

Abstract
BACKGROUND
The healthy human skin with its effective antimicrobial defense system forms an efficient barrier against invading pathogens. There is evidence suggesting that the composition of this chemical barrier varies between diseases, making the easily-collected sweat an ideal candidate for biomarker discoveries.
OBJECTIVE
Our aim was to provide information about the normal composition of the sweat, and to study the chemical barrier found at the surface of skin.
METHODS
Sweat samples from healthy individuals were collected during sauna bathing, and the global protein panel was analyzed by label-free mass spectrometry. SRM-based targeted proteomic methods were designed and stable isotope labeled reference peptides were used for method validation.
RESULTS
95 sweat proteins were identified, 20 of them were novel proteins. It was shown that dermcidin is the most abundant sweat protein, and along with apolipoprotein D, clusterin, prolactin inducible protein and serum albumin, they make up 91% of secreted sweat proteins. The roles of these highly abundant proteins were reviewed; all of which have protective functions, highlighting the importance of sweat glands in composing the first line of innate immune defense system, and maintaining the epidermal barrier integrity.
CONCLUSION
Our findings in regards to the proteins forming the chemical barrier of the skin as determined by label free quantification and targeted proteomics methods are in accordance with previous studies, and can be further used as a starting point for non-invasive sweat biomarker research.
Keywords: epidermal barrier, highly abundant sweat proteins, label free quantification, non-immune defense, SRM, targeted proteomics
Introduction
The healthy human skin possesses an effective antimicrobial defense system, formed by a complex physical and chemical epidermal barrier that is in cooperation with other cellular components of the innate immune system and normal microbial flora of the skin [1, 2]. As a constituent of the chemical barrier, expression of antimicrobial peptides by keratinocytes, mast cells, neutrophils, sebocytes and eccrine epithelial cells have been reported [3]. Some of the antimicrobial peptides were shown to be expressed constitutively (e.g. dermcidin, RNase7, psoriasin) while others were found to be inducible upon pathogenic stimuli (e.g. LL-37, hBD2, hBD3) [3–5]. It has been reported that antimicrobial peptides such as dermcidin, lysozyme, lactoferrin, psoriasin, cathelicidin and β-defensins are present in human sweat [4, 6–9] and dermcidin-derived peptides were identified as the principal antimicrobial components [7]. Reduced presence of dermcidin-derived peptides was detected in the sweat of atopic dermatitis patients in association with an impaired cutaneous antimicrobial defense [7], while its increased expression has been demonstrated in different cancer cells [10, 11]. Beside their antibiotic properties, some components of the skin’s chemical barrier are also able to recruit inflammatory cells and induce cytokine release [12].
Little is known about the causes and consequences of qualitative and quantitative alterations of the human sweat proteins, moreover, the composition of the sweat itself has not been fully explored. Currently, intense research efforts are being carried out to discover new diagnostic, prognostic and predictive biomarkers in samples which are easily obtainable from patients using noninvasive methods. Recent advances in analytical techniques now allows for thorough analysis of human sweat proteins, hence, facilitating sweat biomarker studies [13, 14].
Label free quantification of proteins with mass spectrometry is a powerful and sensitive method for protein identification and relative quantification. The protein digests are introduced into high precision mass spectrometers where MS/MS spectra are recorded. Based on the spectra, the proteins/peptides are identified, and taking into account either the number of MS/MS events (spectral counting) or the intensity of the precursor ions, the relative quantification of peptides is performed [15, 16]. Silva and coworkers have also used LCMSE to absolutely quantify proteins. In their method, the average MS signal response for the three most intense tryptic peptides per mole of protein is constant within a coefficient of variation of less than +/− 10%. Moreover, given an internal standard, this relationship can be used to calculate a universal response factor [17]. Targeted proteomics is a challenging part of proteomics, and it is mainly based on the Selected Reaction Monitoring (SRM) scan mode of triple quadrupole-containing mass spectrometers being able to provide specific identification and quantification of the proteins in question [18, 19]. During the scan, the first quadrupole transmits ions only with specified, compound-specific m/z; these ions are fragmented in the second quadrupole functioning as a collision cell, and the third quadrupole selectively transmits only one fragment with a specified m/z. This means that a signal can be detected only in case if two criteria characteristic for a compound are fulfilled in the same time. These SRM transitions provide high specificity in one hand and quantitative data on the other, as the AUC of the specific signal is proportional to the amount of the compound entering the mass spectrometer [19].
In this study our aim was to examine the qualitative and quantitative protein profile of the sweat collected from healthy volunteers, in order to gain more insight into the protein composition of healthy human sweat. The presented data contribute to our knowledge concerning the normal physiological function of human sweat, and provide data for further biomarker research.
Materials and Methods
All reagents used in this study were of analytical or LC-MS grade and were purchased from Sigma-Aldrich unless stated otherwise.
Sample collection
10 male and 10 female healthy young adults (between 22–35 years of age) were recruited into the study. Heat induced sweat was collected in an 80 °C electric Finnish-type sauna in the morning. Volunteers were asked not to use any cosmetics that day, and before entering the sauna, they were required to take a shower using only water, and to dry their skin. Volunteers were instructed to stay in the sauna for 20 minutes, thereafter, take a shower, dry their skin and rest for 10 min, followed by another 20 min sauna time. All sample collection complied with the guidelines of the Helsinki Declaration, approved by the Regional Ethical Committee (DEOEC RKEB/IKEB 4078/2013 and 2885/2008) and the subjects signed an informed consent. The collected samples were transported to the laboratory on ice and in less than 30 minutes from sample collection, samples were centrifuged in order to get rid of the shed skin cells and cellular debris. The clear sweat was concentrated by lyophilization, redissolved in ammonium bicarbonate and pooled. The protein concentration of the pooled sweat was determined by Bradford method [20], and the concentrated sample was stored at −70 °C until further processing. Following protein extraction in 0.1M ammonium bicarbonate, pH 7.8, the pooled sample was precipitated twice, using six volumes of ice-cold acetone. The final pellet was solubilized in 0.1M ammonium bicarbonate, pH 7.8, and a micro bicinchoninic acid protein assay [21] was performed according to the manufacturer’s instructions (Pierce, Rockford, IL) to determine the total protein content.
Label free quantification by LCMSE and protein identification by LC-MS/MS
Pooled sweat protein samples (4.2 μg total) were first reduced and alkylated, followed by tryptic digestion overnight at 37 °C. For LCMSE [17], tryptic digests (600 ng in an injection volume of 4 μL containing 50 ng of a T33V Rhodobacter cytochrome c digest as an internal standard), were injected onto a Waters QTOF Premier equipped with a nanoESI source and a NanoAcquity UPLC (Waters Corp., Millford, MA). LC separation of peptides was performed in triplicate using a Symmetry C18 5 μm, 20-mm × 180-μm precolumn and a BEH130 C18 1.7 μm, 100-mm × 100-μm analytical reversed phase column (Waters Corp. Millford, MA). Solvent A was water with 0.1% formic acid and solvent B was 0.1% formic acid in acetonitrile in case of all LC separations. The peptides were separated with a gradient of 2–35% solvent B over 150 min followed by a rise to 95% of solvent B over 2 minutes and a 5-min rinse with 95% of solvent B, after which the system returned to 2% solvent B in 2 minutes. The flow rate was 750 nl/min and the column temperature was 35 °C. The lock mass was delivered from the auxiliary pump of the NanoAcquity pump with a constant flow rate of 500 nl/min at a concentration of 100 fmol of [Glu1]fibrinopeptide B/μl to the reference sprayer of the NanoLockSpray source of the mass spectrometer. The mass spectrometer was operated in the V-mode of analysis with a typical resolving power of at least 10,000 full-width half-maximum. All analyses were performed using positive nanoelectrospray ion mode. The reference sprayer was sampled with a frequency of 60 s. Accurate mass LC-MS data were collected in an alternating low energy and elevated energy mode of acquisition [22–23]. The spectral acquisition time in each mode was 1.5 s with a 0.1-s interscan delay. In low energy MS mode, data were collected at constant collision energy of 4 eV. In elevated energy MS mode, the collision energy was ramped from 15 to 35 eV during each 1.5-s data collection cycle with one complete cycle of low and elevated energy data acquired every 3.2 s. The radiofrequency applied to the quadrupole was adjusted such that ions from m/z 50 to 2000 were efficiently transmitted. High energy MS ions were collected between 100 to 1500 m/z. The digested sweat protein sample was also analyzed by LC-MS/MS using an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an Advion nanomate ESI source (Advion, Ithaca, NY), following ZipTip (Millipore, Billerica, MA) C18 sample clean-up according to the manufacturer’s instructions. Peptides (240 ng) were eluted from a C18 precolumn (100-μm id × 2 cm, Thermo Fisher Scientific) onto an analytical column (75-μm ID × 10 cm, C18, Thermo Fisher Scientific) using a 5% hold of solvent B (acetonitrile, 0.1% formic acid) over 5 minutes, followed by a 5–10% gradient of solvent B over 5 minutes, 10–35% gradient of solvent B over 35 minutes, 35–50% gradient of solvent B over 20 minutes, 50–95% gradient of solvent B over 5 minutes, and finally by a 95% solvent B hold for another 4.6 minutes. All flow rates were at 400 nl/min. Data-dependent scanning was performed by the Xcalibur v 2.1.0 software [24] using a survey mass scan at 60,000 resolution in the Orbitrap analyzer scanning m/z 400–1600, followed by collision-induced dissociation tandem mass spectrometry of the fourteen most intense ions in the linear ion trap analyzer. Precursor ions were selected by the monoisotopic precursor selection setting with selection or rejection of ions held to a +/− 10 ppm window. Dynamic exclusion was set to place any selected m/z on an exclusion list for 45 seconds after a single MS/MS.
Database searching and protein identification
LC-MS/MS spectra were searched against human proteins downloaded from Uniprot on August 06, 2013 (http://www.uniprot.org) using Thermo Proteome Discoverer 1.3 (Thermo Fisher Scientific) considering tryptic peptides with up to two missed cleavages. Iodoacetamide derivatives of cysteines, and oxidation of methionines were specified as variable modifications. At the time of the search, this reference proteome set contained 88,323 protein entries. Proteins were identified at 99% confidence with XCorr score cut-offs [25] as determined by a reversed database search. The protein and peptide identification results from the LC-MS/MS experiment were also interrogated and visualized with Scaffold v 4.0.5 (Proteome Software Inc., Portland OR), a program that relies on various search engine results and uses Bayesian statistics to reliably identify more spectra [26, 27]. Those proteins were accepted that passed the criteria of a minimum of two peptides identified at 0.1% FDR at peptide level and 1% FDR at the protein level.
All LCMSE data were processed and searched using ProteinLynx GlobalServer version 2.4 (Waters Corp., Milford, MA). Protein identifications were obtained with the embedded ion accounting algorithm of the software (IdentityE) and searching a human database downloaded from Uniprot to which the primary sequence of the T33V cytochrome c from Rhodobacter was appended. The combined protein database was randomized once before use. Trypsin was specified as the digestion enzyme allowing for 1 missed cleavage. Iodoacetamide derivatives of cysteines, and oxidation of methionines were specified as fixed and variable modifications, respectively. A minimum of 3 fragment ions matched were required per peptide, and a minimum of 1 peptide was required per protein for identification. The ion detection, clustering, and normalization were performed using ProteinLynx GlobalServer. The principles of the applied data clustering and normalization have been explained in great detail elsewhere [28–30]. IdentityE results of all three replicates were imported into Scaffold v 3.2.0 (Proteome Software Inc., Portland, OR). Peptide identifications were accepted if they could be established at greater than 90% probability as specified by the Peptide Prophet algorithm [26]. Protein identifications were accepted if they could be established at greater than 90% probability and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm [27]. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Design of targeted proteomic method
In silico trypsin digestion of human dermcidin and prolactin inducible protein (PIP) sequences derived from Uniprot database were performed and the tryptic peptides were subjected to Blastp search against the NCBInr database in order to exclude the nonspecific sequences. The SRM transitions characteristic for dermcidin and PIP specific peptide sequences were designed with Skyline software (www.brendanx-uw1.gs.washington.edu). Stable isotope labeled synthetic standard peptides were obtained from JPT Peptide Technologies, Germany and used for collision energy and declustering potential optimization.
The sweat samples were trypsin digested and, following ZipTip (Millipore, Billerica, MA) C18 sample clean-up according to the manufacturer’s instructions, 5 μg of tryptic digest was injected to the HPLC. Peptides were eluted from a Zorbax C18 precolumn (Agilent) onto a Zorbax C18 (150 mm × 75 μm, 3,5µm pore size) analytical column (Agilent) performing a 0–100% gradient of solvent B over 15 minutes, a 100% solvent B hold for 5 minutes and a rapid 0% solvent B change in 2 minutes followed by a 0% solvent B hold for 8 minutes. The flow rate was 300 nl/min. SRM data were recorded on a 4000 QTRAP (ABSciex) mass spectrometer equipped with NanoSpray II (ABSciex) operating in positive ion mode (spray voltage 2800 V, nebulizig gas 50 psi, curtain gas 20 psi, source temperature 70 °C) controlled by Analyst 1.4.2. (ABSciex) software and coupled to Easy nLC nano HPLC (Bruker). Data evaluation and the calculation of the AUC values were done using Skyline.
All the recorded data are available upon request by sending an email to the corresponding author.
Results and Discussion
The feasibility of sweat protein analysis; made possible by the abundance of sweat and the simple non-invasive means of sweat collection, makes it an excellent target for biomarker research. Chemical analysis of the sweat may demonstrate the presence of macromolecules others than those derived from the plasma, making the sweat ideal for the detection of disease-specific biomarkers [31]. Sweat analysis was used for the identification of biomarkers characteristic of schizophrenia, and seventeen proteins were identified whose abundance had changed in the sweat of schizophrenic patients as compared to controls [13]. There is also accumulating evidence that the protein profile of skin surface is altered in pathological conditions like atopic dermatitis [32]. In order to identify disease-specific changes, we ought to first understand the role and composition of the protein panel expressed in the surface of the skin under physiological conditions.
Sweat protein identification and quantification
In this work we have used a sweat pool collected during sauna bathing from healthy adult volunteers and examined it in order to gain more insight into the skin surface proteins. Using consecutive mass spectrometry scans, 95 proteins were identified by high resolution LC-MS/MS based on a minimum of 2 peptides, 20 of them being novel proteins that were not reported in sweat previously (Supplementary Table 1).
The relative levels of individual proteins identified from human sweat were determined based on a spiked internal digest of T33V Rhodobacter cytochrome c (Figure 1a). Dermcidin was the most abundant protein in the sweat, with a concentration of 1.14 pmol/μg, a finding that was reported by other experiments as well [33]. Besides dermcidin, levels of apolipoprotein D and clusterin were also shown to be considerably high. Dermicidin, clusterin, apolipoprotein D and PIP accounted for 46%, 17%, 15%, and 8% of the secreted sweat proteins, respectively, while the serum albumin accounted only for 6% (Figure 1b). To the best of our knowledge, this is the first study to present a broad overview of the sweat protein profile, providing comparative analysis of individual protein levels. Based on our quantification, the five most abundant sweat proteins; dermcidin, clusterin, apolipoprotein D, PIP and serum albumin constituted 91% of secreted sweat proteins.
Figure 1. Sweat protein amounts determined by label free quantification.

The bars show the amount of sweat proteins representing the mean values of three parallel label-free mass spectrometry analyses on QTOF instrument. Error bars indicate standard error of means. Panel A. Proteins identified in the sweat. The spiked Rhodobacter cytochrome C is also indicated. Panel B. Percentage of the eight most abundant secreted sweat proteins.
In order to validate our results obtained by label free quantification, we have designed SRM-based targeted proteomic experiments to analyze dermcidin and PIP levels in human sweat. After the determination of tryptic sequences specific for dermcidin and PIP respectively, the sequences ENAGEDPGLAR for dermcidin and YTACLCDDNPK and TVQIAAVVDVIR for PIP were used to design the SRM transitions. Stable isotope labeled reference peptides were administrated, and the relative amount of dermcidin and PIP in the sweat pool was determined (Figure 2). Both the SRM-based targeted proteomics experiment and the label free quantification support the evidence regarding dermcidin being the most characteristic sweat protein.
Figure 2. Relative amount of dermcidin and PIP determined by SRM-based targeted proteomic approach. Panel A–C.
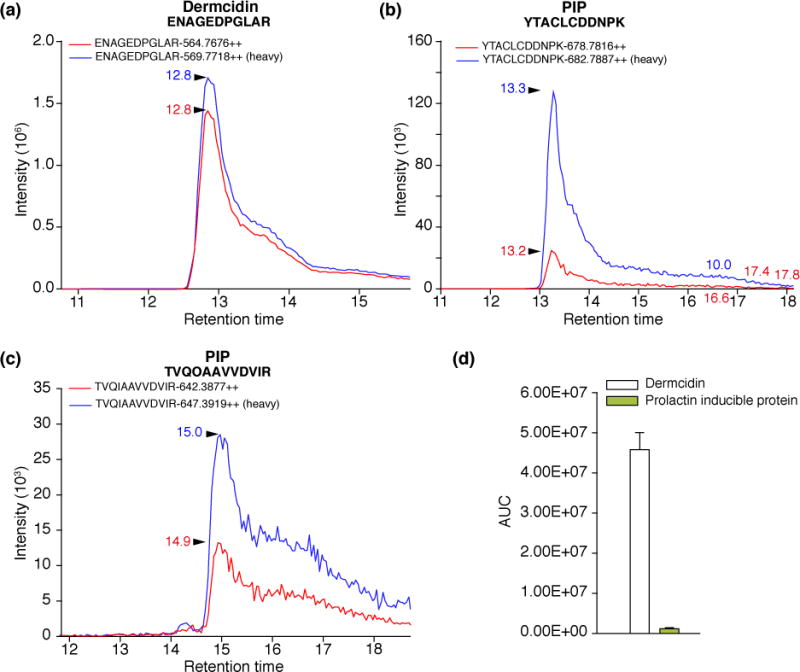
Representative SRM spectra for dermcidin and PIP specific sequences (red spectra) and their synthetic, stable isotope labeled counterparts (blue spectra). The following optimized parameters were used: ENAGEDPGLAR peptide: 564.7/628.3, 564.7/513.3, CE 27.9eV, DP 72.3eV; ENAGEDPGLAR* peptide: 569.7/638.3, 569.7/523.3, CE 27.9eV, DP 72.3eV; YTACLCDDNPK peptide: 678.7/1092.4, 678.7/748.3, CE 34.4eV, DP 80.6eV; YTACLCDDNPK* peptide: 682.7/1100.4, 682.7/756.3, CE 34.4eV, DP 80.6eV; TVQIAAVVDVIR peptide: 642.4/842.5, 642.4/771.5, CE 32.4eV, DP 77.9eV; TVQIAAVVDVIR* peptide: 647.4/852.5, 647.4/781.5 CE 32.4eV, DP 77.9eV. * represents the stable isotope containing amino acid. Panel D. Relative amount of dermcidin and PIP. The bars represent the mean AUC values per μg of total protein in case of two parallel experiments. Error bars indicate standard error of means.
Role of highly abundant proteins in the skin barrier function and non-immune mediated defense
Based on the GeneOnthology (GO) annotation, the number of identified sweat proteins involved in immune system processes and defense against bacteria and fungi is quite high (Figure 3, Supplementary Table 2). To better understand the function of highly abundant sweat proteins, the scientific literature was reviewed. The meticulous analysis of these highly abundant sweat proteins revealed their possible implication in protecting the skin from invading pathogens (Table 1), as part of a chemical barrier whose alterations may lead to an increased rate of skin infections [7, 32–39]. Dermcidin and its peptide derivatives with broad spectrum antibiotic activity were shown to be the main sweat protein/peptides secreted [33]. Their activity is not altered by low pH and high salt concentrations – conditions typically observed in sweat – making them the principal skin antimicrobial peptides [4]. In this study, using label free quantitative MS analysis, we confirmed that the most abundant component of sweat derived from healthy human volunteers is dermcidin (Figure 1). The function of PIP is not clear, it can bind different bacteria, and was identified in human sweat, saliva and tears, practically in all body fluids which are exposed to microbes [34–37]. It was detected in mice at an early embryonic stage, before the development of the immune system, and in human amniotic fluid, most probably providing some protection against bacteria before the development and maturation of the immune system [35]. Secreted form of clusterin belongs to the extracellular chaperones, and it is present in almost all physiological fluids, maintaining fluid-epithelial interface homeostasis and preventing the onset of inflammatory conditions [38, 39]. Recent data suggest that clusterin interacts with MMPs inhibiting their enzymatic activity, and it is highly possible that it is able to reduce keratinocyte damage and inflammation of the skin [38]. The presence of albumin in sweat was demonstrated by other studies as well [31], but its origin is not clear. It may originate from the blood as seen in the case of tears where the albumin content can be used as the measure of transudation [40]. It was shown previously that the DAHK peptide; derived from the N-terminal part of albumin, has a potent antioxidant activity [41]. Considering the presence of different proteases in the sweat [42], (Supplementary Table 1) and the GO function of albumin, (Supplementary Table 2) one can speculate about the possible antioxidant function of this protein in human sweat. The lipocalin family member apolipoprotein D is a multi-ligand binding protein able to transport hydrophobic molecules. It was proven to have a role in preventing lipid peroxidation, especially in the central nervous system, protecting the cells from oxidative stress [43, 44]. Its presence in the sweat may indicate its protective roles as a component of the first line of defense, preventing the skin lipids from damage caused by peroxidation.
Figure 3. GO functions of identified sweat proteins.

Number of sweat proteins having a role in different GO Biological Processes (Panel A) and GO Molecular Functions (Panel B).
Table 1.
| Protein name | Accession Number | Function |
|---|---|---|
| Dermcidin | P81605 | Defense response to bacteria and fungi* Antimicrobial peptide with broad antimicrobial spectrum [4] |
| Prolactin-inducible protein | P12273 | Binding to different species of bacteria showing highest affinity to streptococci [34,36] Binding to IgG, IgG-Fc, CD4-T cell receptor suggesting a wide range of immunological functions [35] |
| Clusterin | P10909 | Chaperone, modulator of MMP9 activity [37,38] |
| Apolipoprotein D | P05090 | Tissue regeneration* Lipid binding* Prevents lipid damage caused by free radicals [43] |
| Serum albumin | P02768 | Response to stress* Transport, binding, antioxidant role* [40] |
Taking into account our findings and the information available in the scientific literature, concerning the function of highly abundant proteins in human sweat, it seems evident that these highly abundant proteins are essential to the formation of the chemical barrier. Dermcidin and PIP most probably play a pivotal role in antibacterial defense, while clusterin functions as a chaperone and MMP inhibitor. It seems obvious that the primary role of apolipoprotein D in the sweat is to inhibit lipid peroxidation, while the presence of albumin might hint to either a scavenger function; by binding different molecules preventing their enrichment on the skin surface, or an antioxidant effect through its N-terminal tetrapeptide.
Concluding remarks
Utilizing label free quantification and SRM-based validation, we were able to identify and quantify dermcidin, clusterin, apolipoprotein D, PIP and serum albumin as highly abundant sweat proteins. Based on data from the literature, it appears that they are all implicated in the formation of the chemical barrier of the skin. Moreover, dermcidin and PIP probably play a vital role; as part of the innate immune response, in defense against potential pathogens. We hope that our findings may shed some light on the protein composition of the sweat, thereby providing insight into the constituents of the skin`s chemical barrier. This in turn will serve as valuable asset to the ever-growing, rapidly-advancing field of biomarker studies.
Supplementary Material
Acknowledgments
We are grateful for the funding of TÁMOP-4.2.2.A-11/1/KONV-2012-0045, TÁMOP-4.2.4.-A/2-11-1-2012-0001 and Training Grant Number - ES007091. Label free mass spectrometry data were acquired by the Arizona Proteomics Consortium supported by NIEHS grant ES06694 to the SWEHSC, NIH/NCI grant CA023074 to the AZCC and by the BIO5 Institute of the University of Arizona. The Thermo Fisher LTQ Orbitrap Velos mass spectrometer was provided by grant 1S10 RR028868-01 from NIH/NCRR. The help of Andrea Kulcsár and Dr. Mohamed Mahdi is greatly acknowledged.
Abbreviations
- MMP
matrix metalloproteinase
- hBD
human beta defensin
- PIP
prolactin inducible protein
Footnotes
The authors have no conflict of interest to declare.
Presented data are MIAPE-compliant.
References
- 1.Borkowski AW, Gallo RL. The coordinated response of the physical and antimicrobial peptide barriers of the skin. J Invest Dermatol. 2011;131:285–287. doi: 10.1038/jid.2010.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakatsuji T, Gallo RL. Antimicrobial peptides, old molecules with new ideas. J Invest Dermatol. 2012;132:887–895. doi: 10.1038/jid.2011.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metz-Boutigue MH, Shooshtarizadeh P, Prevost G, et al. Antimicrobial peptides present in mammalian skin and gut are multifunctional defence molecules. Curr Pharm Des. 2010;16:1024–1039. doi: 10.2174/138161210790963823. [DOI] [PubMed] [Google Scholar]
- 4.Wiesner J, Vilcinskas A. Antimicrobial peptides, the ancient arm of the human immune system. Virulence. 2010;1:440–464. doi: 10.4161/viru.1.5.12983. [DOI] [PubMed] [Google Scholar]
- 5.Harder J, Schröder JM, Gläser R. The skin surface as antimicrobial barrier, present concepts and future outlooks. Exp Dermatol. 2013;22:1–5. doi: 10.1111/exd.12046. [DOI] [PubMed] [Google Scholar]
- 6.Park JH, Park GT, Cho IH, et al. An antimicrobial protein, lactoferrin exists in the sweat, proteomic analysis of sweat. Exp Dermatol. 2011;20:369–371. doi: 10.1111/j.1600-0625.2010.01218.x. [DOI] [PubMed] [Google Scholar]
- 7.Rieg S, Steffen H, Seeber S, et al. Deficiency of dermcidin-derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J Immunol. 2005;174:8003–8010. doi: 10.4049/jimmunol.174.12.8003. [DOI] [PubMed] [Google Scholar]
- 8.Rieg S, Seeber S, Steffen H, et al. Generation of multiple stable dermcidin-derived antimicrobial peptides in sweat of different body sites. J Invest Dermatol. 2006;126:354–365. doi: 10.1038/sj.jid.5700041. [DOI] [PubMed] [Google Scholar]
- 9.Murakami M, Ohtake T, Dorschner RA, et al. Cathelicidin anti-microbial peptide expression in sweat, an innate defense system for the skin. J Invest Dermatol. 2002;119:1090–1095. doi: 10.1046/j.1523-1747.2002.19507.x. [DOI] [PubMed] [Google Scholar]
- 10.Chang WC, Huang MS, Yang CJ, et al. Dermcidin identification from exhaled air for lung cancer diagnosis. Eur Respir J. 2010;35:1182–1185. doi: 10.1183/09031936.00169509. [DOI] [PubMed] [Google Scholar]
- 11.Stewart GD, Skipworth RJ, Pennington CJ, et al. Variation in dermcidin expression in a range of primary human tumours and in hypoxic/oxidatively stressed human cell lines. Br J Cancer. 2008;99:126–132. doi: 10.1038/sj.bjc.6604458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hata TR, Gallo RL. Antimicrobial peptides, skin infections, and atopic dermatitis. Semin Cutan Med Surg. 2008;27:144–150. doi: 10.1016/j.sder.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raiszadeh MM, Ross MM, Russo PS, et al. Proteomic analysis of eccrine sweat, implications for the discovery of schizophrenia biomarker proteins. J Proteome Res. 2012;11:2127–2139. doi: 10.1021/pr2007957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burian M, Velic A, Matic K, et al. Quantitative proteomics of the human skin secretome reveal a reduction in immune defense mediators in ectodermal dysplasia patients. J Invest Dermatol. 2015;135:759–767. doi: 10.1038/jid.2014.462. [DOI] [PubMed] [Google Scholar]
- 15.Bantscheff M, Schirle M, Sweetman G, et al. Quantitative mass spectrometry in proteomics, a critical review. Anal Bioanal Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 16.Ishihama Y, Oda Y, Tabata T, et al. Exponentially modified protein abundance index emPAI, for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Silva JC, Gorenstein MV, Li G-Z, et al. Absolute Quantification of Proteins by LCMSE. Mol Cell Proteomics. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.James A, Jorgensen C. Basic design of MRM assays for peptide quantification. Methods Mol Biol. 2010;658:167–185. doi: 10.1007/978-1-60761-780-8_10. [DOI] [PubMed] [Google Scholar]
- 19.Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Chem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 22.Bateman RH, Langridge JI, McKenna T, et al. Methods and apparatus for mass spectrometry. U S Patent 2. 2006;385:918A. [Google Scholar]
- 23.Bateman RH, Carruthers R, Hoyes JB, et al. A novel precursor ion discovery method on a hybrid quadrupole orthogonal acceleration time-of-flight mass spectrometer for studying protein phosphorylation. J Am Soc Mass Spectrom. 2002;13:792–803. doi: 10.1016/S1044-0305(02)00420-8. [DOI] [PubMed] [Google Scholar]
- 24.Andon NL, Hollingworth S, Koller A, et al. Proteomic characterization of wheat amyloplasts using identification of proteins by tandem mass spetrometry. Proteomics. 2002;2:1156–1168. doi: 10.1002/1615-9861(200209)2:9<1156::AID-PROT1156>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 25.Qian WJ, Liu T, Monroe ME, et al. Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: the human proteome. J Proteome Res. 2005;4:53–62. doi: 10.1021/pr0498638. [DOI] [PubMed] [Google Scholar]
- 26.Keller A, Nesvizhskii AI, Kolker E, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 28.Silva JC, Denny R, Dorschel CA, et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77:2187–2200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 29.Hughes MA, Silva JC, Geromanos SJ, et al. Quantitative proteomic analysis of drug-induced changes in mycobacteria. J Proteome Res. 2006;5:54–63. doi: 10.1021/pr050248t. [DOI] [PubMed] [Google Scholar]
- 30.Vissers JPC, Langridge JI, Aerts JMF. Serum markers and protein signatures for Gaucher disease. Mol Cell Prot. 2007;6:755–766. doi: 10.1074/mcp.M600303-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Nakayashiki N. Sweat protein components tested by SDS-polyacrylamide gel electrophoresis followed by immunoblotting. Tohoku J Exp Med. 1990;161:25–31. doi: 10.1620/tjem.161.25. [DOI] [PubMed] [Google Scholar]
- 32.Leung DYM. New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int. 2013;62:151–161. doi: 10.2332/allergolint.13-RAI-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagawa K, Kimura A, Saito Y, et al. Production and characterization of a monoclonal antibody for sweat-specific protein and its application for sweat identification. Int J Legal Med. 2003;117:90–95. doi: 10.1007/s00414-002-0341-8. [DOI] [PubMed] [Google Scholar]
- 34.Nistor A, Bowden G, Blanchard A, et al. Influence of mouse prolactin-inducible protein in saliva on the aggregation of oral bacteria. Oral Microbiol Immunol. 2009;24:510–513. doi: 10.1111/j.1399-302X.2009.00543.x. [DOI] [PubMed] [Google Scholar]
- 35.Lee B, Bowden GH, Myal Y. Identification of mouse submaxillary gland protein in mouse saliva and its binding to mouse oral bacteria. Arch Oral Biol. 2002;47:327–332. doi: 10.1016/s0003-9969(01)00113-3. [DOI] [PubMed] [Google Scholar]
- 36.Hassan MI, Waheed A, Yadav S, et al. Prolactin inducible protein in cancer, fertility and immunoregulation: structure, function and its clinical implications. Cell Mol Life Sci. 2009;66:447–459. doi: 10.1007/s00018-008-8463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schenkels LC, Walgreen-Weterings E, Oomen LC, et al. In vivo binding of the salivary glycoprotein EP-GP identical to GCDFP-15, to oral and non-oral bacteria detection and identification of EP-GP binding species. Biol Chem. 1997;378:83–88. doi: 10.1515/bchm.1997.378.2.83. [DOI] [PubMed] [Google Scholar]
- 38.Jeong S, Ledee DR, Gordon GM, et al. Interaction of clusterin and matrix metalloproteinase-9 and its implication for epithelial homeostasis and inflammation. Am J Pathol. 2012;180:2028–2039. doi: 10.1016/j.ajpath.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizzi F, Bettuzzi S. The clusterin paradigm in prostate and breast carcinogenesis. Endocr Relat Cancer. 2010;17:R1–17. doi: 10.1677/ERC-09-0140. [DOI] [PubMed] [Google Scholar]
- 40.Tözsér J, Berta A. Lactate dehydrogenase activity in pathological human tears obtained with glass capillaries correlates with the albumin content. Int Ophthalmol. 1998;22:289–292. doi: 10.1023/a:1006378613666. [DOI] [PubMed] [Google Scholar]
- 41.Gum ET, Swanson RA, Alano C, et al. Human serum albumin and its N-terminal tetrapeptide (DAHK) block oxidant-induced neuronal death. Stroke. 2004;35:590–595. doi: 10.1161/01.STR.0000110790.05859.DA. [DOI] [PubMed] [Google Scholar]
- 42.Baechle D, Flad T, Cansier A, et al. Cathepsin D is present in human eccrine sweat and involved in the postsecretory processing of the antimicrobial peptide DCD-1L. J Biol Chem. 2006;281:5406–5415. doi: 10.1074/jbc.M504670200. [DOI] [PubMed] [Google Scholar]
- 43.Bajo-Grañeras R, Sanchez D, Gutierrez G, et al. Apolipoprotein D alters the early transcriptional response to oxidative stress in the adult cerebellum. J Neurochem. 2011;117:949–960. doi: 10.1111/j.1471-4159.2011.07266.x. [DOI] [PubMed] [Google Scholar]
- 44.Ganfornina MD, Do Carmo S, Lora JM, et al. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell. 2008;7:506–515. doi: 10.1111/j.1474-9726.2008.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.