

Abstract
Alkaloids, secondary metabolites that contain basic nitrogen atoms, are some of the most well-known biologically active natural products in chemistry and medicine1. Although the efficient laboratory syntheses of alkaloids would enable researchers to study and optimize their biological properties,2 the basicity and nucleophilicity of nitrogen, its susceptibility to oxidation, and its ability to alter reaction outcomes in unexpected ways – for example, through stereochemical instability and neighboring group participation – complicates their preparation in the laboratory. Efforts to address these issues have led to the invention of a large number of protecting groups that temper the reactivity of nitrogen3; however, the use of protecting groups typically introduce additional steps and obstacles into the synthetic route. Alternatively, the use of aromatic nitrogen heterocycles as synthetic precursors can attenuate the reactivity of nitrogen and streamline synthetic strategies4. In this manuscript, we use such an approach to achieve a synthesis of the complex anti-HIV alkaloid (+)-batzelladine B in nine steps (longest-linear sequence) from simple pyrrole-based starting materials. The route employs several key transformations that would be challenging or impossible to implement using saturated nitrogen heterocycles and highlights some of the advantages conferred by the use of aromatic starting materials.
The retrosynthetic conversion of a saturated nitrogen heterocycle to a heteroaromatic exchanges a reactive, basic functional group with one that is lower in energy, non-basic, non-nucleophilic, and more easily-manipulated. For example, analysis of well-appreciated physical organic scales of basicity and nucleophilicity shows that the six-membered heterocycle piperidine is much more basic (pKb = 3.1, DMSO)5 and nucleophilic (N = 18.1, H2O)6 than the corresponding aromatic heterocycle pyridine (pKb = 10.6, DMSO7; N = 11.0, H2O6). Moreover, functionalized heteroaromatics are readily elaborated by well-established C–C bond-forming reactions, such as cross-couplings. In the strategy we pursued herein, simple pyrrole-based precursors would serve as sources of partially- or fully-saturated nitrogen heterocycles and would be advanced by carbon–carbon bond-forming and reductive transformations. This approach complements terpene synthesis and biosynthesis, which typically proceeds by oxidation of a complex hydrocarbon template8.
We applied this strategy toward a synthesis of the guanidinium alkaloid (+)-batzelladine B (1, Fig. 1a)9. Structurally, 1 contains a syn-tricyclic guanidine (vessel) connected to a bicyclic guanidine (anchor) via an alkyl ester. At least 15 batzelladine alkaloids have been isolated9–12 and several members of this family inhibit the binding of HIV glycoprotein gp120 to human CD4 receptor cells (IC50 of 1 = 31 μM)9, thereby preventing viral induction. The absolute stereochemistries of the vessel and anchor of 1 were established by Overman13 and Gin14, respectively, and syntheses and synthetic studies of other batzelladines have been reported (for selected examples, see refs. 15–22). Notably, Overman has developed a tethered Biginelli condensation strategy that has provided access to several batzelladines and related alkaloids23. Gin17 and Nagasawa16 have reported enantioselective synthetic routes to (+)-batzelladine A (2), but a route to 1 has not been described.
Figure 1. Structure and synthetic analysis of (+)-batzelladine B (1).

a, The chemical structures of (+)-batzelladine B (1) and (+)-batzelladine A (2), with embedded pyrrole substructures shown. b, The strategy we pursued calls for elaboration of the pyrrole-based starting materials 3 and 6 to the vessel and anchor substructures of 1 (5 and 8, respectively) via the intermediates 4 and 7.
We envisioned that the vessel and anchor fragments of 1 (Fig. 1b) could be derived from the pyrrole-based precursors 3 and 6, respectively, if suitable methods for carbon–carbon bond formation and controlled reduction in oxidation state could be achieved. A rhodium-catalyzed formal [4+3] cycloaddition between 3 and a donor–acceptor carbene24 was envisioned to provide entry to the dehydrotropane 4, which contains all of the functional group handles required for synthesis of the vessel fragment. We envisioned that the pyrrole 6 could serve as a precursor to the anchor of 1 by a Mannich addition25, followed by cyclization and controlled adjustment of oxidation state, with concomitant isomerization.
The N-amidinylpyrrole 3 (Fig. 2a) was prepared in two steps and 75% yield from commercial reagents (see Supporting Information). Extensive experimentation was required to realize the formal [4+3] cycloaddition with high yield and stereoselectivity. Ultimately, we found that use of the (S)-pantolactonyl α-diazo ester 926 and dirhodium(II) tetrakis[N-phthaloyl-(S)-tert-leucinate] as catalyst (0.5 mol%) provided the dehydrotropane 10 in 93% yield and >95:5 diastereoselectivity. Formal cycloaddition between 3 and ent-9 using the same catalyst provided 10 with 76:24 diastereoselectivity (81% yield), demonstrating that the former substrate–catalyst pair is stereochemically-matched (an example of double asymmetric synthesis27). Formal cycloaddition between 3 and achiral diazoesters afforded the corresponding adducts in 45–93% yield and 60–86% ee (Table S1). The yield of 10 was essentially unaffected (87%) when the catalyst loading was reduced to 0.1 mol%. With this key step accomplished, the pyrroline ring was selectively reduced by treatment with chlorotris(triphenylphosphine)rhodium under dihydrogen (10→11). Exposure of the reduction product 11 to n-tetrabutylammonium fluoride and 1-[(trimethylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TMS-EBX)28 at −78 °C provided the α-alkynyl-β-ketoester 12 as a single diastereomer (1H NMR analysis). The first three steps of this sequence were readily telescoped to provide 12 in 80% overall yield after one purification. The relative stereochemistry of 12 was unequivocally established by 7-endo-dig hydroguanylation29 (12→18, Fig. 2b) followed by carbamate cleavage (18→19) and X-ray analysis.
Figure 2. Synthesis of the vessel fragment of (+)-batzelladine B (1) and determination of stereochemistry.

a, Synthesis of the vessel precursor 17. Reagents and conditions: 1. Rh2[(S)-pttl]4 (0.5 mol%), pentane, 36 °C, 93%, >95:5 dr, or Rh2[(S)-pttl]4 (0.1 mol%), pentane, 36 °C, 87%, >95:5 dr. 2. H2 (30 atm), ClRh(PPh3)3 (2.0 mol%), i-PrOH, 23 °C. 3. TBAF, TMS-EBX, THF–CH2Cl2 (8:1), −78 °C, 80% (from 3 + 9). 4. n-BuLi, then lithium benzyl octanoate, THF, then DMPU, −78 °C. 5. H2 (1 atm), Pd/C (10 mol%), THF, 23 °C, 49% (two steps). 6. LiOH, THF–H2O (2:1), 0 °C, 75%. b, The relative stereochemistry of 12 was established by cyclization and deprotection, followed by X-ray analysis. Reagents and conditions: 7. AgOAc, AcOH, CH2Cl2, 24 °C, >99%. 8. TFA, CH2Cl2, 0→23 °C, >99%.
We then investigated the ring-opening of the bicyclic skeleton of 12 by cleavage of the β-ketoester. As 12 presents four acidic sites, a careful balance between the protonation state of the substrate and the basicity of the incoming nucleophile was essential to achieving the desired mode of reactivity. After intensive experimentation and optimization, we found that deprotonation of 12 with n-butyllithium (1.0 equiv) followed by addition of lithium benzyl octanoate (1.8 equiv) afforded the bicyclic pyrrolidine 15. This cascade sequence is thought to proceed by 1,2-addition to the β-ketoester, retro-aldol ring-opening, and proton transfer to provide the enolyne 13. Isomerization of 13 to the acylallene 14 followed by Michael addition of the guanidinyl anion and neutralization of the resulting enolate may then provide 15. The addition of 1,3-dimethyl-3,4,5,6-tetrahydro-2-pyrimidinone (DMPU) was necessary to promote the retro-aldol ring-opening. Other nucleophiles, such as sodium methoxide, morpholine, ethanethiol, and Grignard or organozinc reagents, were also investigated, but in most instances complex mixtures of products were obtained. The addition–rearrangement product 15 exists as a mixture of diastereomers and tautomers; consequently we cleaved the β-ketoester of the unpurified product with palladium on carbon under dihydrogen, to form the ketone 16 (49% from 12). Saponification of the pantolactonyl ester (lithium hydroxide) afforded the keto acid 17 (75%; 29% overall from 3).
The anchor fragment was assembled by the sequence shown in Fig. 3 and begins with a highly-diastereoselective Mannich addition25 to form the β-aminoester of the target. Treatment of 6 with lithium diisopropylamide and chloro tris(isopropoxy)titanium, followed by addition of the sulfinimine 20, provided the product 21 in 99% yield. The addition product 21 was formed as a single detectable C-2 stereoisomer and an inconsequential (~94:6) mixture of C-1 stereoisomers (1H NMR analysis). The C-1 and C-2 stereocenters were assigned by analogy to related products25 and the C-2 stereochemistry was confirmed by derivatization (see Supporting Information). Notably, attempts to functionalize saturated analogs of 6 by a Mannich addition would be complicated by issues of diastereoselectivity and β-elimination. Owing to the presence of the alkyne and the difficulties associated with handling the vinylogous carbamate of the target17, reduction of the pyrrole ring was postponed until later in the sequence. The tert-butanesulfinyl substituent of 21 was cleaved by treatment with hydrochloric acid in methanol, and the resulting product was cyclized in the presence of bis(chlorodibutyltin)oxide, to provide the urea 22 (78%, two steps). O-Selective ethylation formed an iso-urea (90%, not shown) that was treated with 2,4-(dimethoxy)benzyl (DMB) amine hydrogen chloride to provide the guanidine 23 (71%). The ester was then cleaved (trimethylsilyl trifluoromethanesulfonate, 2,6-lutidine) and the resulting carboxylic acid was coupled with the alcohol 24, to provide 25, which contains the complete carbon framework of the anchor (75%). Anti-Markovnikov reductive hydration30 of the terminal alkyne of 25 mediated by the ruthenium catalyst 26 (15 mol%) formed the alcohol 27 (71%; 26% overall from 6). The addition of p-toluenesulfonic acid (PTSA) to quantitatively protonate the guanidine was essential in this step.
Figure 3. Synthesis of the (+)-batzelladine B (1) anchor.
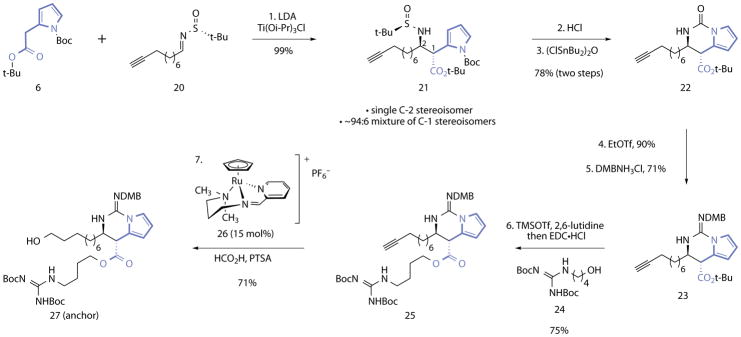
Reagents and conditions: 1. LDA, Ti(Oi-Pr)3Cl, THF, −78 °C, 99%, >20:1 mixture of C-1 stereoisomers, ~94:6 mixture of C-2 stereoisomers. 2. HCl, CH3OH–1,4-dioxane (4.4:1), 0 °C. 3. (ClSnBu2)2O, toluene, 100 °C, 78% (two steps). 4. EtOTf, 2,4,6-tri-tert-butyl-pyrimidine, CH2Cl2, 23 °C, 90%. 5. DMBNH3Cl, 3 Å MS, EtOH, 70 °C, 71%. 6. TMSOTf, 2,6-lutidine, CH2Cl2, 0→23 °C, then 24, DMAP, EDC•HCl, CH2Cl2, 0→23 °C, 75%. 7. 26 (15 mol%), PTSA (1.0 equiv), HCO2H, NMP–H2O (4:1), 23 °C, 71%.
The vessel and anchor fragments 17 and 27 were coupled using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC•HCl) to provide the penultimate intermediate 28 (77%), and the synthesis was completed by the carefully-optimized sequence shown in Fig. 4. First, a dry mixture of palladium on carbon and the coupling product 28 was suspended in trifluoroacetic acid under argon for 2 h at 24 °C. Under these conditions, the four tert-butoxycarbonyl protecting groups were cleaved, the liberated vessel domain underwent cyclodehydration, and the 1,1-disubstituted enamide was isomerized into conjugation with the ester (28→29). Upon completion of this step (as judged by UPLC/MS analysis), the atmosphere within the reaction vessel was replaced with dihydrogen. Stirring the resulting mixture for 18 h at 24 °C effected stereoselective reduction of the trisubstituted eneguanidine of the vessel (>20:1 dr, see Supporting Information)15, controlled semireduction of the anchor pyrrole with tandem isomerization of the resulting dihydropyrrole, and cleavage of the DMB substituent, to provide 1 in 40% isolated yield (45% by NMR).
Figure 4. Coupling of 17 and 27 and completion of the synthesis of (+)-batzelladine B (1).
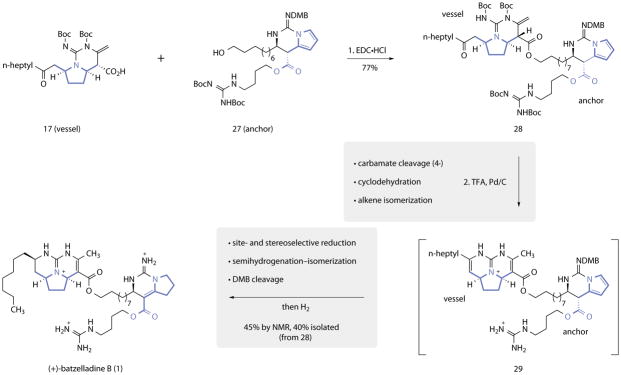
Reagents and conditions: 1. EDC•HCl, DMAP, CH2Cl2, 24 °C, 77%. 2. TFA, Pd/C, argon, 0 °C, then H2, 24 °C, 45% (NMR), 40% (isolated).
Prior approaches to batzelladine alkaloids and related natural products have employed non-aromatic (aliphatic) nitrogen precursors, followed by stepwise adjustments (typically, increases) of oxidation state. The approach we have presented proceeds in the opposite direction and begins with oxidized nitrogen heteroaromatics, followed by C–C bond-forming reactions and controlled reduction to achieve the saturation patterns of the target. This approach brings to light additional synthetic pathways not apparent or viable when starting from aliphatic nitrogen building blocks, and tempers nitrogen’s promiscuous and often problematic reactivity. An added virtue of this strategy derives from its dependence on late-stage C–H bond-forming reactions, which are among the most reliable classes of transformations.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (NRSA fellowship GM110898-01A1 to B.T.P, Chemistry Biology Interface Training Program T32GM067543 to C.E.) and Yale University is gratefully acknowledged. We thank Dr. Brandon Mercado for X-ray crystallographic analysis of 19 and Dr. Kung-pern Wang for assistance with HPLC purification of 1. The authors dedicate this manuscript to the memory of Grant R. Krow.
Footnotes
Author Contributions: B.T.P. and C.E. performed and analyzed the experiments. All authors contributed to the design of experiments and composition of the manuscript.
Crystallographic data for 19 have been deposited at The Cambridge Crystallographic Data Center as CCDC 1400311. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
The authors declare no competing financial interests.
Supplementary Information including detailed experimental procedures and characterization of all new compounds (1H and 13C NMR, IR, HRMS) are available in the online version of this manuscript.
References
- 1.Alkaloids: Biochemistry, Ecology, and Medicinal Applications. Plenum Press; 1998. [Google Scholar]
- 2.Wender PA, Miller BL. Synthesis at the molecular frontier. Nature. 2009;460:197–201. doi: 10.1038/460197a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3. Wiley; 1999. [Google Scholar]
- 4.Lipshutz BH. Five-membered heteroaromatic rings as intermediates in organic synthesis. Chem Rev. 1986;86:795–819. [Google Scholar]
- 5.Crampton RM, Robotham AI. Acidities of some substituted ammonium ions in dimethyl sulfoxide. J Chem Res, Synop. 1997:22–23. [Google Scholar]
- 6.Mayr H, Ofial Armin R. Kinetics of electrophile-nucleophile combinations: A general approach to polar organic reactivity. Pure Appl Chem. 2005;77:1807. [Google Scholar]
- 7.Bordwell FG. Equilibrium acidities in dimethyl sulfoxide solution. Acc Chem Res. 1988;21:456–463. [Google Scholar]
- 8.Chen K, Baran PS. Total synthesis of eudesmane terpenes by site-selective c-h oxidations. Nature. 2009;459:824–828. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 9.Patil AD, et al. Novel alkaloids from the sponge batzella sp.: Inhibitors of hiv gp120-human CD4 binding. J Org Chem. 1995;60:1182–1188. [Google Scholar]
- 10.Patil AD, et al. Batzelladines F–I, novel alkaloids from the sponge batzella sp.:_ Inducers of p56lck-CD4 dissociation. J Org Chem. 1997;62:1814–1819. [Google Scholar]
- 11.Hua HM, et al. Batzelladine alkaloids from the caribbean sponge monanchora unguifera and the significant activities against HIV-1 and AIDS opportunistic infectious pathogens. Tetrahedron. 2007;63:11179–11188. [Google Scholar]
- 12.Laville R, et al. Bioactive guanidine alkaloids from two caribbean marine sponges. J Nat Prod. 2009;72:1589–1594. doi: 10.1021/np900244g. [DOI] [PubMed] [Google Scholar]
- 13.Franklin AS, Ly SK, Mackin GH, Overman LE, Shaka AJ. Application of the tethered Biginelli reaction for enantioselective synthesis of batzelladine alkaloids. Absolute configuration of the tricyclic guanidine portion of batzelladine B. J Org Chem. 1999;64:1512–1519. doi: 10.1021/jo981971o. [DOI] [PubMed] [Google Scholar]
- 14.Duron SG, Gin DY. Synthesis and determination of absolute configuration of the bicyclic guanidine core of batzelladine A. Org Lett. 2001;3:1551–1554. doi: 10.1021/ol015848m. [DOI] [PubMed] [Google Scholar]
- 15.Snider BB, Chen J, Patil AD, Freyer AJ. Synthesis of the tricyclic portions of batzelladines A, B and D. Revision of the stereochemistry of batzelladines A and D. Tetrahedron Lett. 1996;37:6977–6980. [Google Scholar]
- 16.Shimokawa J, Shirai K, Tanatani A, Hashimoto Y, Nagasawa K. Enantioselective total synthesis of batzelladine A. Angew Chem, Int Ed. 2004;43:1559–1562. doi: 10.1002/anie.200353200. [DOI] [PubMed] [Google Scholar]
- 17.Arnold MA, Day KA, Durón SG, Gin DY. Total synthesis of (+)-batzelladine A and (−)-batzelladine D via [4 + 2]-annulation of vinyl carbodiimides with n-alkyl imines. J Am Chem Soc. 2006;128:13255–13260. doi: 10.1021/ja063860+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen F, Overman LE. Evolution of a strategy for the synthesis of structurally complex batzelladine alkaloids. Enantioselective total synthesis of the proposed structure of batzelladine F and structural revision. J Am Chem Soc. 2006;128:2594–2603. doi: 10.1021/ja0574320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen F, Overman LE. Enantioselective total synthesis of batzelladine F and definition of its structure. J Am Chem Soc. 2006;128:2604–2608. doi: 10.1021/ja057433s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans PA, Qin J, Robinson JE, Bazin B. Enantioselective total synthesis of the polycyclic guanidine-containing marine alkaloid (−)-batzelladine D. Angew Chem, Int Ed. 2007;46:7417–7419. doi: 10.1002/anie.200700840. [DOI] [PubMed] [Google Scholar]
- 21.Butters M, et al. Synthesis and stereochemical determination of batzelladine C methyl ester. Org Biomol Chem. 2009;7:5001–5009. doi: 10.1039/b914744f. [DOI] [PubMed] [Google Scholar]
- 22.Babij NR, Wolfe JP. Asymmetric total synthesis of (+)-merobatzelladine B. Angew Chem, Int Ed. 2012;51:4128–4130. doi: 10.1002/anie.201201001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aron ZD, Overman LE. The tethered Biginelli condensation in natural product synthesis. Chem Commun. 2004:253–265. doi: 10.1039/b309910e. [DOI] [PubMed] [Google Scholar]
- 24.Reddy RP, Davies HML. Asymmetric synthesis of tropanes by rhodium-catalyzed [4 + 3] cycloaddition. J Am Chem Soc. 2007;129:10312–10313. doi: 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]
- 25.Tang TP, Ellman JA. Asymmetric synthesis of β-amino acid derivatives incorporating a broad range of substitution patterns by enolate additions to tert-butanesulfinyl imines. J Org Chem. 2002;67:7819–7832. doi: 10.1021/jo025957u. [DOI] [PubMed] [Google Scholar]
- 26.Davies HML, Ahmed G, Churchill MR. Asymmetric synthesis of highly functionalized 8-oxabicyclo[3.2.1]octene derivatives. J Am Chem Soc. 1996;118:10774–10782. [Google Scholar]
- 27.Masamune S, Choy W, Petersen JS, Sita LR. Double asymmetric synthesis and a new strategy for stereochemical control in organic synthesis. Angew Chem, Int Ed. 1985;24:1–30. [Google Scholar]
- 28.Fernández González D, Brand JP, Waser J. Ethynyl-1,2-benziodoxol-3(1_h)-one (ebx): An exceptional reagent for the ethynylation of keto, cyano, and nitro esters. Chem –Eur J. 2010;16:9457–9461. doi: 10.1002/chem.201001539. [DOI] [PubMed] [Google Scholar]
- 29.Gainer MJ, Bennett NR, Takahashi Y, Looper RE. Regioselective rhodium(ii)-catalyzed hydroaminations of propargylguanidines. Angew Chem, Int Ed. 2011;50:684–687. doi: 10.1002/anie.201006087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng M, Li L, Herzon SB. A highly active and air-stable ruthenium complex for the ambient temperature anti-markovnikov reductive hydration of terminal alkynes. J Am Chem Soc. 2014;136:7058–7067. doi: 10.1021/ja501738a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.