

Abstract
Objective
Genome-wide association studies (GWAS) have so far identified 159 significant and suggestive loci for coronary artery disease (CAD). We now report comprehensive bioinformatics analyses of sequence variation in these loci to predict candidate causal genes.
Approach and Results
All annotated genes in the loci were evaluated with respect to protein coding SNPs and gene expression parameters. The latter included expression quantitative trait loci, tissue specificity, and miRNA binding. High priority candidate genes were further identified based on literature searches and our experimental data. We conclude that the great majority of causal variations affecting CAD risk occur in non-coding regions, with 41 % affecting gene expression robustly versus 6% leading to amino acid changes. Many of these genes differed from the traditionally annotated genes, which was usually based on proximity to the lead SNP. Indeed, we obtained evidence that genetic variants at CAD loci affect 98 genes which had not been linked to CAD previously.
Conclusions
Our results substantially revise the list of likely candidates for CAD and suggest that GWAS efforts in other diseases may benefit from similar bioinformatics analyses.
Keywords: coding SNP, eQTL, miRNA, systems biology
Introduction
The most recent meta-analysis of genome-wide association studies (GWAS) for coronary artery disease (CAD) identified 46 genome-wide significant and 104 genome-wide suggestive loci associated with increased risk1. Together these loci explain ~10% of the heritability. While quantitatively the effects of common risk alleles identified by GWAS, e.g. in the HMGCoR, LDLR or PCSK9 genes, are modest, the pathways tagged by these genes have utmost clinical importance as they constitute prime targets for preventive medication. Accordingly, the largest scientific relevance of GWAS discoveries is seen in elucidation of yet unknown causal mechanisms leading to CAD in the human population.
Twelve of the genome-wide significant loci are associated with blood lipid levels and five with blood pressure, suggesting that they function through these intermediate phenotypes to increase the risk for CAD1. However, the precise genetic mechanisms at the CAD loci affecting either intermediary traits or as of today unknown pathways leading to disease are largely unknown. Thus, translating GWAS loci into genes and pathways will help to provide novel insight into disease susceptibility and ultimately lead to novel treatments for CAD patients that may also be tailored to the genetic and molecular makeup of individual patients2.
In theory, the process of moving from an associated genetic variant to a disease mechanism seems straight-forward and linear: First, identify the causal (rather than the associated) variant, next identify how the causal variant alters gene function of the putative causal gene, and then work out how the altered function of an affected gene perturbs processes at the molecular, cellular, physiological and whole organism levels that ultimately promote the development of atherosclerosis. However, barring a few notable examples where relevant intermediary phenotypes (e.g. an effect on plasma cholesterol) already pointed to a pathway3, 4, experience has shown that dissecting the mechanism is very complex. Most lead single-nucleotide polymorphisms (SNPs) identified by GWAS map outside protein-coding regions. Rather, accumulation of lead SNPs was found in regulatory elements that have been identified, for example, in the Encyclopedia of DNA Elements (ENCODE) project5–7. These results suggest that common genetic variants causally involved in CAD alter gene expression rather than protein sequence.
A further challenge lies in the identification of the causal gene out of multiple candidates in the vicinity of a lead SNP. For example, gene targeting studies in a rat model of hypertension showed that 5 out of the 6 genes at the Agtrap-Plod1 hypertension locus affected blood pressure8. Recent evidence from the Genotype-Tissue Expression (GTEx) project also showed that numerous GWAS loci are associated with the expression of a gene that is not the physically most proximal gene to the locus9. Even if a single causal gene is identified, defining the function of the gene in the disease context if the gene is poorly annotated is also difficult.
A systematic attempt predicting candidate causal genes and their functional mechanisms in all 159 CAD genome-wide significant and suggestive loci has not yet been carried out. In this study, we employed a bioinformatics approach (Figure 1) in order to (i) catalog all the transcript coding genes in these loci, (ii) evaluate structural SNPs in the protein coding genes, (iii) identify expression quantitative trait loci (eQTL) that overlap CAD loci and (iv) prioritize candidate genes with respect to their likely functional relevance based on evidence from the literature and experimental results from our previous studies.
Figure 1.

Candidate SNP and gene prioritization pipeline. (A) SNPs in 159 CAD GWAS loci were interrogated for their effects on amino acid sequence, gene expression and possible effects on transcription factor binding due to their presence in regulatory regions identified in ENCODE and NIH Roadmap Epigenome projects24 (B) Genes that were functionally linked to GWAS loci were prioritized based on prior knowledge- or data-driven approaches.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Locus boundaries and genes within loci
As a first step we defined the boundaries of each of the 159 CAD loci by determining the locations of proxy SNPs (r2>0.8) on either side of the lead SNP (Supplemental Figure IA). There were 3,432 proxy SNPs based on the 1,000 Genomes EUR genotypes determined by the SNAP bioinformatics tool10. The SNPs were located in 135 non-overlapping regions. Nine lead SNPs loci had no proxy SNPs suggesting that they probably represent the causal SNP (SupplementaL Table I). The size of the remaining regions ranged from 488 bp to 566 kb with an average of 76.5 kb.
To catalogue the genes within these CAD loci, we searched the latest release of the ENSEMBL database (release 75) which contains gene model annotations from RefSeq11, GENCODE12, and ENSEMBL/HAVANA13 databases. Collectively, within the boundaries of the CAD loci, there were 183 protein coding genes along with genes for 29 antisense transcripts, 27 long intervening non-coding RNAs, 15 miscellaneous RNAs, 12 miRNAs, eight small nuclear RNAs, eight small nucleolar RNAs, three intronic sense RNAs, three processed transcripts that do not contain an open reading frame, two long non-coding transcripts that contain a coding gene in their introns on the same strand, and one ribosomal RNA (Supplemental Figure IB).
SNP prioritization pipeline overview
To identify the most plausible causal SNP for each locus, we developed a SNP prioritization pipeline (Figure 1). The pipeline consists of two main parts: identification of candidate SNPs (Figure 1A) and the identification of genes functionally related to those SNPs (Figure 1B). We considered the 159 peak GWAS SNPs and the 3,432 SNPs in high linkage disequilibrium (LD) with the peak SNPs. We assessed the functional implication of each of these SNPs based on three main criteria: (i) we checked whether the SNP cause a deleterious amino acid (AA) change; (ii) we identified all SNPs that have an eQTL effect, and (iii) we identified SNPs that lie within known regulatory regions of the genome. We further analyzed the SNPs that fulfilled at least of one these criteria as “potential causal SNPs.” The results of the annotation are shown in SupplementaL Table II.
Each potential causal SNP assembled by these criteria and the gene it affects either due to AA change or eQTL were next analyzed in the second part of the pipeline. To establish a link between the disease and the candidate genes, we assessed all relevant information from published resources as well as experimental evidence from our laboratories.
CAD loci with predicted non-synonymous/deleterious mutations
To determine SNPs that have protein altering effects, we annotated all SNPs using ANNOVAR software14. The gene annotation was based on RefGene, KnownGene and several functional prediction scores such as SIFT, PolyPhen and Mutation Taster using the annovar LJB2 database15 as well as the CADD database16.
Of the 159 lead SNPs, 33 (or 20.7%) are exonic: 11 synonymous and 22 non-synonymous (either the lead SNP itself or its proxy SNPs, see Supplemental Table III). 66.7% of all exonic SNPs lead to non-synonymous AA changes in CAD loci. This percentage was not statistically significantly enriched compared to all known non-synonymous common SNPs (>1% MAF) in exonic regions as identified in the 1,000 genomes data (48.9% of all exonic SNPs, Fisher’s p-value=0.053). Next, we focused on the deleterious SNPs since they are likely to be causal (Table 1). Five lead SNPs (rs3184504, rs3825807, rs867186, rs2571445 and rs11556924) cause amino acid (AA) changes. Three of these are predicted to be deleterious. In addition, two of these had proxy SNPs that also cause deleterious AA changes. Further, for seven other lead SNPs, we identified proxy SNPs that are also predicted to be deleterious. In total, 12 SNPs predicted to be deleterious represent 10 independent loci.
Table 1.
Predicted deleterious CAD SNPs. Of the 29 CAD related non-synonymous SNPs, 12 are predicted to have a functional effect. Highlighted SNPs indicate that the lead SNP is the potential functional SNP.
| Proxy SNP | Lead SNP | Chr | SNP Position (hg19) |
Gene | Transcript ID | Amino Acid Change |
|---|---|---|---|---|---|---|
| rs35107735 | rs12125501 | 1 | 169390957 | CCDC181 | NM_021179 | p.F238I |
| rs2820312 | rs2820315 | 1 | 201869257 | LMOD1 | NM_012134 | p.T295M |
| rs35212307 | rs2351524 | 2 | 203765756 | WDR12 | NM_018256 | p.I75V |
| rs2571445 | rs2571445 | 2 | 218683154 | TNS1 | NM_022648 | p.W1197R |
| rs1137524 | rs7642590 | 3 | 47956424 | MAP4 | NM_001134364 | p.V628L |
| rs1060407 | rs7642590 | 3 | 47958037 | MAP4 | NM_001134364 | p.S427Y |
| rs11556924 | rs11556924 | 7 | 129663496 | ZC3HC1 | NM_016478 | p.R363H |
| rs11528010 | rs7074064 | 10 | 88635779 | BMPR1A | NM_004329 | p.P2T |
| rs1169288 | rs2244608 | 12 | 121416650 | HNF1A | NM_000545 | p.I27L |
| rs4584886 | rs4299203 | 17 | 17896205 | LRRC48 | NM_001130092 | p.R191W |
| rs11906160 | rs867186 | 20 | 33565755 | MYH7B | NM_020884 | p.A25T |
| rs867186 | rs867186 | 20 | 33764554 | PROCR | NM_006404 | p.S219G |
Our results demonstrate the complexity for some of the loci. For example, two SNPs (rs1137524, rs1060407) in the chromosome 3p21.31 locus, affect the gene MAP4 complicating the identification of the causal SNP. Further, the lead SNP, rs867186, in the 20q11.22 locus causes a deleterious AA change in the PROCR gene. This SNP is also in high LD with rs11906160 (r2= 0.92) that causes a deleterious AA change in the MYH7B gene making it difficult to identify if one or both of these genes are causal for CAD in this locus.
CAD loci with regulatory effects on gene expression
To determine SNPs that have effects on gene expression, we interrogated several eQTL results that are part of the Genome-Wide Repository of Associations between SNPs and Phenotypes (GRASP) database17, Stockholm Atherosclerosis Gene Expression (STAGE)18 study, MGH liver/adipose study19, Cardiogenics consortium monocyte/macrophages study20, and aortic endothelial cells study21. These eQTL results are from more than 50 tissues and cell types, some are highly relevant to atherosclerosis, such as liver, adipose, and vessel wall as well as monocytes, macrophages, and endothelial cells. We looked for significant association between CAD loci and nearby gene expression (within 1 MB of the lead SNP). We eliminated the spurious associations by assessing if the most significantly associated expression SNP (eSNP) is among the 3,591 CAD SNPs. The significant associations and the source of the eQTL association are presented in detail in Supplementary Table 2. In total, we found significant associations between 66 CAD lead SNPs and nearby gene expression. This is in contrast to 10 CAD loci that are predicted to harbor common coding variants predicted to be deleterious, suggesting that the mechanism of majority of the CAD loci is by regulating nearby gene expression.
AA-changes and eQTLs
Our analyses lead to the prediction of complex mechanisms in some of the loci. For example, CAD SNP rs2246833 at the 10q23.31 locus has a proxy SNP (rs1051338, r2=0.89) which leads to an amino acid change in the LIPA gene (Thr16Pro). The same SNP is also associated with the expression level of LIPA gene in the Cardiogenics monocyte eQTL dataset from 758 individuals (p=3.4×10−130). The CAD risk allele (T) is associated with increased expression of the gene (β=0.46), consistent with earlier reports22. The risk allele also shows significant associations with increased LIPA expression in the MGH dataset for liver, subcutaneous and omental adipose tissue, with the most significant association in the liver (p=2×10−44, n=741 individuals), followed by omental fat (p=2×10−15, n=567 individuals) and subcutaneous fat (p=1×10−3, n=612 individuals).
Multiple eQTL genes in CAD loci
Another example of complexity was the presence of multiple eQTL genes in CAD loci. In more than half of the loci (38 out of 66) the risk SNP affected expression of multiple genes suggesting that several mechanisms, perhaps functioning in different tissues, could be influencing the disease susceptibility. For example, CAD risk SNP rs17514846 in the 5q26.1 locus is located in the intron of the FURIN gene and is associated with its expression but also with two nearby genes, FES and MAN2A2.
eQTLs re-assign gene annotation for CAD loci
For some loci, we found that some of the risk SNPs located in non-genic regions are not associated with the expression of genes that are in the immediate physical proximity. For example, CAD risk SNP rs12936587 in the chromosome 17p11.2 locus lies between genes RAI1, PEMT and RASD1 but is associated with the expression of TOM1L2 located ~200 kb away from the SNP. Also the CAD risk SNP rs9608859 (22q12.2) that has been traditionally linked to its closest genes OSM or GATSL3 is associated with the expression of another gene, SF3A1, ~65kb away.
In addition to SNPs located in non-genic regions, we also found SNPs located within a gene but associated with the expression of another nearby gene. For example, rs2681472 (12q21.33) is located in the intron of ATP2B1 but is associated with the expression of GALNT4 ~1kb away. Another example is the CAD SNP rs2895811 (14q32.2), located in the intron of HHIPL1, which is associated with the expression of the YY1 gene in CD19+ B cells that lies ~500kb away23. The region spans more than a dozen genes and miRNAs but the only identified potential disturbed mechanism of this locus is the expression level of YY1.
eSNPs located in promoters
SNPs located in promoter or enhancer regions are likely causal variants for regulating the gene expression levels. 61 CAD SNPs with eQTL effects are located in promoter histone marks based on HaploReg annotation (Supplemental Table II)24. For example, the CAD risk SNP rs590121 in the chromosome 11q13.4 locus is associated with the expression of SERPINH1 and lies in the promoter of the same gene suggesting that the risk SNP alters the binding of transcription factors (TFs) affecting SERPINH1 gene expression levels and is likely the causal SNP. On the other hand, rs2028900, in high LD (r2=0.93) with the CAD risk SNP rs1561198 (2p11.2 locus), lies in the promoter region of MAT2A and is associated with its expression but it is also associated with the expression of nearby genes VAMP8, VAMP5, and GGCX, making it difficult to predict the causal relationship between the risk SNP and nearby genes.
Tissue specific eQTL effects
In two loci, we observed tissue-specific effects of the risk SNPs. SNP rs602633, located between PSRC1 and CELSR2 genes, in the 1p13 locus has been associated with CAD and lipid levels25. This locus regulates the expression level of PSCR1, CELSR2 and SORT1 genes in the human liver. In a recent study, hepatic expression of SORT1 has been shown to regulate lipid levels and therefore this gene has been predicted to be the causal gene in this locus3. We observed that the lipid lowering T allele (of rs602633) is associated with the higher expression of CELSR2 in liver tissue but is associated with lower expression levels in adipose tissue (Figure 2A) suggesting that tissue specific eQTL effects need to be considered when dissecting the mechanisms of GWAS loci.
Figure 2.
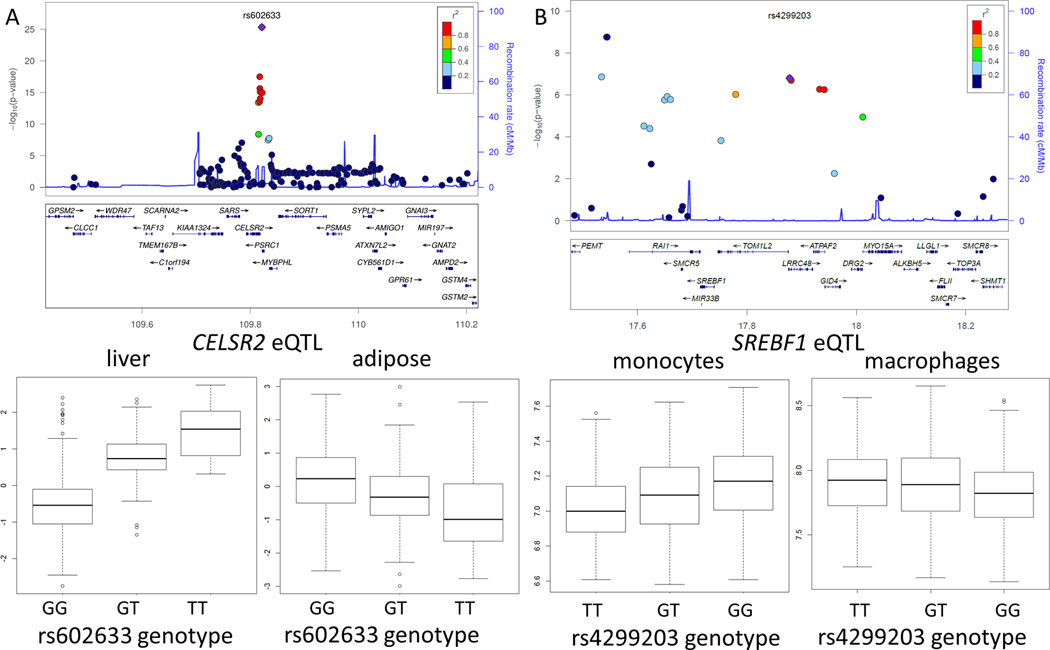
Tissue-specific eQTL effects in CAD loci. (A) Regional association plot of CAD GWAS in the 1p13 locus. Risk allele (G) of SNP rs602633 is associated with the lower expression of CELSR2 in liver tissue but is associated with higher expression levels in adipose tissue. (B)) Regional association plot of CAD GWAS in the 17p11 locus
Similarly, in the chromosome 17p11 locus, rs4299203 has a suggestive association with CAD (Figure 2B). Expression levels of five genes, DRG2, C17orf39, MYO15A, TOM1L2, SREBF1, are associated with this locus in various tissues (Supplemental Table II). The CAD risk allele (G) is associated with higher level SREBF1 expression in monocytes but is associated with lower expression in macrophages (Figure 2B). SREBPF1 encodes the sterol regulatory element binding protein (SREBP-1), one of the two major transcription factors that regulate cellular cholesterol levels. This locus is not associated with plasma lipid levels25 suggesting that the SNP effect on SREBPF1 is independent of the possible effects of the locus on lipid levels.
CAD SNPs affecting miRNA binding
A possible mechanism by which risk SNPs affect gene expression is altering the affinity of miRNA binding to the 3’ untranslated regions (UTRs) of disease genes. For example, rs12190287 in the 6q23.2 locus resides in the 3’ UTR of the TCF21 gene and affects binding of miR-22426. Therefore, we interrogated the CAD SNPs mapping to 3’ UTR region of genes using the microSNIPER database to assess whether they could reside in a predicted target miRNA binding site27. We restrained our analysis to miRNAs with predicted seed length of 7mers or more. Fifty five 3’ UTR CAD SNPs from thirty three distinct genes were predicted to lie within a miRNA binding site for a total of 254 distinct miRNAs (SupplementaL Table IV). The predicted number of miRNAs targeting the 3’ UTR region of a gene ranged from one (for BCAP29, MAP4, RND3 and WDR12) to 29 (for MRAS). Of note, 23 miRNAs were predicted to bind more than one candidate CAD gene. For example, hsa-miR-130a-5p was predicted to bind UBE2Z (with the 3’ UTR SNP rs15563) and SLC22A3 (with the 3’ UTR SNP rs3088442), and hsa-miR-4722-5p was predicted to bind APOA5 (with the 3’ UTR SNP rs2266788) and ICA1L (with the 3’ UTR SNP rs72932707). In accordance with the expected effect on APOA5 rs2266788 was significantly associated with plasma triglyceride levels25.
Of the 55 SNPs affecting miRNA binding, 13 are also associated with the expression of the same gene. At the 11p15.4 locus, rs360137 affects the binding of hsa-miR-3198 to the 3’UTR of the SWAP70 gene and is also associated with the expression of the same gene. Similarly, rs1058588 at the 2p11.2 locus affects the binding of hsa-miR-5197-3p to the 3’ UTR of VAMP8 and is also associated with the VAMP8 expression. SNP rs12733378 at the 1q32.1 locus affects the binding of five miRNAs at the 3’ UTR of CAMSAP2 and is also associated with the expression of the same gene. These examples suggest that eQTL effect may be due to altering miRNA binding to the target genes.
SNPs affecting miRNA binding and promoter regions
We also observed that SNPs that altered miRNA binding sites in UBE2Z and MAP4 were in high LD with SNPs in their promoter regions. rs6442101 is predicted to be in the promoter region of MAP4 in various cell types and tissues examined in the ENCODE and NIH RoadMap Epigenome project24. This SNP is in high LD with rs1061003 (r2=0.97) which is predicted to affect the binding of miR-378a-5p in the 3’ UTR of MAP4. Similarly, rs999474, located in the promoter region of UBE2Z is in high LD with rs15563 (r2=0.84) which is predicted to affect the binding of eight different microRNAs (Supplemental Table IV). Therefore, it is plausible that the MAP4 and UBE2Z loci affect the expression of these genes by either altering the affinity of TF binding in the promoter region or miRNA binding in the 3’ UTR region. By studying the expression patterns of TFs or miRNAs whose binding would be altered, it might be possible to predict the tissue which these genes would be functional in the context of CAD.
Candidate gene prioritization and prediction of novel CAD genes
The CAD GWAS loci have been typically annotated based on their proximity to a gene, yielding a total of 161 genes. However, recent literature evidence9, 28, 29 suggests that the nearest gene is often not the target of a given GWAS association. Instead, the identification of eQTLs can be used for predicting the target genes. In this work, annotating the 159 CAD risk SNPs led to a list of 151 CAD candidate risk genes based on non-synonymous AA changes and eQTL effects. Of note, we were not able to assign a gene to all loci. We compared our list of genes with the GWAS genes reported in the literature1 and identified 98 CAD genes hitherto not considered to be involved in the genetics of CAD for which our bioinformatics data provide suggestive evidence (Supplemental Table V and Figure 3A). Of the previously considered GWAS genes, 98 do not overlap with our annotation. These genes might be unrelated to CAD or missed by our annotation efforts. We attempted to prioritize the 98 novel genes using literature and database based approaches (prior knowledge) or using analyses performed in this manuscript and data from our laboratories (data-driven).
Figure 3.

Comparison of the new annotation of the CAD loci with previous annotations. (A) Circos plot of CAD loci for which our annotation efforts predicted candidate causal genes. Red colored genes indicate novel predictions, black colored genes show the genes consistent with the published prediction and our prediction and blue colored genes show the reported genes in the original GWAS publications1. (B) Previous annotations are typically based on physical distance of a gene to the lead SNP of association in a given locus. Using various approaches we identified novel candidate causal genes.
For the prior knowledge approach, we first used a statistical text mining approach, Gene Relationships Among Implicated Loci (GRAIL)30, that assesses the degree of relatedness among putative candidate genes within disease regions using PubMed article abstracts. Second, we utilized another integrative tool, Data-driven Expression Prioritized Integration for Complex Traits (DEPICT)31, that predicts gene functions from manually curated pathways, protein-protein interaction screens and phenotypes from mouse gene knock-out studies to prioritize the most likely causal genes at each associated loci, as well as performs pathways enrichment analysis and identifies tissues and cell types where genes from the associated loci are highly expressed. Third, we used the functional annotation information available from the public databases: (1) Mouse Phenotypes from the Mouse Genome Database (MGD)32; (2) Functional Disease Ontology (FunDO)33; (3) Biochemical Pathway information, as collected from the ConsensusPathDB database34 and the Gene Ontology (GO)annotation35, 36. If a gene was predicted to be a causal gene (e.g., p≤0.05 assigned by GRAIL and/or DEPICT) or its functional annotation (e.g., Mouse Phenotype, Biochemical Pathways, and/or Disease/GO annotation) was CAD-related (see Methods and Data for the definition of CAD-relatedness), we assigned a score of one for a total of six (SupplementaL Table VI). Of note, the prior knowledge driven approaches are biased because of the availability of literature-based information on well-studied genes. As a result, using prior-knowledge driven approach, four genes reached the maximum score of six: LPL, CDKN2B, ALDH2 and PROCR, all of them being among the 604 genes with CAD-related evidence manually extracted from scientific publications and deposited in the Coronary Artery Disease Gene Database (CADgene) V2.016.
Because of the biased prior knowledge scoring, we also used an alternative, data-driven, approach in order to look for novel candidate genes. For the data driven approach, we assigned scores to the genes if they harbored non-synonymous SNPs, had eQTLs, had promoters with CAD SNPs, were members of a CAD-relevant Bayesian Network constructed from CAD-relevant tissue gene expression studies or had a significant correlation with aortic-root lesion size in a systems genetics study of atherosclerosis in a mouse population37. Hence, the total score a gene could achieve was five based on the data driven approach. We present these prioritization results in Supplemental Table VI. For 69% of the genes there was evidence from both the prior-knowledge based or data-driven approaches. However, for 31% of the genes, only the data-driven approach provided evidence for the involvement in CAD pathogenesis. The results of the prioritization approach can be found in the Supplemental Table VI. Here, we highlight some of the new potential CAD genes that were prioritized based on our data-driven approach. REST, GIP, and TMEM116 received the top three scores.
SNP rs17087335 at the 4q12 locus is located on the NOA1 gene but has proxy SNPs that lead to non-synonymous amino acid changes in the REST gene. This CAD locus is also associated with the expression of REST in lung tissue and the CAD SNPs are located in its promoter. Further, the aortic expression of REST is significantly correlated with lesion size in mice37. REST encodes for a transcriptional repressor that has been shown to play a role in the phenotypic modulation of vascular smooth muscle cells38. REST binds to the promoter of the potassium channel KCa3.1 and represses its expression during intimal hyperplasia. In humans, there appears to be an inverse correlation between REST expression and vascular smooth muscle cell proliferation38. Consistently, in our mouse dataset37, we observed an inverse correlation between aortic expression of Rest and lesion size (r = −0.24, p = 0.03).
SNP rs15563 at the 17q21.32 locus is located on the UBE2Z gene but has a proxy SNP that lead to non-synonymous amino acid changes in the GIP gene. The locus is also associated with the expression of this gene and CALCOCO2, DLX4, SPAG9, ATP5G1, D6RB11 and UBE2Z. GIP is the highest ranked gene for this locus based on our scoring. It is an incretin hormone that belongs to the glucagon superfamily and is associated with insulin secretion39. Mouse knock out models of Gip showed reduced obesity and insulin resistance40, 41. In our mouse dataset37 we also found positive correlation between Gip expression in the aorta and lesion size (r=0.23, p=0.05).
SNP rs3809274 at the 12q24.13 locus has previously been annotated with the ATXN2 gene1. It is also in high LD with a SNP increasing the expression of the transmembrane protein 116 (TMEM116). Although little is known about this gene, in our data driven approach, it is one of the highest ranked genes implying an influence on CAD. We do not observe an eQTL effect on ATXN2 but we observe an eQTL effect on TMEM116, C12orf30, SH2B3, BRAP, ALDH2, MAPKAPK5-AS1, HECTD4, MAPKAPK5. We prioritized TMEM116 based on our data-driven approach since it harbored SNPs with non-synonymous AA change and its expression level was associated with the CAD locus in multiple tissues (Supplemental Table VI).
In addition, we also highlight MYH7B here. The gene is linked to rs867186 at the 22q11.2 by a SNP predicted to cause a deleterious AA change. The lead SNP lies in the PROCR gene and also affects the expression of this gene. It is also associated with the expression of eight other genes including the MYH7B gene in his locus; however, rs867186 is located in the promoter of MYH1B. MYH7B encodes the heavy chain of myosin II and is expressed in heart and skeletal muscle42. It is also reported to be expressed in smooth muscle cells in mice43. Therefore, we prioritized MYH7B as the causal candidate gene out of the eight genes in this locus.
Finally, to assess the information gain of our annotation effort, we compared the genes previously assigned to the loci1 and our annotations (Figure 3B and Supplemental Table V). Of the 159 CAD GWAS loci, 15 loci (9%) showed identical annotation. For 25 loci (16%), we found no overlap between our new and the traditional annotation. We could not identify any functional evidence for the genes traditionally assigned to the loci, but we could link other genes with functional evidence. For 32 loci (20%), we gained additional knowledge using our annotation. Of these, eight loci (5%) are traditionally annotated with at least one gene that cannot be validated by our annotation and hence might be wrong. For the other 24 loci (15%), we annotate additional genes that are not reported for the loci so far. For 87 loci, we did not find any functional links and hence no gene assignment.
Discussion
Recent genome-wide association efforts to understand the genetic architecture of coronary atherosclerosis and myocardial infarction have led to the identification of numerous novel DNA variants associated with disease risk. The main challenge for gaining biological insights from genetic associations is identifying which genes and pathways explain the associations. Only few studies partially identified the susceptibility mechanisms affected by CAD loci and thereby offer blueprints for subsequent efforts to explain disease etiology. These include CAD risk alleles at the 1p13, 6q23 and 4q32 loci, which displayed functional links to gene expression and related disease mechanisms involving SORT1, TCF21, and GUCY1A33, 26, 44, 45. The most robustly associated chromosomal region, the 9p21 locus, still remains a mystery after almost a decade of studies46.
More comprehensive efforts are needed to translate the GWAS loci into actionable genes and pathways. Cell-type-specific expression quantitative trait loci or coding (non-synonymous) variants in strong LD with associated variants can potentially link these variants to genes involved in atherosclerosis. Here we queried publically available databases and our own experimental datasets to predict the functional genes in the CAD genome-wide significant and suggestive loci.
We observed that majority of the GWAS loci affect gene expression as opposed to leading to amino-acid changes (41% vs 6%). This is in agreement with previous studies that predicted 70–80% of GWAS SNPs to be regulatory5, 47. Among the loci that lead to changes in gene expression, we revealed that several are associated with differential expression of multiple genes in multiple tissues. Moreover, we observed at a few loci both protein alterations and eQTL effects for significantly associated SNPs. The finding that one locus might harbor several proxy SNPs that have eQTL effects on different genes adds further to the complexity of elucidating genetic mechanisms underlying CAD. This is for example true for the 19q13.32 locus where the lead SNP is in LD with a missense SNP (in the APOC4 gene) and in LD with SNPs affecting the expression of three genes (APOC4, APOC2, APOE). Second, we identified multiple SNPs that alter promoter and enhancer sequences. ENCODE data indicate that the average number of target genes of a distal regulatory element is 2.5, suggesting that the expression of more than one gene is affected29. A potential example of this mechanism is the 10q22.3 locus. The CAD SNP lies in a potential enhancer and is associated with the expression of two genes, ANXA11 and MAT1A. Third, we observed loci that affect the expression of a transcription factor leading to changes in the expression of nearby genes. An example is the 17p11 locus which regulates the expression of the transcription factor SREBF1, as well as four other nearby genes. Hence, our annotation efforts show that the downstream effects of a locus may be highly complex, not fitting into one pathway or function (for example, the 9p21 locus), and that some loci may contain multiple causal genes. Further efforts to analyze pathways and gene networks affected by individual loci will be useful to understand if more than one gene is functional in a locus.
We relied heavily on eQTL data to identify likely causal genes at the CAD loci. Since a large fraction of the variation underlying common diseases appears to be regulatory5, 6, 47 this is a sound strategy. But we note that the sample sizes in eQTL studies vary considerably hence, there may have been insufficient power to detect the eQTLs at the CAD loci. Additionally, there may have been confounding factors contributing to the detection of an eQTL, such as population structure or experimental heterogeneity. Importantly, only few studies utilized tissues relevant to atherosclerosis, such as endothelial cells21 or the vascular wall48, to detect eQTLs as these tissue resources are difficult to obtain. Ongoing projects such as the Genotype-Tissue Expression (GTEx)9, will contribute to the identification of eQTLs in CAD-relevant tissues.
The complex expression patterns of multiple genes regulated by significantly associated SNPs at a single locus make it challenging to dissect the principle mechanism of the locus altering disease risk. A SNP affecting several genes (either the expression or the protein sequence) might increase the risk of the disease in an additive fashion. However, the disease might also only be caused by only one of the altered functions. Hence, it is not straightforward to identify the underlying disease mechanisms. In silico prediction can help to establish a link between an identified genetic effect and the disease. However, functional characterization using molecular biology and genetic approaches are required to understand the mechanisms in more detail.
Traditionally for locus annotations, the nearest gene to the identified risk SNP is reported. However, recent evidence suggests that due to the 3D chromosomal confirmation, genomic locations that appear to physically distant can interact with each other47. One such example is the FTO obesity locus that was shown to interact, at megabase distance, with the enhancer of transcription factor IRX3 and regulate its expression49. The majority of GWAS-identified variants fall in noncoding regions of the genome, the most frequent elements affected being transcriptional enhancers and silencers, which are typically located more than 1 kb from their target genes and regulate transcription through long-range interactions28. In fact, recent analysis by the ENCODE Consortium demonstrated that only ~27% of the distal regulatory elements have an interaction with the nearest promoter29, suggesting that the nearest gene is often not the target of a given GWAS association. Therefore, we used local eQTL results from various resources and protein-altering information, and identified 98 genes that had not been linked to the CAD loci previously. However, this analysis typically provides indirect evidence of an association, and the overlap of an eQTL with a disease locus may be coincidental. Our annotation is also limited due to lack of results from 5C or other chromatin capture methodology to assess long-range genomic interactions in CAD-relevant tissues. It is crucial to consider disease-relevant tissue types as some eQTLs are tissue dependent50, 51 and trait-associated variants tend to exert more tissue-specific effects52, 53. Additional functional assays would be required for confirming the mechanistic relevance of these eQTLs to the disease or trait54.
Gene assignment without functional evidence demonstrates the misleading potential of GWAS reports. It is biased by the biological relevance (and reported phenotypic effects) of the neighboring genes. For instance the locus on chromosome 19p13.2 with the lead SNP rs1122608 spanning the SMARCA4 gene is also assigned to the neighboring LDLR gene which has a well-established role in regulating plasma LDL levels. In this work, we only identify an eQTL link between the locus and the SMARCA4 but not the LDLR gene. Our results imply that the disease-causing effect underlying the locus could be the altered expression of the SMARCA4 gene rather than influencing the LDLR. Alternatively, the tissue samples evaluated here for studying eQTL effects missed the interaction of the SNP and LDLR expression which is less likely because LDLR is primarily expressed in the liver and our eQTL databases included ample liver eQTLs from multiple studies. After all, the LDLR gene is clearly a causal CAD gene, just perhaps not underlying this GWAS signal. Another example is the lead SNP at locus 12q24.12, rs3809274 (see Figure 4). It is located between the genes ATXN2 (upstream) and BRAP (downstream) and was traditionally assigned to ATXN2. However, based on our annotation effort, we do not find a link between the locus and ATXN2, but instead on six other genes (Supplemental Table II). Another lead SNP, rs3184504, downstream of ATXN2 and located within SH2B3 is, however, associated with the expression of ATXN2 gene. Of note, rs3184504 and rs3809274 are not in LD. rs3184504 was assigned to SH2B3, but we do not find a functional link between rs3184504 and SH2B3. There is, however, an eQTL association between rs3809274 and SH2B3. In other words, while ATXN2 and SH2B3 are both CAD GWAS candidate genes, the previous gene locus assignment is not supported by our data.
Figure 4.

Gene reassignment based on eQTL effects. The lead SNP rs3809274 was traditionally assigned to ATXN2. This link was not verified by our annotation effort. rs3809274 is associated with the expression of SH2B but not of ATXN2. The lead SNP rs3184504 was traditionally linked to SH2B, but the functional effect identified in this work, links the SNP to the ATXN2 gene.
We used prior knowledge and data driven approaches to prioritize the novel candidate genes. However, we note that using prior biological knowledge about the candidate genes undermines the agnostic nature of the GWAS approach54. In addition, both the number of functional annotations per gene and the number of genes per functional annotation demonstrate scale-free properties33, 55, meaning that there is a small number of genes with a large number of functional annotations, whereas for a very large number of genes there are only very few or no functional annotations available. Hence, these approaches are limited by incomplete information about gene functions. From our pipeline, most functional annotations could be retrieved when searching for Biochemical Pathways in the ConsensusPathDB database and for the Gene Ontology (GO) annotations, where 75/154 or ~49% genes could be mapped to at least one annotation term. However, this also indicates that about half (~51%) of the candidate genes lacked any functional annotation and therefore could not be considered for prioritization here. Through our prioritization pipeline, we would again select only well-annotated genes for further studies (the “rich get richer principle”), whereas the biological function of under-investigated and under-annotated genes would further remain enigmatic; therefore, we highlighted some of the novel genes with top scores from only the data driven prioritization approach.
Finally, we note that, while we were as comprehensive as possible in annotating the CAD loci, we are limited by the available datasets from previous studies. CAD loci may harbor coding variants that are not presently in databases or regulatory variants that may affect gene expression in a tissue or cell type that has not been examined. For example, the CAD loci, as defined by SNPs in high LD with the lead SNP contain a total of 291 genes, ~40% of which are non-coding. Recent large RNA sequencing studies and integrative projects such as ENCODE suggest that noncoding RNAs constitute up to 60% of transcribed RNAs. Moreover, in recent years functional studies suggest that they play an important role in the regulation of transcription and translation56, 57 We do not have microarray probes measuring the expression levels of all protein coding or non-coding genes. Further, most eQTL studies were of moderate sample size17; therefore, the power to detect significant associations is limited. Additionally, we only considered SNPs in high LD (r2≥0.8) with the peak SNP. It is possible that the GWAS lead SNP imperfectly tags the causal SNP which is moderate LD with the lead SNP, i.e. lower than 0.8. This could be the case if the causal variant has slightly different MAF compared to the lead SNP. Then by focusing on SNP with r2≥0.8 with the lead SNP may lead to improper conclusion about the functional variant and the "causal" gene. On the other hand, if we were to reduce the LD threshold, this could have led to spurious associations even though the eQTL and CAD locus are actually independent from each other but in low LD.
Ultimately, for a full understanding, each CAD locus will have to be individually investigated using tools such as experimental organisms and iPS cells. In the present study we have employed some relatively standard tools to refine the list of candidates. Additional approaches that could be useful at present include chromosome conformation analyses58, application of novel algorithms for causal SNP analysis59, network analyses, and identification of rare variants. Looking forward, new resources and tools, such as noncoding RNA annotation, RNA binding maps, splicing variants and code annotation, and detailed enhancer and transcription maps in a variety of cell types relevant to atherosclerosis, will greatly assist such efforts.
Supplementary Material
Significance.
Coronary artery disease (CAD) remains the leading cause of death in the Western world despite significant advances in early detection and extensive use of lipid-lowering and anti-hypertensive drugs. The pathogenesis of atherosclerosis involves environmental factors, hundreds of genetic variants, and their interactions, each of which exerting a relatively small effect on disease susceptibility. A more complete understanding of the disease susceptibility is urgently needed to develop additional diagnostics and therapeutics. Genome-wide association studies (GWAS) identified numerous genetic loci associated with CAD. Translating these findings into therapies will require the identification of causal genes in the associated loci. In this study, we used publically available and in-house functional information to systematically review evidence of the involvement of genes in and near the associated loci. Using this approach, we identified 98 possible novel candidate genes to be involved in the pathology of CAD.
Acknowledgements
Acknowledgements: None
Sources of Funding
This work was supported by: Transatlantic Networks of Excellence Award (12CVD02) from Foundation Leducq (all authors), US National Institutes of Health grants HL28481, HL114437, HL126753, and HL123295 (AJL). Funding is provided by the German Federal Ministry of Education and Research (BMBF) in the context of the e:Med program (e:AtheroSysMed and sysINFLAME), the FP7 European Union project and CVgenes@target (261123). Further grants were received by the local focus program “Medizinische Genetik” of the Universität zu Lübeck. This study was also supported through the Deutsche Forschungsgemeinschaft (DFG) cluster of excellence ‘Inflammation at Interfaces’. MC is supported by an NIH Pathway to Independence Award (HL121172). YZ and XY are supported by an American Heart Association Scientist Development Grant (13SDG17290032). NJS and TRW are funded by the British Heart Foundation. NJS is a UK National Institute for Health Research (NIHR) Senior Investigator. SEH is funded by the NIHR Leicester Cardiovascular Biomedical Research Unit. JB is supported by grants from Astra-Zeneca Translational Science Centre-Karolinska Institutet and by the University of Tartu (SP1GVARENG).
Disclosures
Dr. Björkegren is the founder, main shareholder, and chairman of the board for Clinical Gene Networks (CGN). Dr. Schadt is a board member of CGN.
Abbreviations
- CAD
Coronary artery disease
- SNP
Single nucleotide polymorphism
- eQTL
expression quantitative trait loci
- LD
Linkage disequilibrium
- GO
Gene ontology
- GWAS
Genome-wide association studies
- AA
Amino acid
Footnotes
Supplemental Information (SI)
Supplemental Information (SI) is available at the journal website
References
- 1.Consortium CAD, Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature genetics. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjorkegren JL, Kovacic JC, Dudley JT, Schadt EE. Genome-wide significant loci: How important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol. 2015;65:830–845. doi: 10.1016/j.jacc.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via sort1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen H, Samani NJ, Schunkert H. Mendelian randomization studies in coronary artery disease. Eur Heart J. 2014;35:1917–1924. doi: 10.1093/eurheartj/ehu208. [DOI] [PubMed] [Google Scholar]
- 5.Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated snps are more likely to be eqtls: Annotation to enhance discovery from gwas. PLoS genetics. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome research. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flister MJ, Tsaih SW, O'Meara CC, Endres B, Hoffman MJ, Geurts AM, Dwinell MR, Lazar J, Jacob HJ, Moreno C. Identifying multiple causative genes at a single GWAS locus. Genome research. 2013;23:1996–2002. doi: 10.1101/gr.160283.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Consortium GT. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. SNAP: A web-based tool for identification and annotation of proxy snps using hapmap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pruitt KD, Brown GR, Hiatt SM, et al. Refseq: An update on mammalian reference sequences. Nucleic acids research. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrow J, Frankish A, Gonzalez JM, et al. Gencode: The reference human genome annotation for the ENCODE project. Genome research. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flicek P, Amode MR, Barrell D, et al. Ensembl 2014. Nucleic acids research. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: A database of human non-synonymous snvs and their functional predictions and annotations. Human mutation. 2013;34:E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, Liu W, Liao Y, Cheng L, Liu Q, Ren X, Shi L, Tu X, Wang QK, Guo AY. Cadgene: A comprehensive database for coronary artery disease genes. Nucleic acids research. 2011;39:D991–D996. doi: 10.1093/nar/gkq1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eicher JD, Landowski C, Stackhouse B, Sloan A, Chen W, Jensen N, Lien JP, Leslie R, Johnson AD. GRASP v2.0: An update on the genome-wide repository of associations between snps and phenotypes. Nucleic acids research. 2015;43:D799–D804. doi: 10.1093/nar/gku1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foroughi Asl H, Talukdar HA, Kindt AS, et al. Expression quantitative trait loci acting across multiple tissues are enriched in inherited risk for coronary artery disease. Circulation. Cardiovascular genetics. 2015 doi: 10.1161/CIRCGENETICS.114.000640. [DOI] [PubMed] [Google Scholar]
- 19.Zhong H, Beaulaurier J, Lum PY, et al. Liver and adipose expression associated snps are enriched for association to type 2 diabetes. PLoS genetics. 2010;6:e1000932. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rotival M, Zeller T, Wild PS, et al. Integrating genome-wide genetic variations and monocyte expression data reveals trans-regulated gene modules in humans. PLoS genetics. 2011;7:e1002367. doi: 10.1371/journal.pgen.1002367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erbilgin A, Civelek M, Romanoski CE, Pan C, Hagopian R, Berliner JA, Lusis AJ. Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. Journal of lipid research. 2013;54:1894–1905. doi: 10.1194/jlr.M037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wild PS, Zeller T, Schillert A, et al. A genome-wide association study identifies LIPA as a susceptibility gene for coronary artery disease. Circulation. Cardiovascular genetics. 2011;4:403–412. doi: 10.1161/CIRCGENETICS.110.958728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, Dilthey A, Ellis P, Langford C, Vannberg FO, Knight JC. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nature genetics. 2012;44:502–510. doi: 10.1038/ng.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward LD, Kellis M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Global Lipids Genetics C. Willer CJ, Schmidt EM, et al. Discovery and refinement of loci associated with lipid levels. Nature genetics. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller CL, Haas U, Diaz R, et al. Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation. PLoS genetics. 2014;10:e1004263. doi: 10.1371/journal.pgen.1004263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barenboim M, Zoltick BJ, Guo Y, Weinberger DR. MicroSNiPer: A web tool for prediction of SNP effects on putative microRNA targets. Human mutation. 2010;31:1223–1232. doi: 10.1002/humu.21349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sexton T, Bantignies F, Cavalli G. Genomic interactions: Chromatin loops and gene meeting points in transcriptional regulation. Seminars in cell & developmental biology. 2009;20:849–855. doi: 10.1016/j.semcdb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raychaudhuri S, Plenge RM, Rossin EJ, Ng AC, International Schizophrenia C. Purcell SM, Sklar P, Scolnick EM, Xavier RJ, Altshuler D, Daly MJ. Identifying relationships among genomic disease regions: Predicting genes at pathogenic SNP associations and rare deletions. PLoS genetics. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pers TH, Karjalainen JM, Chan Y, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nature communications. 2015;6:5890. doi: 10.1038/ncomms6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, Mouse Genome Database G The mouse genome database (MGD): Comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic acids research. 2012;40:D881–D886. doi: 10.1093/nar/gkr974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osborne JD, Flatow J, Holko M, Lin SM, Kibbe WA, Zhu LJ, Danila MI, Feng G, Chisholm RL. Annotating the human genome with disease ontology. BMC genomics. 2009;10(Suppl 1):S6. doi: 10.1186/1471-2164-10-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamburov A, Stelzl U, Lehrach H, Herwig R. The Consensus PathDB interaction database: 2013 update. Nucleic acids research. 2013;41:D793–D800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nature genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gene Ontology C. Gene ontology consortium: Going forward. Nucleic acids research. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghazalpour A, Rau CD, Farber CR, et al. Hybrid mouse diversity panel: A panel of inbred mouse strains suitable for analysis of complex genetic traits. Mammalian genome : official journal of the International Mammalian Genome Society. 2012;23:680–692. doi: 10.1007/s00335-012-9411-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheong A, Bingham AJ, Li J, Kumar B, Sukumar P, Munsch C, Buckley NJ, Neylon CB, Porter KE, Beech DJ, Wood IC. Downregulated rest transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Molecular cell. 2005;20:45–52. doi: 10.1016/j.molcel.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 39.Thorens B. Glucagon-like peptide-1 and control of insulin secretion. Diabete Metab. 1995;21:311–318. [PubMed] [Google Scholar]
- 40.Miyawaki K, Yamada Y, Ban N, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. 2002;8:738–742. doi: 10.1038/nm727. [DOI] [PubMed] [Google Scholar]
- 41.Nasteska D, Harada N, Suzuki K, Yamane S, Hamasaki A, Joo E, Iwasaki K, Shibue K, Harada T, Inagaki N. Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high-fat diet conditions. Diabetes. 2014;63:2332–2343. doi: 10.2337/db13-1563. [DOI] [PubMed] [Google Scholar]
- 42.Nagase T, Kikuno R, Ishikawa K, Hirosawa M, Ohara O. Prediction of the coding sequences of unidentified human genes. Xvii. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7:143–150. doi: 10.1093/dnares/7.2.143. [DOI] [PubMed] [Google Scholar]
- 43.Warkman AS, Whitman SA, Miller MK, Garriock RJ, Schwach CM, Gregorio CC, Krieg PA. Developmental expression and cardiac transcriptional regulation of Myh7b, a third myosin heavy chain in the vertebrate heart. Cytoskeleton (Hoboken) 2012;69:324–335. doi: 10.1002/cm.21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erdmann J, Stark K, Esslinger UB, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. 2013;504:432–436. doi: 10.1038/nature12722. [DOI] [PubMed] [Google Scholar]
- 45.Miller CL, Anderson DR, Kundu RK, Raiesdana A, Nurnberg ST, Diaz R, Cheng K, Leeper NJ, Chen CH, Chang IS, Schadt EE, Hsiung CA, Assimes TL, Quertermous T. Disease-related growth factor and embryonic signaling pathways modulate an enhancer of TCF21 expression at the 6q23.2 coronary heart disease locus. PLoS genetics. 2013;9:e1003652. doi: 10.1371/journal.pgen.1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:196–206. doi: 10.1161/ATVBAHA.111.232678. [DOI] [PubMed] [Google Scholar]
- 47.Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16:197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- 48.Foroughi Asl H, Talukdar HA, Kindt AS, et al. Expression quantitative trait loci acting across multiple tissues are enriched in inherited risk for coronary artery disease. Circulation. Cardiovascular genetics. 2015;8:305–315. doi: 10.1161/CIRCGENETICS.114.000640. [DOI] [PubMed] [Google Scholar]
- 49.Smemo S, Tena JJ, Kim KH, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dimas AS, Deutsch S, Stranger BE, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nica AC, Parts L, Glass D, et al. The architecture of gene regulatory variation across multiple human tissues: The MuTHER study. PLoS genetics. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown CD, Mangravite LM, Engelhardt BE. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eqtls. PLoS genetics. 2013;9:e1003649. doi: 10.1371/journal.pgen.1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu J, Wolfs MG, Deelen P, et al. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS genetics. 2012;8:e1002431. doi: 10.1371/journal.pgen.1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edwards SL, Beesley J, French JD, Dunning AM. Beyond gwass: Illuminating the dark road from association to function. American journal of human genetics. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 56.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 57.Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15:423–437. doi: 10.1038/nrg3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dostie J, Dekker J. Mapping networks of physical interactions between genomic elements using 5C technology. Nat Protoc. 2007;2:988–1002. doi: 10.1038/nprot.2007.116. [DOI] [PubMed] [Google Scholar]
- 59.Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, Beer MA. A method to predict the impact of regulatory variants from DNA sequence. Nature genetics. 2015 doi: 10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.