doi: 10.3389/fendo.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi-Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, Lefkowitz RJ. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee MM, Blomenrohr M, van der Doelen AA, Wat JW, Smits N, Hanson BJ, van Koppen CJ, Zaman GJ. Pharmacological characterization of receptor redistribution and beta-arrestin recruitment assays for the cannabinoid receptor 1. Journal of biomolecular screening. 2009;14:811–823. doi: 10.1177/1087057109337937. [DOI] [PubMed] [Google Scholar]
- Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends in pharmacological sciences. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. The Journal of biological chemistry. 2011;286:24288–24299. doi: 10.1074/jbc.M110.213835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- Wess J, Nakajima K, Jain S. Novel designer receptors to probe GPCR signaling and physiology. Trends in pharmacological sciences. 2013;34:385–392. doi: 10.1016/j.tips.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Xie X. Tools for GPCR drug discovery. Acta pharmacologica Sinica. 2012;33:372–384. doi: 10.1038/aps.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]