

Abstract
Platelet Endothelial Aggregation Receptor 1 (PEAR1) is a newly identified membrane protein reported to be involved in multiple vascular and thrombotic processes. While most studies to date have focused on the effects of this receptor in platelets, PEAR1 is located in multiple tissues including the endothelium, where it is most highly expressed. Our first objective was to evaluate the role of PEAR1 in endothelial function by examining flow-mediated dilation of the brachial artery in 641 participants from the Heredity and Phenotype Intervention Heart Study. Our second objective was to further define the impact of PEAR1 on cardiovascular disease computationally through meta-analysis of 75,000 microarrays, yielding insights regarding PEAR1 function, and predictions of phenotypes and diseases affected by PEAR1 dysregulation. Based on the results of this meta-analysis we examined whether genetic variation in PEAR1 influences endothelial function using an ex vivo assay of endothelial cell migration. We observed a significant association between rs12041331 and flow-mediated dilation in participants of the Heredity and Phenotype Intervention Heart Study (P = 0.02). Meta-analysis results revealed that PEAR1 expression is highly correlated with several genes (e.g. ANG2, ACVRL1, ENG) and phenotypes (e.g. endothelial cell migration, angiogenesis) that are integral to endothelial function. Functional validation of these results revealed that PEAR1 rs12041331 is significantly associated with endothelial migration (P = 0.04). Our results suggest for the first time that genetic variation of PEAR1 is a significant determinant of endothelial function through pathways implicated in cardiovascular disease.
Introduction
Platelet endothelial aggregation receptor 1 (PEAR1; also known as JEDI and MEGF12) is a recently identified transmembrane receptor expressed in a number of different tissues, with highest expression in endothelial cells and megakaryocytes [1]. While little is currently known regarding the molecular mechanism(s) of this receptor, prior investigations suggest that PEAR1 is important in a diverse range of biological functions, including sustained platelet aggregation through glycoprotein αIIbβ3 [2], altered megakaryopoiesis and thrombopoiesis via PI3K/PTEN pathways [3], and apoptotic neuron clearance through endocytosis-dependent activities in dorsal root ganglia [4]. In addition to these mechanism-based investigations, several studies have examined the role of genetic variation in PEAR1, most notably the intronic single nucleotide polymorphism (SNP) rs12041331. These studies have implicated rs12041331 genotype in differential PEAR1 expression as well as platelet aggregation, both at baseline and in the presence of therapeutic agents such as aspirin and prasugrel [5–11]. However, a seemingly paradoxical effect of rs12041331 on cardiovascular phenotypes has been observed; the allele associated with better aspirin response, as measured by platelet function testing, is also associated with higher adverse cardiovascular event rates in patients with coronary artery disease on aspirin, potentially suggesting an alternative role for PEAR1 in cardiovascular disease progression [10].
Given that PEAR1 is most highly expressed in endothelial cells [1], we first explored the effects of genetic variation in PEAR1 on endothelial function. Specifically, we tested the impact of rs12041331 on flow-mediated dilation (FMD) of the brachial artery in 641 participants of the Heredity and Phenotype Intervention (HAPI) Heart Study. In an attempt to further define the role of PEAR1 in cardiovascular biology we used a bioinformatics approach called GAMMA (Global Microarray Meta-Analysis) [12] to identify genes consistently correlated with PEAR1 expression across 75,000 human 1-color microarray experiments from within the publicly available datasets in National Center for Biotechnology Information’s Gene Expression Omnibus. Based on our meta-analysis’s results, we extended our findings by evaluating and confirming the effect of the PEAR1 rs12041331 variant on endothelial cell migration using functional ex vivo assays of human umbilical vein endothelial cells (HUVECs) derived from de-identified umbilical cords.
Materials and Methods
HAPI Heart Study Participants
The HAPI Heart Study recruited 868 healthy Old Order Amish (OOA) participants aged 20 years or older from 2003 to 2006 as previously described [13]. This report evaluates 641 HAPI Heart Study participants in whom brachial artery FMD measurements were recorded. Briefly, all study participants discontinued the use of medications, vitamins, and supplements 7 days prior to their initial clinic visit. Physical examinations, anthropometric measures, medical and family histories, and other phenotype information were collected at the Amish Research Clinic in Lancaster, Pennsylvania after an overnight fast. Individuals were excluded if any of the following criteria were met: pregnancy, coexisting malignancy, severe hypertension (blood pressure > 180/105 mmHg), serum creatinine > 2.0 mg/dl, AST or ALT greater than twice the upper limit of normal, hematocrit < 32%, TSH < 0.4 or > 5.5 mIU/l, or inability to safely discontinue prescription and nonprescription medications.
Complete blood count and serum lipid concentrations were assayed by Quest Diagnostics (Horsham, Pennsylvania), and levels of LDL-cholesterol were calculated using the Friedewald equation. Any participant with an LDL-cholesterol greater than 160 mg/dl or taking prescription cholesterol-lowering medications was designated hyperlipidemic. Individuals were described as hypertensive if they had one or more of the following criteria: systolic blood pressure (SBP) ≥ 140 mmHg, diastolic blood pressure (DBP) ≥ 90 mmHg, or requirement of prescription blood pressure lowering medications. Diabetes and current smoking status (cigarette, cigar, or pipe) were obtained by self-report.
Study protocols were approved by the Institutional Review Board at the University of Maryland School of Medicine, and the study was conducted according to the principles expressed in the Declaration of Helsinki. Written informed consent was obtained from each HAPI Heart Study participant; participants were compensated for their participation. Data from the HAPI Heart Study pertaining to the measurements used for analysis are available upon request to preserve the anonymity of OOA participants.
Flow-Mediated Dilation (FMD)
Assessment of endothelial function was evaluated by FMD of the brachial artery. All brachial artery measurements were obtained after an overnight fast. Briefly, the subject’s left arm was immobilized in the extended position and the left brachial artery was imaged above the antecubital fossa in the longitudinal plane by continuous 2D gray-scale imaging with an 11 MHz ultrasound (HDI 5000CV [Phillips, Andover, Massachusetts]) by a trained sonographer. A baseline rest image was acquired and blood flow was estimated by time-averaging the pulsed Doppler velocity signal obtained from a mid-artery sample volume. Arterial occlusion was created by cuff inflation to suprasystolic pressure (50 mmHg above systolic pressure) for 5 minutes, after which the cuff was deflated. The longitudinal image of the artery was recorded continuously from 30 seconds before to 2 minutes after cuff deflation. Flow images were captured on videotape, and read in a blinded fashion. From longitudinal images, the boundaries for diameter measurement were identified manually with electronic calipers at the lumen-intima interface. Five evenly spaced arterial diameter measurements were taken within a 5 cm segment of vessel at baseline and one minute after cuff deflation, and averaged for the brachial artery width measurement.
Genotyping
PEAR1 rs12041331 SNP genotyping in HAPI Heart Study participants was performed using a TaqMan SNP genotyping assay (Applied Biosystems/Life Technologies, Foster City, California). The mean genotype concordance rate for this polymorphism in a subset of duplicate samples was 100% and the genotype call rate was 98.3%.
GAMMA (Global Microarray Meta-Analysis)
GAMMA [12] was used to identify consistent transcriptional correlations across 75,000 publicly available one-color microarrays from the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) database in order to identify a set of 30 genes highly correlated with PEAR1 across a broad range of experimental conditions. Microarrays were quantile-normalized and processed with an automated quality checking process that includes comparison of parametric expression distributions of individual experiments to expected distributions. Unlike traditional meta-analytic approaches which evaluate gene expression under specific experimental conditions, and control for cell or tissue type, GAMMA utilizes heterogeneous conditions in order to identify general co-expression patterns to more accurately identify common biological responses. In other words, its goal is to identify the strongest gene-gene correlations regardless of experimental condition, tissue, or other variables. Therefore, examination of PEAR1 co-expression patterns in specific cell types (e.g. endothelial cells) or conditions was not explored. The set of genes highly correlated with PEAR1 can then be considered as related to it in a broad, biological sense and queried for statistically significant biological associations they may share. This approach is useful for genes with little or no annotation. Because the use of Gene Ontology for this purpose has fallen under recent criticism [14] and Gene Ontology covers only biological processes, molecular functions, and cellular components, we also used literature mining [15] to identify published commonalities for the genes that were most highly correlated with PEAR1 and had entries in MEDLINE (25 of 30 genes); this includes other categories such as disease relevance, phenotype, and other genes predicted to be relevant to PEAR1’s genetic neighborhood. GAMMA’s performance has been previously benchmarked by predicting Gene Ontology annotations for 5,000 genes and then comparing the predictions to their known Gene Ontology annotations [12, 16]. Importantly, predicted phenotypes and functions from GAMMA have also been validated experimentally in several published studies [16–21].
Cell Culture
Fifty-five de-identified umbilical cords were obtained from the University of Maryland Medical Center Division of Maternal and Fetal Medicine under an IRB exempt protocol due to their de-identified nature. Upon receipt of umbilical cords, HUVECs were harvested as described previously [22]. HUVECS were maintained at 37°C in a 5% CO2 incubator using Endothelial Basal Media 2 (Lonza, Catalog #CC-3156 & CC-4176, Walkersville, Maryland) containing 2% FBS, 0.04% hydrocortisone, 0.4% hFGF, 0.1% VEGF, 0.1% R3-IGF, 0.1% ascorbic acid, 0.1% hEGF, 0.1% gentamicin-amphotericin-B [GA-1000], and 0.1% heparin. DNA from each cell line was extracted using a Gentra Puregene Cell Kit (Qiagen, Valencia, California) as recommended by the manufacturer. PEAR1 rs12041331 genotype was determined using a TaqMan SNP genotyping assay (Applied Biosystems/Life Technologies, Foster City, California), which resulted in the identification of 28 major allele homozygotes (GG), 25 heterozygotes (GA), and 2 minor allele homozygotes.
Endothelial Cell Migration Assay
We evaluated the impact of PEAR1 rs12041331 genotype on ex vivo endothelial cell migration using methods described previously [23]. Briefly, 10 primary HUVEC lines were randomly chosen within each genotype (4 rs12041331 major allele homozygotes [GG], 4 heterozygotes [GA], and 2 minor allele homozygotes [AA]) and split at passage 3 onto a gelatin-coated 6-well plate containing Endothelial Basal Media 2. Confluent HUVEC monolayers were scraped in a uniform manner using a P1000 pipette tip to generate the scratch, which was followed by replacement of growth media. Cells were photographed at 0 and 6 hours using an Axiocam MRc 5 camera (Carl Zeiss Microscopy, Pleasanton, California) mounted on a Lumar.V12 microscope (Carl Zeiss Microscopy, Pleasanton, California). The area of the scratch was measured using ImageJ [24] and used to calculate endothelial cell migration as described in the Statistical Analysis section.
Statistical Analyses
HAPI Heart Study
Summary statistics and frequencies for the OOA HAPI Heart Study were calculated using SAS version 9.2 (SAS Institute Inc., Cary, North Carolina). Measures of Hardy-Weinberg equilibrium were calculated using a χ2 test. For HAPI Heart Study-related analyses, P-values less than 0.05 were considered statistically significant. All statistical tests were 2-sided.
Clinical correlates of FMD response were evaluated using a regression-based approach as implemented in SOLAR version 4.07 (Texas Biomedical Research Institute, San Antonio, Texas). Given the unique ancestral history of the Lancaster OOA community, all participants are related and their relationships were accounted for using the extensive genealogical records of the OOA [25] by including a polygenic component as a random effect as previously described [26]. Triglyceride levels were logarithm-transformed for analysis and back-transformed for presentation. The relationship between smoking and FMD was only measured in sex-stratified analyses to account for the OOA community’s cultural norms that limit smoking to males. Association analyses with PEAR1 rs12041331 and FMD were performed under an additive model using a variance component method that assesses the effect of genotype on the quantitative trait. Analyses were adjusted for age, sex, body mass index (BMI), diabetes, SBP, DBP, and the aforementioned polygenic component, which was modeled using the relationship matrix derived from the complete OOA pedigree structure available through the Anabaptist Genealogy Database [25, 27]. Secondary analyses were adjusted for the same covariates above in addition to baseline brachial artery width (Dbase) and heart rate (HR) to account for changes in FMD caused by these variables [28]. Heritability of FMD response corresponds to the proportion of the trait variance accounted for by the polygenic component, and the heritability estimate was created with adjustments for age and sex.
GAMMA
Statistical analyses implemented by GAMMA have been previously described [12, 29]. Briefly, 1-color microarrays were processed to create a gene vs experiment expression matrix in which subsets can be extracted to perform meta-analyses and identify gene-gene correlations. Function, phenotype and disease relevance are predicted by identifying a set of 30 genes that are most correlated in their expression patterns with a query gene of interest. Of these 30 co-expressed genes, 25 had MEDLINE entries and were analyzed by literature-mining software [15] for what they have in common (essentially a “guilt by association” approach). Microarrays were quantile normalized and noise thresholds were used to identify transcription levels that were statistically significant.
Endothelial Cell Migration
Each of 10 HUVEC lines was plated into 6-well plates, and 3 equidistant photographs were taken per well at 0 and 6 hours after scratch generation of the endothelial monolayer, resulting in 18 area measurements per cell line at each time point. Mean endothelial cell migration distance was calculated by dividing the area of the scratch by the height of the frame. Differences in endothelial cell migration distance between PEAR1 rs12041331 genotype groups were assessed using two-tailed analysis of variance. Genotype-specific differences in endothelial cell migration distance were assessed 0 and 6 hours post-scratch generation. P-values < 0.05 were considered statistically significant.
Results
We evaluated the effect of PEAR1 rs12041331 on in vivo endothelial function in 641 subjects of the HAPI Heart Study. Characteristics of the HAPI Heart Study participants are shown in Table 1. Subjects were generally healthy, middle-aged (mean age = 43.2 years), drug-naïve, and had low prevalence of disease (e.g. diabetes [0.78%], hypertension [12.8%], hypercholesterolemia [16.9%]), and obesity [mean BMI = 26.3]). FMD was normally distributed in this population (S1 Fig). Poorer FMD response was associated with increasing age (0.9% of the variance; P = 0.007), male sex (17.6% of the variance; P = 3.53 x 10−30), increased Dbase (29.2% of the variance; P = 2.5 x 10−54), increased DBP (0.6% of the variance; P = 0.026), increased SBP (1.5% of the variance; P = 0.001), decreased HR (7.5% of the variance, P = 2.73 x 10−12), and presence of hypertension (0.7% of the variance; P = 0.025) (Table 2). Initially, smoking was also significantly associated with FMD (1.9% of the variance; P = 1.38 x 10−3), but after stratifying for sex, since only men smoke in the Amish community, no association was observed (P = 0.817). Also, we found that the variance caused by sex was driven by Dbase, as the effect of sex was diminished (P = 0.85) when adjusting for Dbase. The estimated residual heritability of FMD after adjustment for age and sex was 0.16 ± 0.09 (P = 0.03).
Table 1. Characteristics of HAPI Heart Study Participants.
| Characteristic (Units) | Men | Women |
|---|---|---|
| Number (n) | 365 | 276 |
| Age ± SD (years) | 42.2 ± 13.7 | 44.5 ± 14.1 |
| BMI ± SD (kg/m2) | 25.6 ± 3.2 | 27.3 ± 4.8 |
| Systolic blood pressure ± SD (mm Hg) | 122.0 ± 12.8 | 120.4 ± 16.3 |
| Diastolic blood pressure ± SD (mm Hg) | 78.0 ± 8.7 | 75.4 ± 8.4 |
| No. with hypertension (%) * | 42 (11.5) | 40 (14.5) |
| Total cholesterol ± SD (mg/dl) | 202.3 ± 44.7 | 212.7 ± 48.4 |
| LDL cholesterol ± SD (mg/dl) | 136.4 ± 40.9 | 139.6 ± 45.8 |
| HDL cholesterol ± SD (mg/dl) | 53.3 ± 13.0 | 59.0 ± 14.4 |
| Triglycerides ± SD (mg/dl) † | 62.8 ± 37.9 | 70.9 ± 45.5 |
| No. with hypercholesterolemia (%) ‡ | 57 (15.7) | 51 (18.6) |
| No. with self-reported diabetes (%) | 3 (0.8) | 2 (0.7) |
| Hematocrit ± SD (%) | 43.2 ± 2.5 | 38.5 ± 2.5 |
| White blood cell count ± SD (n x 1000) | 5.4 ±1.2 | 5.2 ± 1.0 |
| Platelet count ± SD (n x 100,000) | 231.4 ± 52.3 | 240.8 ± 50.5 |
| No. of current smokers (%) § | 73 (20.2) | 0 (0) |
| No. taking aspirin (%) | 14 (3.8) | 5 (1.8) |
| No. taking lipid-lowering medications (%) | 5 (1.4) | 2 (0.7) |
| No. taking anti-hypertensive medications (%) | 1 (0.3) | 0 (0) |
| Brachial artery width pre-occlusion ± SD (mm) | 4.1 ± 0.5 | 3.1 ± 0.4 |
| Brachial artery width post-occlusion ± SD (mm) | 4.4 ± 0.5 | 3.5 ± 0.4 |
Abbreviations: BMI, body mass index; HAPI, Heredity and Phenotype Intervention; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SD, standard deviation.
SI conversion factors: To convert HDL-cholesterol, LDL-cholesterol, and total cholesterol values to mmol/L, multiply by 0.0259; triglycerides to mmol/L, multiply by 0.0113.
*Defined as systolic blood pressure greater than 140 mm Hg or diastolic blood pressure greater than 90 mm Hg or taking prescription medication for previously diagnosed hypertension.
†Logarithm-transformed for analysis and back-transformed for presentation.
‡Defined as LDL-cholesterol greater than 160 mg/dl or taking prescription medication for previously diagnosed hypercholesterolemia.
§Self-reported history of smoking cigarette, pipe, or cigar. Only men report smoking in the OOA community.
Table 2. Predictors of Variance in Flow-Mediated Dilation in HAPI Study Participants.
| Predictor (Units) | Beta | Standard Error | P-value | Variance of Significant Predictor (%) |
|---|---|---|---|---|
| Age (years) | -0.05 | 0.02 | 0.007 | 0.9% |
| Sex (female) | 5.07 | 0.42 | 3.5 x 10−30 | 17.6% |
| BMI (kg/m2) | 0.04 | 0.06 | 0.448 | |
| Current smoking (%) * | 0.15 | 0.66 | 0.819 | |
| Self-reported diabetes (%) | -4.72 | 2.66 | 0.076 | |
| Brachial artery width pre-occlusion (mm) | -0.65 | 0.04 | 2.5 x 10−54 | 29.2% |
| HDL cholesterol (mg/dl) | 0.02 | 0.02 | 0.369 | |
| LDL cholesterol (mg/dl) | 0 | 0.01 | 0.937 | |
| Total cholesterol (mg/dl) | 0 | 0.01 | 0.578 | |
| Triglycerides (mg/dl) | ||||
| Diastolic blood pressure (mm Hg) | -0.06 | 0.03 | 0.026 | 0.6% |
| Systolic blood pressure (mm Hg) | -0.05 | 0.02 | 0.001 | 1.5% |
| Mean arterial pressure (mm Hg) | -0.07 | 0.02 | 0.003 | 1.2% |
| Heart rate (beats/min) | 0.17 | 0.02 | 2.7 x 10−12 | 7.5% |
| Hypertension (%) | -1.54 | 0.68 | 0.025 | 0.7% |
| rs12041331 genotype (A allele) | 1.22 | 0.62 | 0.047 | 0.5% |
Abbreviations: BMI, body mass index; HAPI, Heredity and Phenotype Intervention; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
*Observed using a sex-stratified analysis to account for the OOA community’s male-only smoking cohort.
The minor allele (A) frequency of PEAR1 rs12041331 in the HAPI Heart Study was 0.09, similar to that reported in other populations of European descent [7, 8, 10], resulting in 535 major allele homozygotes (GG), 101 heterozygotes (GA), and 5 minor allele homozygotes (AA), and conformed to expectations of Hardy-Weinberg equilibrium (P = 0.92). Characteristics of study participants by rs12041331genotype are shown in S1 Table. In our primary model accounting for clinical characteristics including age, sex, diabetes, SBP, DBP, and BMI, FMD was significantly higher in carriers of the PEAR1 rs12041331 A-allele when compared to subjects who did not carry this allele (GG = 10.2 ± 0.3, GA = 10.8 ± 0.6, AA = 16.5 ± 5.4, P = 0.019). PEAR1 rs12041331 remained significantly associated with FMD after also accounting for variation in Dbase (GG = 10.2 ± 0.2, GA = 11.0 ± 0.4, AA = 14.1 ± 2.8, P = 0.032), HR (GG = 10.2 ± 0.1, GA = 11.1 ± 0.3, AA = 13.7 ± 2.3, P = 0.019), and both Dbase and HR (GG = 10.2 ± 0.2, GA = 11.0 ± 0.4, AA = 13.9 ± 2.8, P = 0.034).
Using the approach implemented in GAMMA, we identified genes that were most significantly associated with PEAR1 expression (S2 Table) and built a genetic neighborhood of protein-protein interactions shared by the co-expressed genes (Fig 1). Of the 30 most significant co-expressed gene-pairs, using the 25 with MEDLINE entries, the top phenotypes predicted to be affected by changes in PEAR1 gene expression were “endothelial cell migration,” “vasculogenesis,” and “angiogenesis” (Table 3). Similarly, the disease most highly predicted to be influenced by alterations in PEAR1 gene expression was “vascular disease” (Table 3). Full lists of the phenotypes and diseases that were predicted to be influenced by PEAR1 expression are shown in S3 and S4 Tables, respectively.
Fig 1. PEAR1 Genetic Network.
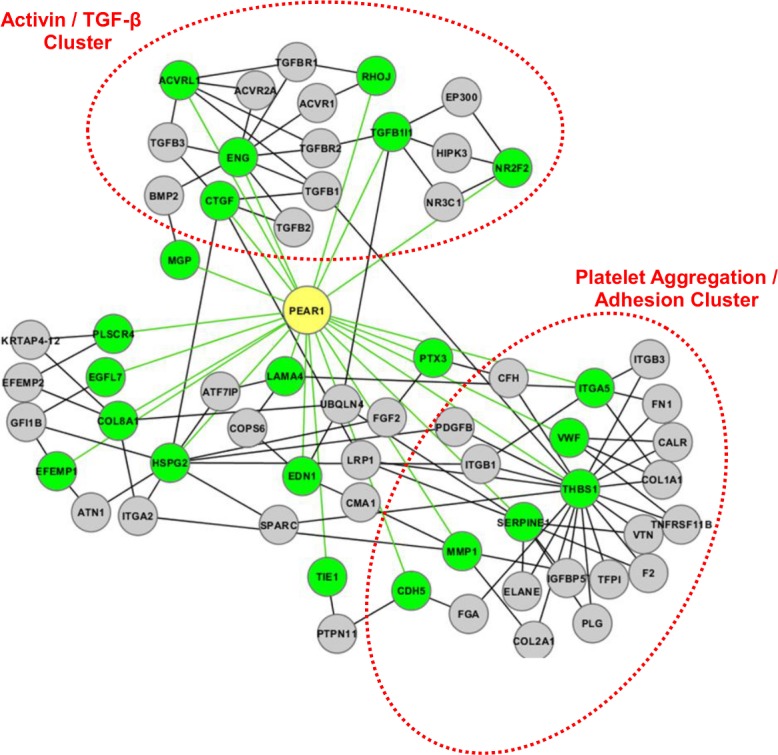
Genes highly correlated with PEAR1 (green nodes) were evaluated for protein-protein interactions (gray nodes) that were shared by at least 2 of the 30 genes analyzed. Green lines indicate a co-expression relationship; black lines indicate a physical protein-protein interaction. Genetic neighborhoods of similar pathway or function have been highlighted and labeled.
Table 3. Meta-Analysis Results of Publicly Available Microarray Datasets.
| Top predicted phenotypes | # Shared Relations * | Score † |
|---|---|---|
| Endothelial cell migration | 12 | 136 |
| Vasculogenesis | 11 | 103 |
| Angiogenesis | 20 | 86 |
| Lymphangiogenesis | 8 | 70 |
| Neovascularization | 11 | 57 |
| Endothelial cell proliferation | 9 | 55 |
| Platelet aggregation | 7 | 55 |
| Cell adhesion | 15 | 52 |
| Top predicted diseases | ||
| Vascular disease | 12 | 59 |
| Non-small cell lung carcinoma | 14 | 55 |
| Osteoarthritis | 11 | 46 |
| Preeclampsia | 10 | 42 |
| Pancreatic cancer | 12 | 40 |
| Diabetic nephropathy | 9 | 39 |
| Colorectal cancer | 14 | 39 |
* Using the 25 genes with MEDLINE entries that are most highly correlated with PEAR1 expression (see S2 Table), predicted phenotypes, diseases, and genes were identified with Global Microarray Meta-Analysis (GAMMA), and the number of shared relations represents how many of the 25 genes were related to term (left column).
†Score reflects the relative enrichment of observed connections within the analyzed network relative to a random network with the same number of connections per gene, enabling prediction to be prioritized [15].
Given our GAMMA results, we functionally tested whether the well-described PEAR1 rs12041331 variant significantly influenced endothelial cell migration. Genotypic differences in endothelial cell migration distances were assessed at 6 hours post-scratch generation. Consistent with the results of our microarray meta-analysis regarding endothelial cell migration, we observed that the A-allele of PEAR1 rs12041331 was significantly associated with increased endothelial cell migration (P = 0.04; 143.8 ± 58.4 μm for the 4 GG cell lines, 153.1 ± 39.8 μm for the 4 GA cell lines, and 168.4 ± 34.8 μm for the 2 AA cell lines [Fig 2]). Moreover, this association remained statistically significant when we grouped cells line containing the A-allele (i.e. GA and AA genotypes) and compared them to GG homozygotes (P = 0.048; 143.8 ± 58.4 μm for the 4 GG cell lines and 158.2 ± 38.7 μm for cell lines that carried the A-allele [N = 6])
Fig 2. The Impact of PEAR1 rs12041331 on Endothelial Cell Migration in Human Umbilical Vein Endothelial Cells (HUVECs).
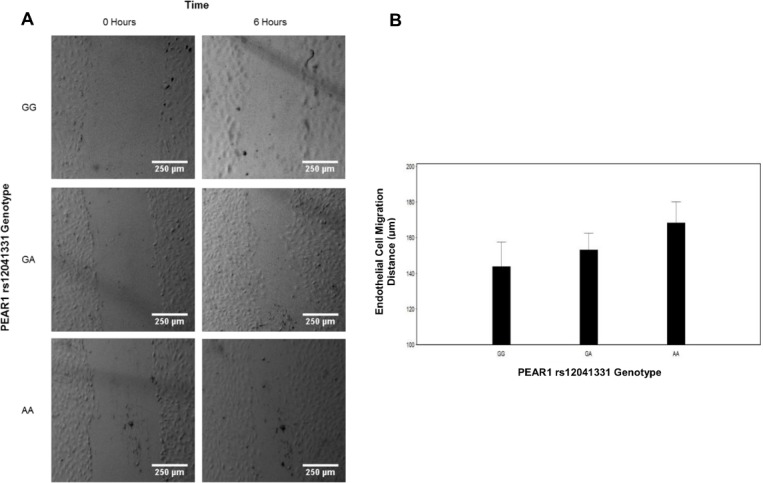
(A) Representative phase-contrast images of rs12041331-stratified HUVECs at 0 and 6 hours post-scratch generation during an ex vivo endothelial cell migration assay. Scale bar, 250 μm. (B) Quantitative depiction of mean HUVEC migration. Endothelial cell migration distance was calculated by dividing the area of the scratch by the height of the frame using ImageJ. Mean endothelial cell migration distance was calculated based on 72, 72, and 36 independent measurements for GG, GA, and AA genotypes, respectively, as described in the Materials and Methods section.
Discussion
PEAR1 was identified in 2005 by Nanda and colleagues as a type I membrane protein that is highly expressed in platelets and endothelial cells, and is involved in platelet aggregation through secondary signaling via the αIIbβ3 integrin following platelet-platelet contact [1]. Several subsequent studies, including our own [10], have focused almost exclusively on the functional or phenotypic effects of this gene in platelets and megakaryocytes [1–3, 5–9, 11]. Taken together, these investigations have yielded insights into the molecular signaling of PEAR1 as a tyrosine kinase that signals through PI3K and Akt [1, 2], as well as the role of this receptor in megakaryopoiesis [3], platelet aggregation [1, 2, 5–11], antiplatelet therapy response [8–11], and cardiovascular events [10].
In this investigation, we measured FMD of the brachial artery, the most widely used noninvasive test of endothelial function, in order to estimate its heritability, establish clinical predictors, and assess the potential effect of genetic variation in PEAR1 on endothelial function in vivo. In relatively healthy participants of the HAPI Heart Study, we found FMD to be normally distributed (S1 Fig). Gender and Dbase accounted for over 45% of the variation in FMD, with age, smoking, SBP, DBP, hypertension, and HR jointly accounting for another 13–14%. Similar to previous reports [30, 31], our heritability estimates suggest that approximately 16% of the variation in FMD is attributable to relatedness, for which genetic factors likely contribute. To our knowledge, this investigation describes the first reported relationship between PEAR1 and clinical measures of endothelial function. Specifically, our data show that the minor allele of a common intronic variant (rs12041331) in this gene is significantly associated with greater brachial artery FMD. While the biological mechanism behind this association is not understood currently, future investigations probing the previously established relationship between PEAR1 and AKT signaling [2], an important mediator of FMD and nitric oxide production, seem warranted.
In an attempt to gain a more comprehensive view regarding phenotypes and diseases that may be influenced by PEAR1, we also used in silico genetic and statistical methodologies [12] to conduct a meta-analysis of publicly available microarray datasets. As expected, our PEAR1 genetic network identified several genes critical in platelet adhesion and aggregation including von Willebrand factor (vWF), thrombospondin 1 (THBS1), and plasminogen activator inhibitor 1 (SERPINE1). Interestingly, we also identified several PEAR1 co-expressed genes that are part of the TGF-β signaling system. These genes are important in blood vessel formation (ACVRL1) [32], endothelial cell migration and angiogenesis (RhoJ) [33], endothelial cell proliferation, smooth muscle cell recruitment, and vascular remodeling (ENG) [34, 35], as well as endothelial cell adhesion and survival (CTGF) [36]. Furthermore, several genes that were significantly co-expressed with PEAR1 have also been previously implicated in endothelial-related pathological conditions including pulmonary fibrosis (CTGF) [37] and hemorrhagic telangiectasia (ACVRL1 and ENG) [38]. While, to date, no investigation has evaluated the potential relationship between PEAR1 and any of these genes and conditions, our results suggest that future studies may be warranted.
Through GAMMA, we also generated a list of phenotypes that are most likely to be influenced by changes in PEAR1 expression. Interestingly, 6 of the 8 most significantly associated phenotypes predicted by GAMMA are critical in vascular endothelial function (endothelial cell migration, vasculogenesis, angiogenesis, neovascularization, endothelial cell proliferation, and cell adhesion). The two most significantly associated phenotypes that were not related to endothelial function were “platelet aggregation,” which has been previously validated [5–11], and “lymphangiogenesis,” which, at this time, has never been evaluated in the context of PEAR1. Similarly, a list of diseases predicted to be influenced by changes in PEAR1 expression was generated using the same approach. Consistent with our previous report [10], “vascular disease” was most strongly related to changes in PEAR1 expression. Preeclampsia, a condition characterized by high blood pressure and endothelial dysfunction, was also predicted to be influenced by PEAR1. Intriguingly, other diseases predicted to be related to PEAR1 included several types of cancer (e.g. colorectal, pancreatic, and non-small cell lung carcinoma), osteoarthritis, and diabetic nephropathy. While there is currently no available data suggesting a relationship between PEAR1 and osteoarthritis, diabetic nephropathy, aortic aneurisms, or cancer risk, it is interesting to speculate given the role of the TGF-β signaling system in the progression of these disorders.
In order to experimentally validate, in part, the results of our microarray meta-analysis, we tested whether a well-described genetic variant in PEAR1 (rs12041331) significantly influenced endothelial cell migration, the most highly predicted phenotype to be affected by PEAR1 based on our GAMMA results, through the use of the well-described endothelial cell migration assay. Indeed, we observed that PEAR1 rs12041331 was significantly associated with endothelial migration distance. Our results indicate that HUVECs homozygous for the A-allele of PEAR1 rs12041331 have approximately 117% better endothelial cell migration capabilities compared to cells homozygous for the major allele (G). This novel observation not only strengthens our confidence in the results obtained by GAMMA, but also provides at least one phenotype by which PEAR1 influences endothelial cell biology.
To our knowledge, the only other investigation to date that has evaluated PEAR1 in the context of endothelial cell biology was a recently reported publication by Vandenbriele and colleagues [39]. In that study, it was observed that lentiviral-mediated knockdown of PEAR1 resulted in enhanced endothelial cell proliferation through the Akt/p21/CDC2 pathway, and, consistent with the current investigation, observed that PEAR1 knockdown resulted in increased endothelial cell migration, potentially suggesting that PEAR1 is a negative regulator of these phenotypes. Given that prior evidence has shown that PEAR1 influences AKT/PTEN signaling [2, 39], the increased endothelial migration observed with the rs12041331 minor allele as well as in PEAR1 knockdown studies could suggest decreased action of PTEN, leading to an increase in AKT phosphorylation, a key mediator in endothelial cell migration. Given the observed impact of PEAR1 on endothelial cell proliferation and migration, the authors extended these findings showing that PEAR1 knockdown also resulted in increased endothelial tube length, number, and branch points using a matrigel assay. Interestingly, they also showed that Pear1 -/- mice have increased neoangiogenesis compared to wild-type mice using a hind limb ischemia ligation model as well as significantly decreased wound size and closure time in an independent skin wound healing model. Importantly, all of these findings are consistent with the aforementioned top phenotypes predicted to be associated with PEAR1 by GAMMA. Therefore, the data shown by Vandenbriele and colleagues, combined with our data showing that genetic variation in PEAR1 influences flow-mediated dilation in humans suggest that PEAR1 may be a critical determinant of endothelial homeostasis with potential implications in the development of vascular disease.
Most of the investigations of PEAR1 to date have focused almost exclusively on its role in platelets and megakaryocytes. Indeed, prior mechanistic investigations by Kauskot and colleagues suggest that PEAR1 significantly influences sustained platelet aggregation through glycoprotein αIIbβ3 [2] as well as megakaryopoiesis through the PI3K/PTEN pathways [3]. In addition, several genetic investigations have shown that polymorphisms in PEAR1 are associated with ex vivo platelet aggregation in response to several platelet agonists (e.g. ADP, collagen, epinephrine) as well as pre- and post-antiplatelet therapy treatment (i.e. aspirin and prasugrel) [5–11]. In our own previous investigation, we identified PEAR1 rs12041331 as a strong determinant of collagen-stimulated platelet aggregation after dual anti-platelet therapy (DAPT) with aspirin and clopidogrel, as well as decreased 1-year survival in DAPT-treated patients undergoing percutaneous coronary intervention and increased rates of myocardial infarction (MI) in aspirin-treated patients with stable coronary artery disease [10]. However, despite the consistent data that shows PEAR1 significantly influences platelet biology and thrombosis, the current work and the investigation of Vandenbriele and colleagues highlight a novel genotype-phenotype relationship in the endothelium. Therefore, it is important that future investigations aiming to understand the relationship between this gene and cardiovascular disease take into account the pleiotropic nature of PEAR1 in these tissues.
There are some limitations to our study. While the use of FMD is the most commonly used method to non-invasively assess endothelial function, recent work by Atkinson and Batterham have shown that inadequate adjustment of FMD analyses, particularly of a ratio-scaling inconsistency between percent FMD and baseline vessel diameter, leads to biased results [40]. In order to minimize the effect of this potential confounder and others, we tested for association between PEAR1 rs12041331 and FMD using several statistical models that adjusted for different clinical variables that impact FMD measurements (e.g. age, sex, Dbase, and HR). In all models, PEAR1 rs12041331 remained significantly associated with FMD. In addition, given the number of minor allele homozygotes (N = 5) and observed genotypic means, we also tested for association between rs12041331 and FMD using both dominant (GG vs AG/AA; P = 0.03) and recessive (GG/AG vs AA; P = 0.05) genetic models, showing a consistent direction of association. A potential limitation of the approach implemented in GAMMA is that it tends to be biased towards inclusion of genes that change significantly in their expression level and against genes whose transcriptional levels are too low to reliably detect. Although the analysis we performed of multiple genes for their published commonalities tends to reduce the bias towards a small number of genes skewing the results, the scientific literature itself may have its own biases in terms of preferences for what is both studied and reported. Therefore, while we functionally validated one phenotype predicted by GAMMA (i.e. endothelial migration), we believe future functional experiments to evaluate the relationship between PEAR1 and the genes/phenotypes identified by GAMMA are warranted.
The dynamic relationship between platelets and endothelial cells is critical in cardiovascular physiology, and dysfunction of either cell type can lead to a cardiovascular event. There is a growing body of literature indicating that PEAR1 has important effects on platelet-related processes and cardiovascular outcomes. In this investigation, we have established for the first time that genetic variation in PEAR1 significantly impacts endothelial function as well. Looking forward, it is critical to further characterize the function(s) of PEAR1 in both platelet and endothelial cells to elucidate the mechanism by which this gene may contribute to cardiovascular risk. Once understood, PEAR1 may be a novel target for treatment and prevention of cardiovascular disease.
Supporting Information
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLSX)
Acknowledgments
We gratefully acknowledge our Amish liaisons and field workers and the extraordinary cooperation and support of the Amish community, without which these studies would not have been possible. We also acknowledge Dr. Alan Shuldiner for his impactful insights and guidance.
Data Availability
De-identified data is available upon request to the corresponding author or Dr. Alan Shuldiner. Given the unique ancestral relationships among Amish participants and publicly available genealogical records (i.e. Anabaptist Genealogical Data Base), limited data may be provided in order to protect participant confidentiality.
Funding Statement
Support was provided by: U01GM074518 National Institute of General Medical Sciences [http://www.nigms.nih.gov/Pages/default.aspx]; U01HL072515 National Heart, Lung, and Blood Institute [http://www.nhlbi.nih.gov/] to author BDM; P20GM103636 National Institute of General Medical Sciences [http://www.nigms.nih.gov/Pages/default.aspx] to author JDW; U54GM104938 National Institute of General Medical Sciences [http://www.nigms.nih.gov/Pages/default.aspx] to author JDW; K01HL116770 National Heart, Lung, and Blood Institute [http://www.nhlbi.nih.gov/] to author LMYA; K23GM102678 National Institute of General Medical Sciences [http://www.nigms.nih.gov/Pages/default.aspx] to author JPL; P30DK072488 National Institute of Diabetes and Digestive and Kidney Diseases [http://www.niddk.nih.gov/Pages/default.aspx] to author BDM; T32HL072751 National Heart, Lung, and Blood Institute [http://www.nhlbi.nih.gov/] to author ASF. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nanda N, Bao M, Lin H, Clauser K, Komuves L, Quertermous T, et al. Platelet endothelial aggregation receptor 1 (PEAR1), a novel epidermal growth factor repeat-containing transmembrane receptor, participates in platelet contact-induced activation. J Biol Chem. 2005;280(26):24680–9. Epub 2005/04/27. 10.1074/jbc.M413411200 . [DOI] [PubMed] [Google Scholar]
- 2. Kauskot A, Di Michele M, Loyen S, Freson K, Verhamme P, Hoylaerts MF. A novel mechanism of sustained platelet alphaIIbbeta3 activation via PEAR1. Blood. 2012;119(17):4056–65. Epub 2012/03/01. doi: blood-2011-11-392787 [pii]. 10.1182/blood-2011-11-392787 . [DOI] [PubMed] [Google Scholar]
- 3. Kauskot A, Vandenbriele C, Louwette S, Gijsbers R, Tousseyn T, Freson K, et al. PEAR1 attenuates megakaryopoiesis via control of the PI3K/PTEN pathway. Blood. 2013;121(26):5208–17. Epub 2013/05/15. 10.1182/blood-2012-10-462887 . [DOI] [PubMed] [Google Scholar]
- 4. Wu HH, Bellmunt E, Scheib JL, Venegas V, Burkert C, Reichardt LF, et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat Neurosci. 2009;12(12):1534–41. Epub 2009/11/17. 10.1038/nn.2446 nn.2446 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herrera-Galeano JE, Becker DM, Wilson AF, Yanek LR, Bray P, Vaidya D, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol. 2008;28(8):1484–90. Epub 2008/05/31. 10.1161/ATVBAHA.108.168971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones CI, Bray S, Garner SF, Stephens J, de Bono B, Angenent WG, et al. A functional genomics approach reveals novel quantitative trait loci associated with platelet signaling pathways. Blood. 2009;114(7):1405–16. Epub 2009/05/12. 10.1182/blood-2009-02-202614 . [DOI] [PubMed] [Google Scholar]
- 7. Johnson AD, Yanek LR, Chen MH, Faraday N, Larson MG, Tofler G, et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet. 2010;42(7):608–13. Epub 2010/06/08. 10.1038/ng.604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faraday N, Yanek LR, Yang XP, Mathias R, Herrera-Galeano JE, Suktitipat B, et al. Identification of a specific intronic PEAR1 gene variant associated with greater platelet aggregability and protein expression. Blood. 2011;118(12):3367–75. Epub 2011/07/28. doi: blood-2010-11-320788 [pii]. 10.1182/blood-2010-11-320788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim Y, Suktitipat B, Yanek LR, Faraday N, Wilson AF, Becker DM, et al. Targeted deep resequencing identifies coding variants in the PEAR1 gene that play a role in platelet aggregation. PLoS One. 2013;8(5):e64179 Epub 2013/05/25. 10.1371/journal.pone.0064179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lewis JP, Ryan K, O'Connell JR, Horenstein RB, Damcott CM, Gibson Q, et al. Genetic variation in PEAR1 is associated with platelet aggregation and cardiovascular outcomes. Circ Cardiovasc Genet. 2013;6(2):184–92. Epub 2013/02/09. 10.1161/CIRCGENETICS.111.964627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiang Q, Cui Y, Zhao X, Zhao N. Identification of PEAR1 SNPs and their influences on the variation in prasugrel pharmacodynamics. Pharmacogenomics. 2013;14(10):1179–89. Epub 2013/07/19. 10.2217/pgs.13.108 . [DOI] [PubMed] [Google Scholar]
- 12. Wren JD. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics. 2009;25(13):1694–701. 10.1093/bioinformatics/btp290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitchell BD, McArdle PF, Shen H, Rampersaud E, Pollin TI, Bielak LF, et al. The genetic response to short-term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am Heart J. 2008;155(5):823–8. Epub 2008/04/29. 10.1016/j.ahj.2008.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gillis J, Pavlidis P. Characterizing the state of the art in the computational assignment of gene function: lessons from the first critical assessment of functional annotation (CAFA). BMC bioinformatics. 2013;14 Suppl 3:S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wren JD, Bekeredjian R, Stewart JA, Shohet RV, Garner HR. Knowledge discovery by automated identification and ranking of implicit relationships. Bioinformatics. 2004;20(3):389–98. 10.1093/bioinformatics/btg421 . [DOI] [PubMed] [Google Scholar]
- 16. Daum JR, Wren JD, Daniel JJ, Sivakumar S, McAvoy JN, Potapova TA, et al. Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Current biology: CB. 2009;19(17):1467–72. 10.1016/j.cub.2009.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lupu C, Zhu H, Popescu NI, Wren JD, Lupu F. Novel protein ADTRP regulates TFPI expression and function in human endothelial cells in normal conditions and in response to androgen. Blood. 2011;118(16):4463–71. 10.1182/blood-2011-05-355370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clemmensen SN, Bohr CT, Rorvig S, Glenthoj A, Mora-Jensen H, Cramer EP, et al. Olfactomedin 4 defines a subset of human neutrophils. Journal of leukocyte biology. 2012;91(3):495–500. 10.1189/jlb.0811417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Towner RA, Jensen RL, Colman H, Vaillant B, Smith N, Casteel R, et al. ELTD1, a potential new biomarker for gliomas. Neurosurgery. 2013;72(1):77–90; discussion 1. 10.1227/NEU.0b013e318276b29d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Towner RA, Jensen RL, Vaillant B, Colman H, Saunders D, Giles CB, et al. Experimental validation of 5 in-silico predicted glioma biomarkers. Neuro-oncology. 2013;15(12):1625–34. 10.1093/neuonc/not124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gandhapudi SK, Tan C, Marino JH, Taylor AA, Pack CC, Gaikwad J, et al. IL-18 Acts in Synergy with IL-7 To Promote Ex Vivo Expansion of T Lymphoid Progenitor Cells. J Immunol. 2015;194(8):3820–8. Epub 2015/03/18. 10.4049/jimmunol.1301542 jimmunol.1301542 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung AL. Isolation and culture of human umbilical vein endothelial cells (HUVEC). Curr Protoc Microbiol. 2007;Appendix 4:Appendix 4B. Epub 2008/09/05. 10.1002/9780471729259.mca04bs4 . [DOI] [PubMed]
- 23. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–33. Epub 2007/04/05. doi: nprot.2007.30 [pii]. 10.1038/nprot.2007.30 . [DOI] [PubMed] [Google Scholar]
- 24. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5. Epub 2012/08/30. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Agarwala R, Biesecker LG, Hopkins KA, Francomano CA, Schaffer AA. Software for constructing and verifying pedigrees within large genealogies and an application to the Old Order Amish of Lancaster County. Genome Res. 1998;8(3):211–21. Epub 1998/05/16. . [DOI] [PubMed] [Google Scholar]
- 26. Shuldiner AR, O'Connell JR, Bliden KP, Gandhi A, Ryan K, Horenstein RB, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 2009;302(8):849–57. Epub 2009/08/27. 10.1001/jama.2009.1232 302/8/849 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agarwala R, Biesecker LG, Tomlin JF, Schaffer AA. Towards a complete North American Anabaptist genealogy: A systematic approach to merging partially overlapping genealogy resources. American journal of medical genetics. 1999;86(2):156–61. . [DOI] [PubMed] [Google Scholar]
- 28. Atkinson G, Batterham AM. Allometric scaling of diameter change in the original flow-mediated dilation protocol. Atherosclerosis. 2013;226(2):425–7. Epub 2012/12/25. 10.1016/j.atherosclerosis.2012.11.027 . [DOI] [PubMed] [Google Scholar]
- 29. Dozmorov MG, Wren JD. High-throughput processing and normalization of one-color microarrays for transcriptional meta-analyses. BMC bioinformatics. 2011;12 Suppl 10:S2. 10.1186/1471-2105-12-S10-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benjamin EJ, Larson MG, Keyes MJ, Mitchell GF, Vasan RS, Keaney JF Jr, et al. Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham Heart Study. Circulation. 2004;109(5):613–9. Epub 2004/02/11. 10.1161/01.CIR.0000112565.60887.1E . [DOI] [PubMed] [Google Scholar]
- 31. Suzuki K, Juo SH, Rundek T, Boden-Albala B, Disla N, Liu R, et al. Genetic contribution to brachial artery flow-mediated dilation: the Northern Manhattan Family Study. Atherosclerosis. 2008;197(1):212–6. Epub 2007/04/28. 10.1016/j.atherosclerosis.2007.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gu Y, Jin P, Zhang L, Zhao X, Gao X, Ning Y, et al. Functional analysis of mutations in the kinase domain of the TGF-beta receptor ALK1 reveals different mechanisms for induction of hereditary hemorrhagic telangiectasia. Blood. 2006;107(5):1951–4. 10.1182/blood-2005-05-1834 . [DOI] [PubMed] [Google Scholar]
- 33. Leszczynska K, Kaur S, Wilson E, Bicknell R, Heath VL. The role of RhoJ in endothelial cell biology and angiogenesis. Biochemical Society transactions. 2011;39(6):1606–11. 10.1042/BST20110702 . [DOI] [PubMed] [Google Scholar]
- 34. Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, et al. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circulation research. 2010;106(8):1425–33. 10.1161/CIRCRESAHA.109.211037 . [DOI] [PubMed] [Google Scholar]
- 35. Mancini ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Developmental dynamics: an official publication of the American Association of Anatomists. 2009;238(10):2479–93. 10.1002/dvdy.22066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis. 2002;5(3):153–65. . [DOI] [PubMed] [Google Scholar]
- 37. Yan LF, Wei YN, Nan HY, Yin Q, Qin Y, Zhao X, et al. Proliferative phenotype of pulmonary microvascular endothelial cells plays a critical role in the overexpression of CTGF in the bleomycin-injured rat. Experimental and toxicologic pathology: official journal of the Gesellschaft fur Toxikologische Pathologie. 2014;66(1):61–71. 10.1016/j.etp.2013.08.004 . [DOI] [PubMed] [Google Scholar]
- 38. Berg J, Porteous M, Reinhardt D, Gallione C, Holloway S, Umasunthar T, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. Journal of medical genetics. 2003;40(8):585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vandenbriele C, Kauskot A, Vandersmissen I, Criel M, Geenens R, Craps S, et al. Platelet endothelial aggregation receptor-1: a novel modifier of neoangiogenesis. Cardiovascular research. 2015. 10.1093/cvr/cvv193 . [DOI] [PubMed] [Google Scholar]
- 40. Atkinson G, Batterham AM. When will the most important confounder of percentage flow-mediated dilation be reported and adjusted for at the study level? International journal of cardiology. 2014. 10.1016/j.ijcard.2013.12.264 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLSX)
Data Availability Statement
De-identified data is available upon request to the corresponding author or Dr. Alan Shuldiner. Given the unique ancestral relationships among Amish participants and publicly available genealogical records (i.e. Anabaptist Genealogical Data Base), limited data may be provided in order to protect participant confidentiality.