

Abstract
Alterations in glutamatergic transmission onto developing GABAergic systems, in particular onto parvalbumin-positive (Pv+) fast-spiking interneurons, have been proposed as underlying causes of several neurodevelopmental disorders, including schizophrenia and autism. Excitatory glutamatergic transmission, through ionotropic and metabotropic glutamate receptors, is necessary for the correct postnatal development of the Pv+ GABAergic network. We generated mutant mice in which the metabotropic glutamate receptor 5 (mGluR5) was specifically ablated from Pv+ interneurons postnatally, and investigated the consequences of such a manipulation at the cellular, network and systems levels. Deletion of mGluR5 from Pv+ interneurons resulted in reduced numbers of Pv+ neurons and decreased inhibitory currents, as well as alterations in event-related potentials and brain oscillatory activity. These cellular and sensory changes translated into domain-specific memory deficits and increased compulsive-like behaviors, abnormal sensorimotor gating and altered responsiveness to stimulant agents. Our findings suggest a fundamental role for mGluR5 in the development of Pv+ neurons and show that alterations in this system can produce broad-spectrum alterations in brain network activity and behavior that are relevant to neurodevelopmental disorders.
INTRODUCTION
GABAergic interneurons help maintain the physiological balance between excitation and inhibition (E/I) in brain circuits. Normal E/I balance enables neural networks to respond promptly to stimuli and maintain complex patterns of ongoing activity, while preventing uncontrolled epileptic activity.1 Normal E/I balance is also critical during early postnatal brain development to shape mature cortical circuits2 by influencing the experience-dependent refinement of synaptic connections. However, understanding the contribution of GABAergic cells to cortical network development can be challenging because of the diversity of inhibitory cell types and because these neurons are outnumbered by excitatory pyramidal cells with which they form neural circuits. For example, recent data3 showed that the E/I in the visual cortex is cooperatively controlled by excitatory neurons and parvalbumin-positive (Pv+) inhibitory neurons.
Among GABAergic cell types, the subset of Pv+ interneurons provides feedback inhibition onto pyramidal neurons, required for maintaining the oscillatory activity of cortical networks in the gamma frequency range (30–80 Hz).4 Such oscillations are believed to coordinate neuronal communication between circuits and brain regions.5 Accordingly, disruption of normal gamma-band activity is associated with interneuron dysfunction,1 which often accompanies cognitive impairments observed in neuro-developmental disorders.1,5 In rodents, the functional activity of Pv+ cells gradually emerges during postnatal brain development,2,6,7 beginning with the expression of Pv, as well as with the onset of their responsiveness to GABA and glutamate by the end of the first week of life.8 Subsequently, the development of Pv+ cells continues and they functionally mature in the fourth postnatal week, when they assume their full role as fast-spiking inhibitory interneurons.2,7 In the primary visual cortex, the maturation of Pv+ cells marks the end of the sensitive period for synaptic plasticity.9 The temporal dynamics of Pv+ cell development and maturation parallels the refinement of cortical circuits, suggesting that these cells could have critical roles in shaping normal brain development.
Previous studies showed that the activity of Pv+ cells is driven by glutamatergic transmission through ionotropic N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors that develop postnatally in parallel with Pv+ cells.10,11 Deletion of NMDA receptor subunit NR1 on Pv+ cells alters cortical oscillations in the theta and gamma bands, disrupts pyramidal cell firing and induces selective cognitive deficits.12–14 Deletion of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors from hippocampal Pv+ interneurons alters fast-spiking firing and produces deficits in hippocampal-dependent tasks.15 In addition to ionotropic receptors, glutamate exerts its effects through several types of metabotropic glutamate receptors (mGluRs). Group 1 mGluRs (consisting of mGluR1 and 5) are coupled to Gq-proteins and signal through the release of calcium from intracellular stores.16 These receptors are widely expressed throughout the cortex and located on both neurons and glial cells.16 Importantly, Pv+ interneurons express functional metabotropic glutamate receptor 5 (mGluR5)17 that are necessary for their synaptic plasticity.18 Genetic deletion of mGluR5 in all cells (global knockout) confers several phenotypes that are suggestive of interneuron dysfunction.19,20 However, the importance of mGluR5 function for interneuron maturation, circuit formation and behavioral outcomes remains unknown.
To determine mGluR5 function in the modulation and development of Pv+ cells, we created a mutant mouse in which mGluR5 was ablated from Pv+ interneurons during their postnatal maturation, analyzed how such manipulation affected the overall physiology of neural networks and investigated behavioral phenotypes pertinent to neurodevelopmental disorders. We found that the activity of mGluR5 in Pv+ neurons is critical for both the normal maturation of these cells and the intact function of mature inhibitory circuits. Our data also show that the disruption of this signaling system is responsible for a variety of behavioral deficits that are reminiscent to defects commonly seen in human neurodevelopmental disorders.
METHODS
Generation of Pv-mG5−/− mice
All animal procedures were conducted in accordance with the guidelines of the American Association for the Accreditation of Laboratory Animal Care and were approved by the Salk Institute for Biological Studies and University of California San Diego Institutional Animal Care and Use Committees. mGluR5-floxed (control) animals21 were backcrossed to C57BL/6 for at least six generations, and then crossed to the Pv-Cre line to generate mGluR5-deficient animals carrying the deletion only in Pv neurons. Deletion of mGluR5 from Pv+ interneurons was confirmed by in-situ hybridization coupled to immunohistochemistry (see below). The time course of Cre-mediated recombination in Pv+ neurons was assessed by crossing the Pv-Cre line with mice carrying Cre-dependent tdTomato fluorescence (Ai14 line,22 Jackson Laboratories, Sacramento, CA, USA).
Determination of Pv+ neuron numbers
Determination of Pv immunoreactive neurons was performed on 50-μm coronal sections of male mice at 3 months of age using anti-parvalbumin antibody (Swant, Bellizona, Switzerland) and Vectastain as described.23 Coordinates for regions analyzed were as follows: Prelimbic (bregma = 1.9 to 2.4), dorsal caudate putamen (bregma = 0.5 to 1.5) and dorsal hippocampus (bregma = − 1.6 to − 2.2). Pv+ cells in the prelimbic cortex and dorsal caudate putamen were also counted in age-matched female mice. Cell counts across all slices for each region and animal were corrected using Abercrombie’s algorithm,24 and expressed as mean ± s.e.m.
In-situ hybridization
In-situ hybridization on 20-μm cryostat sections was carried out as we previously described.25 For detailed methods, please see Supplementary Materials.
Fluorescence immunodetection and confocal imaging
Determination of Pv and GAD67 immunoreactivity in coronal slices was performed as previously described26 using a Zeiss LSM780 confocal microscope (Carl Zeiss, Ontario, CA, USA) and a × 63 oil immersion objective on coronal slices as described above. Images were taken across 1.2 μm at 0.2 μm Z-steps. To determine levels of parvalbumin and GAD67 in synaptic contacts, all Pv+ synaptic boutons in the image were selected and mean fluorescence levels for parvalbumin and GAD67 were determined using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Electrophysiological recordings in vitro
Slices (350–400 μm thick) that contained the hippocampus were cut from 7- to 9-week-old mice for miniature inhibitory post-synaptic current (mIPSC) recordings or 4- to 11-week-old mice for fEPSPs studies. Electrophysiological recordings were performed in the CA1 region. Long-term potentiation (LTP) was induced by high-frequency stimulation (100 Hz for 1 s repeated five times at 10 s intervals) or a theta-burst protocol (4 stimuli at 100 Hz repeated 10 times at 200 ms intervals); long-term depression was induced by low-frequency stimulation (1000 stimuli at 1 Hz). Paired-pulse facilitation was evoked with pairs of stimuli delivered at 25–300 ms intervals and synaptic fatigue was accessed by 12 stimuli at 40 Hz. mIPSCs were recorded in voltage-clamp mode (Vh = − 60 mV) and single events larger than 6 pA were detected off-line using Minianalysis software (Synaptosoft, Fort Lee, NJ, USA). All data were acquired using a Multiclamp 700B amplifier and pCLAMP 9 software (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA) for recording mIPSCs or the WinLTP program27 for fEPSP studies.
Auditory event-related activity
Event-related potentials (ERPs)
For the recording of epidural ERPs, passive field speakers mounted on the ceiling of the recording chamber produced 10 ms Gaussian white noise ‘clicks’ 25 dB above a 65-dB white-noise background every 2–3 s. 960 stimuli were presented in the 40 min protocol stimulation.
Event-related oscillatory activity
The evoked power gain from baseline, accounting for the phase-locked changes in oscillatory activity pre- and post-stimulus, was determined. The induced oscillatory activity was also calculated, referring to those oscillations not phase-locked to the stimulus, by subtracting the evoked power from each trial of the post-stimulus power before converting again into a baseline power gain for each trial. Principal component analysis and leave-one-out cross-validation were implemented on the resulting principal components.
Social preference/recognition
A three-chambered box, similar to one previously described,28 was used. After habituation (10 min), social preference was assessed by recording durations of interaction with an empty wire cup (chrome Galaxy pencil cup, Spectrum Diversified, Streetsboro, OH, USA) or one containing an unfamiliar mouse for 10 min. After an inter-trial interval (1, 5 or 10 min), social recognition was assessed by recording the duration spent interacting with the now familiar mouse and another novel mouse (10 min).
Novel object/novel place recognition
Novel object recognition was performed in the same three-chambered box, described above. After habituation, animals were allowed to explore two identical cups, either chrome with vertical bars (Galaxy pencil cup, Spectrum Diversified) or black with mesh grating (Nestable jumbo mesh pencil cup, WebOfficeMart). After a 1-, 5- or 10-min inter-trial interval, one of the identical cups was replaced with a novel cup and duration of interaction was assessed in a 10-min trial. Novel place recognition was conducted in an identical manner, with the exception that one of the identical cups was moved to a novel location.
Light/dark box
Anxiety levels were assessed in a two-chambered box. One compartment was brightly illuminated and the other was dark. Time spent in each chamber was determined in a 5-min session.
Barnes maze
A circular maze (75 cm diameter) with 20 holes located around the perimeter was used. One hole contained an escape tunnel. After training (9 days), the escape tunnel was removed and time spent in the target quadrant was determined in the probe trial.
Marble burying test
Marble burying test was conducted in a clean home cage as previously described.29 Twenty marbles were arranged on sawdust bedding and the amount buried after a 30-min session was recorded.
Prepulse inhibition (PPI) of acoustic startle response
The session began with 5 min habituation to background white noise (70 dB white noise). The PPI session began with the presentation of six pulse-alone trials (120 dB, 40 ms), followed by a series of pulse-alone and prepulse trials (74, 78, 82 dB; 20 ms, 100 ms inter-stimulus interval, 120 dB pulse-trial). Electromyography (EMG) was also used to assess PPI. A startle stimulus (50 ms 94 dB pulse), a prepulse stimulus (50 ms 70 dB pulse) and a combination of pulse and prepulse (100 ms inter-stimulus interval) were presented 333 times each in a pseudorandom order in a 50-min session.
Pharmacology
Open field
Activity of mice was assessed in a novel open-field arena (42 × 42 × 30 cm3). Mice were administered either phencyclidine (PCP; 0–15 mg kg−1, i.p.) or amphetamine (0–4 mg kg−1, i.p.), and immediately placed in the open field for a period of 90 min.
PPI
Assessment of the effects of PCP or amphetamine on sensorimotor gating was conducted in acoustic startle sessions identical to the one described above. Mice were administered either PCP (0–10 mg kg −1, i.p.) or amphetamine (0–4 mg kg −1, i.p.) and PPI was assessed after a predetermined pretreatment time (10 min, PCP;30 15 min, amphetamine31).
RESULTS
Postnatal ablation of mGluR5 from Pv+ neurons disrupted GABAergic cell development and inhibitory circuits
Conditional ablation of mGluR5 from Pv+ neurons (Pv-mG5−/− mice) was achieved by crossing mGluR5-LoxP21 animals with a mouse line expressing Cre-recombinase only in Pv-expressing cells (Pv-Cre line32). Conditional deletion of mGluR5 function by Cre-mediated recombination in the mGluR5-LoxP line leads to deletion of exon 7 of the mGluR5 gene.20 Decreased mGluR5 protein expression has been successfully reported previously by breeding the mGluR5-LoxP mouse line to a protamine-driven Cre-recombinase line,21 by viral injection of Cre,33 and by pyramidal neuron-specific deletion using a Nex-Cre line.34,35 In the Pv-Creline used here, recombination begins at around postnatal week 2, reaching a plateau by 6 weeks, as assessed by Beta-Gal expression.12 We have assessed specificity of mGluR5 deletion in Pv+ cells by using combined in-situ hybridization for mGluR5 and Pv immunostaining. Double labeling on 12-week-old brain sections confirmed the deletion of mGluR5 from Pv+ interneurons, showing that ~ 80% of cells in control mice were double-positive for Pvalb and Grm5, whereas only ~ 15% of cells remained double-positive in Pv-mG5−/− mice (Supplementary Figure 1a). Normal distribution and staining intensities of mGluR5 in Pv-negative cells confirmed that deletion of mGluR5 was restricted to Pv+ cells. To determine the time course of Cre-mediated recombination, we crossed the fluorescent mouse reporter line Ai14 (ref. 22) to Pv-Cre and confirmed that recombination begins to plateau at around 6 weeks of age, as previously described12 (Supplementary Figure 1b).
Deletion of mGluR5 from Pv+ neurons reduced the number of Pv+ cells in the prelimbic cortex, caudate putamen and CA3 region of the hippocampus in male mice (Figure 1a), suggesting that mGluR5 function is required for normal Pv+ cell maturation. The abundance of Pv+ cells in the prelimbic cortex was also reduced in female Pv-mG5−/− mice (P<0.001). We found a genotype × sex interaction (F(1,16) = 5.24, P <0.05) for the number of Pv+ cells in the striatum. Compared with controls, there was a significant reduction in male Pv-mG5−/− mice (P<0.001), which was less pronounced in female Pv-mG5−/− mice (P = 0.17).
Figure 1.

Postnatal deletion of metabotropic glutamate receptor 5 (mGluR5) in Pv+ interneurons produced lasting alterations in interneuron development and function. (a) Reduced numbers of Pv+ cells were observed in the prelimbic cortex (genotype: F(1,8) = 41.45, P <0.001), CA3 region of the hippocampus (genotype: F(1,8) = 63.12, P <0.001) and caudate putamen (genotype: F(1,9) = 72.12, P <0.001). No significant reduction of Pv+ cells was observed in the dentate gyrus (genotype: F(1,8) = 0.6, P = 0.4) or CA1 region of the hippocampus (genotype: F(1,8) = 2.54, P = 0.1). Control: n = 5 for the DG, CA3, CA1 and Prl; n = 6 for the Cpu. Pv-mG5 −/−: n =5 for all regions. (b) Representative images of prelimbic cortex and hippocampal slices showed reduced Pv+ basket-type synaptic contacts (red dot-like structures, arrows in image) in Pv-mG5−/− mice. Scale bar =12 μm. (c) Quantification of the remaining Pv+ synaptic contacts in Pv-mG5−/− mice showed decreased expression of parvalbumin and GAD67 compared with control mice in the CA1 (genotype: GAD67, F(1,7) = 10.97, P <0.05; Pv, F(1,7) = 20.67, P <0.01) and CA3 (genotype: GAD67, F(1,7) = 9.06, P <0.05; Pv, F(1,7) = 12.77, P <0.01) regions of the hippocampus. (d) Representative miniature inhibitory postsynaptic currents (mIPSCs) recorded in pyramidal CA1 neurons of control and Pv-mG5−/− mice in the presence of the Na+ channel blocker TTX (0.5–1 μM), the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist CNQX (25 μM) and the NMDA receptor antagonist APV (10 μM). High Cl− concentration in the internal solution made mIPSCs inward (scale bars =25 pA and 1 s). (e) Summary plots of mIPSCs show that mean frequency but not mean amplitude was decreased in Pv-mG5−/− mice (P<0.05, Student’s t-test). (e and f) Pooled cumulative distributions for the inter-event intervals and amplitudes of mIPSCs in Pv-mG5−/− mice are shown in f and g, respectively. Data were obtained from 7- to 12-week-old male mice. Bar graphs depict means ± s.e.m. APV, (2R)-amino-5-phosphonovaleric acid; CA1, Cornu Ammonis region 1; CA3, Cornu Ammonis region 3; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; Cpu, caudate putamen; DG, dentate gyrus; Prl, prelimbic region; TTX, tetrodotoxin.
Cortical Pv+ cells form synaptic contacts, which mature slowly during the first 4 weeks of postnatal life in mice.2 Normal mGluR5 function is required for synaptic plasticity in these inhibitory neurons.18 In accordance to these previous studies, we found an apparent reduction in Pv+ contacts around putative pyramidal neurons (Figure 1b, arrows) in the prelimbic cortex, as well as in CA1 and CA3 hippocampal regions. To determine whether the remaining basket-type Pv+ synaptic contacts in Pv-mG5−/− brains also had altered expression of Pv and glutamic acid decarboxylase isoform 67 (GAD67), we quantified the content of Pv and GAD67 by fluorescence immunohistochemistry followed by confocal microscopy. There was a decrease in Pv and GAD67 expression in the remaining contacts surrounding putative pyramidal neurons in the hippocampal region (Figure 1c) but not in the prelimbic region (GAD67, F(1,8) = 1.3, not significant (NS); Pv, F(1,8) = 3.39, NS; n = 5). These results suggest that even those synaptic contacts that remained Pv+ were still altered with respect to controls in some brain regions. To test the functional disruption of inhibitory neurotransmission further, we recorded mIPSCs in CA1 pyramidal neurons. We found a significant reduction in the frequency, but not in the amplitude, of mIPSCs (Figures 1d and e). This effect was paralleled by a shift to the right of the cumulative probability plots for the inter-event intervals of mIPSCs in Pv-mG5−/− animals (Figure 1f), whereas the cumulative probability curves for amplitude largely overlapped (Figure 1g). These results indicate a reduction in the number of inhibitory presynaptic inputs, consistent with the observed decrease in synaptic contacts (Figure 1b).
In vivo neural network activity was compromised in Pv-mG5−/− mice
Pv+ interneuron-mediated inhibition of principal cells is critical for rhythmic cortical-oscillatory activity and has been linked to information transmission between cortical areas.4,5 We hypothesized that the disruption of inhibitory circuitry observed in Pv-mG5−/− animals may lead to deficits in auditory-evoked oscillatory activity. To test this hypothesis, we characterized network activity in vivo by recording auditory ERPs through electrocorticography. The ERP waveform of Pv-mG5−/− mice displayed specific alterations in the grand-averages of its individual components, including increased amplitude at 20 and 200 ms post-stimulus, and decreased amplitude at 40 ms (Figures 2a and b). Males of both genotypes displayed a shorter latency for the 20–60 ms component for the frontal channel (sex: F(1,70) = 9.32, P<0.01). Female Pv-mG5−/− mice exhibited a longer latency in the 20–60 ms window for the parietal component compared with female controls and males of both genotypes. Further, mutants in both sexes displayed a shorter latency for the 200 ms window in the frontal channel (Supplementary Figure 2a). No significant differences in latency were observed for the 200 ms component in the parietal cortex.
Figure 2.

Pv-mG5−/− mice have altered auditory event-related potentials (ERPs) and evoked and induced power. (a and b) Auditory ERPs in Pv-mG5−/− mice had increased amplitude in the 20 ms (genotype: Frontal, F(1,70) = 12.70, P <0.001; Parietal, F(1,68) = 10.53, P <0.01) and 200 ms (genotype: Frontal, F(1,70) = 18.39), P <0.001; Parietal, F(1,68) = 6.57, P <0.05) minima and decreased amplitude in the 40 ms maxima (genotype: Frontal, F(1,70) = 11.77, P <0.01; Parietal, F(1,68) = 12.12, P <0.001). (c–f) Auditory EEG power, measured as a gain relative to the pre-stimulus baseline and analyzed in a 500-ms post-stimulus window, was significantly altered in several key frequencies in Pv-mG5−/− mice. (c) Evoked power gain significantly increased in the low-frequency bands (2–10 Hz) and the gamma band (40–54 Hz). (d) Induced power gain was increased in both channels in the gamma range, whereas it was diminished in the low-frequency bands (2–6 Hz) in the parietal channel and increased in the beta bands (12–16 Hz) in the frontal channel. (e) Evoked and induced power in the theta, beta and gamma bands segregate individual animals by genotype. (f) Receiver operating characteristic (ROC) analysis showed discriminability of control vs Pv-mG5−/− animals using the best frontal or parietal frequency band or using an optimized combination of frequencies (best principal component (PC)). Classification rates indicated performance of a linear discriminant under leave-one-out cross-validation. Asterisk denotes difference between control and Pv-mG5−/− mice (*P<0.05, **P<0.01, ***P<0.001). Significance between genotypes in a–d was determined by the Wilcoxon rank-sum test. Bar graphs depict means ± s.e.m. The data in the traces depict mean ± standard deviation. Control: n =38 (frontal channel) and n =37 (parietal channel); Pv-mG5−/− : n =36 (frontal channel) and n =35 (parietal channel).
Power spectral analysis revealed no differences in baseline activity (Supplementary Figures 2b and c), whereas the stimulus-evoked (that is, phase-locked) power gain was increased in the 2–10 Hz and 40–54 Hz bands for both regions (Figure 2c). We also observed increases in stimulus-induced power gain in the gamma band in both brain regions, and increases within the delta–theta band and beta band for the parietal and frontal regions (Figure 2d). The separation between genotypes in induced power gain can be observed both qualitatively (Figure 2e) and quantitatively via receiver operating characteristic curves (Figure 2f). The frequency bands that best separate the genotypes were the 50–52 Hz band in the parietal region (63% correct classification using leave-one-out cross-validation; see Supplementary Methods) and 12–14 Hz band in the frontal region (65% correct classification; Figure 2f). We used principal component analysis to integrate information from all frequency bands and channels and found that seven principal components could achieve 79% correct classification performance in our cross-validated tests. These findings demonstrate that disruption of Pv-mediated inhibitory neurotransmission via conditional mGluR5 ablation altered ERPs and increased the power in oscillatory activity.
Postnatal mGluR5 ablation induced domain-selective memory deficits
Alterations in oscillatory activity, as well as in the amplitude and latency of key components of ERPs, are a common feature of neurodevelopmental disorders with associated cognitive and social deficits, including schizophrenia and autism. We thus aimed to identify potential behavioral phenotypes in Pv-mG5−/− mice, particularly those behavioral abnormalities that are pertinent to cognitive and social functions. Pv-mG5−/− mice showed a marked reduction in the preference for social novelty (Figure 3a and Supplementary Figure 3a), similar to what is observed in models of schizophrenia36 and autism.37 To determine whether this deficit was restricted to social cognition, we tested mice in the novel object recognition test. We found that Pv-mG5−/− mice showed reductions in the novel object exploration (Figure 3b and Supplementary Figure 3b), suggesting that postnatal Pv-mGluR5 ablation induced generalized impairment in recognition memory.
Figure 3.

Postnatal metabotropic glutamate receptor 5 (mGluR5) ablation impaired recognition memory and increased repetitive behaviors. (a) Performance in the social recognition task was impaired in Pv-mG5−/− mice, with the ability to discriminate between a novel and familiar mouse abolished (genotype: F(1,119) = 45.61, P <0.001). Social discrimination was also sensitive to an increase in inter-trial interval (ITI), independent of genotype (ITI: F(2,119) = 5.43, P <0.001), with superior performance after a 1 min ITI (1 vs 5 min, P <0.01; 1 vs 10 min, P <0.05; data not shown). (b) Object recognition was impaired in Pv-mG5−/− mice (genotype: F(1,120) = 44.98, P <0.001), with no effect on any other variables. (c) The latency to find the escape tunnel was increased (day × genotype: F(8,384) = 6.02, P <0.001) during days 1 and 2 (P <0.001) (d), coupled with an increase in the number of errors made (day × genotype: F(8,384) = 4.41, P <0.001) on days 1 and 2 (P <0.001). (e) These effects are attributable to the increased perseverative responding (day × genotype: F(8,384) = 4.16, P <0.001) that was also evident on days 1 and 2 (P<0.001). (f) Sociability was preserved in mice, and both groups spent more time interacting with the novel mouse compared with the empty cup (side: F(1,127) = 206.63, P <0.001). The analysis of variance (ANOVA) also indicated that Pv-mG5−/− mice spent more time interacting with cups, independent of side (genotype: F(1,127) = 6.25, P <0.05). (g, left) There was no difference in the frequency of interactions initiated, (g, right) but the duration of each bout was increased in Pv-mG5−/− mice (genotype: F(1,127) = 9.03, P <0.01). (h) The duration of grooming was increased in Pv-mG5−/− mice during the assessment of sociability (genotype: F(1,127) = 14.56, P <0.001). (i) The increased number of marbles buried further indicates that repetitive behavior was increased in Pv-mG5−/− mice (genotype: F(1,31) = 8.99, P <0.01). Numbers over bars represent n numbers. Performance was analyzed with repeated-measures ANOVA followed, when appropriate, by Fisher’s LSD post hoc test. Light bars reflect control mice, and dark bars reflect Pv-mG5−/− mice. Asterisk denotes difference between control and Pv-mG5−/− mice (*P<0.05, **P<0.01). Plus denotes effect side (+++P<0.001). Bar graphs depict means ± s.e.m.
To further investigate whether memory deficits were restricted to recognition memory, or if they extended to other memory domains, we tested our mice in a hippocampus-dependent spatial memory task, the Barnes maze. Mice of both genotypes showed intact memory of a previously located escape tunnel during the probe trial (Supplementary Figure 4a) and after a 2-week retention period (latency: F(1,98) = 0.07, NS; errors: F(1,98) = 0.19, NS), suggesting no spatial memory deficits in Pv-mG5−/− mice. Further, no performance deficits were observed across genotypes in the novel place recognition procedure (Supplementary Figures 4b and 5), supporting the conclusion that spatial memory was unaffected in Pv-mG5−/− mice. These observations were consistent with normal hippocampal synaptic plasticity, as suggested by field-potential studies in acute slices. In particular, high-frequency stimulation (100 Hz), known to evoke postsynaptic depolarization and induce LTP, resulted in a similar potentiation of synaptic transmission in control and Pv-mG5−/− animals (Supplementary Figure 4c). Similarly, theta-burst stimulation-induced LTP was also unchanged in Pv-mG5−/− mice (Supplementary Figure 4d). Furthermore, the magnitude of long-term depression, produced by prolonged low-frequency stimulation, was not significantly different between Pv-mG5−/− and control animals (Supplementary Figure 4e). Paired-pulse facilitation, a measure of short-term modifications of the efficacy of hippocampal synapses when presynaptic neurons are repetitively stimulated, showed no significant differences in Pv-mG5−/− animals compared with controls for each inter-stimuli interval tested (Supplementary Figure 6a). Furthermore, synaptic fatigue triggered by a burst of 12 stimuli separated by 25 ms intervals showed similar profiles in animals of both genotypes (Supplementary Figure 6b). These findings demonstrate that hippocampal-dependent spatial memory and its in vitro correlates are unaffected in Pv-mG5−/− mice, supporting the notion that the observed memory defects may be restricted to recognition memory, rather than a result of a generalized memory failure.
Repetitive behaviors were increased in Pv-mG5−/− mice
Mouse models of autism commonly assess alterations in sociability, social recognition and repetitive behaviors.37 In addition to deficits in social recognition, Pv-mG5−/− mice displayed elevations in repetitive behaviors in a number of procedures. During the acquisition phase of the Barnes maze, male Pv-mG5−/− mice showed a transient increase of the escape latency (Figure 3c). This initial performance deficit was unlikely due to abnormal anxiety levels, because there was no apparent performance difference in the light/dark box test between genotypes (time in light compartment: F(1,92) = 1.11, P = 0.29). It was also unlikely that learning impairments, as seen in the global mGluR5 knockout mice during Morris water maze acquisition,20 accounted for the increased escape latency of Pv-mG5−/− mice as it would be expected that deficits would emerge as controls learned the escape location. Rather, the increased escape latency was accompanied by an increase in errors made (Figure 3d) that was likely mediated by an increase in perseverative errors (Figure 3e). We found this effect to be sex specific. Female Pv-mG5−/− mice exhibited an increased latency but only a modest increase in perseverative errors (Supplementary Figures 7a–c). Perseverative responding was also observed in the social preference test. Although both control and mutant mice displayed social preference, Pv-mG5−/− mice displayed increased interaction levels (Figure 3f). The frequency of individual interactions was unchanged but the average duration of each bout was increased (Figure 3g). In addition, Pv-mG5−/− mice spent significantly more time grooming during social preference assessment (Figure 3h), and during social, object and place recognition trials (Supplementary Figures 8a–c). Collectively, these results show that Pv-mG5−/− mice engage in longer bouts of interactive behavior and exhibit increased repetitive behaviors. To test this possibility in more detail, we assessed marble burying behavior. Consistent with the results reported above, Pv-mG5−/− mice buried more marbles than control animals (Figure 3i). In summary, this collection of behavioral abnormalities across numerous behavioral tests clearly demonstrates increased repetitive behaviors in Pv-mG5−/− mice.
Sensorimotor gating was disrupted after postnatal mGluR5 ablation
Neurodevelopmental disorders are often accompanied by alterations in sensorimotor gating, as assessed by prepulse inhibition (PPI) of the startle response.38–40 We found that PPI was increased in Pv-mG5−/− mice (Figure 4a) independent of alterations in startle magnitude (Figure 4b). This increased PPI is in contrast to the PPI deficits observed in mGluR5 global knockout mice.19 To investigate these contradicting results in more detail, we complemented the traditional methodology of PPI assessment by combining it with EMG recordings. EMG-PPI revealed a modest inhibition of the startle response in control mice that was significantly greater in Pv-mG5−/− mice (Figure 4c). Further, the startle response did not reveal differences between genotypes (Figure 4d). A heat map of the EMG response showed PPI is a complex phenomenon. After the initial response (10–20 ms post-stimulus), there is a resurgence of activity 50–80 ms post-stimulus (Supplementary Figure 9). Collectively, these experiments demonstrate that PPI is increased in Pv-mG5−/− mice. This alteration is robust and evident by using independent methodologies that are commonly used to assess sensorimotor gating.
Figure 4.
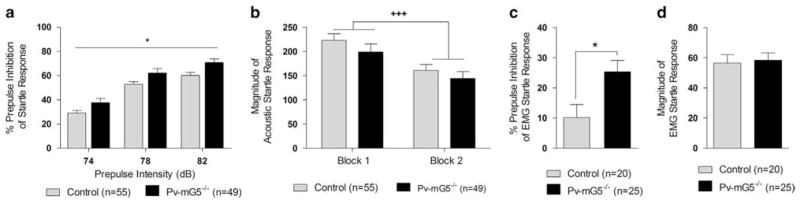
Prepulse inhibition (PPI) was increased in Pv-mG5−/− mice. (a) PPI increased as prepulse intensity was increased (prepulse: F(2,200) = 339.37, P <0.001). PPI was increased in Pv-mG5−/− mice (genotype: F(1,100) = 6.78, P <0.05). (b) Startle magnitude was not different between control and Pv-mG5−/− mice, but habituation to the startle response was evident (block: F(1,100) = 60.43, P <0.001). (c) Increased PPI was also evident in Pv-mG5−/− mice when the electromyography (EMG) response was assessed (genotype: F(1,41) = 5.95, P <0.05; d) and occurred without alterations in the startle response (genotype: F(1,41) = 0.01, not significant (NS)). Performance was analyzed by either repeated-measures or one-way analysis of variance followed, when appropriate, by Fisher’s LSD post hoc test. Light bars reflect control mice, and dark bars reflect Pv-mG5−/− mice. Asterisk denotes difference between control and Pv-mG5−/− mice (*P<0.05, **P<0.01, ***P<0.001). Bar graphs depict means ± s.e.m.
Pv-mG5−/− mice displayed a diminished response to PCP but an increased response to amphetamine
Psychotic disorders, such as schizophrenia, are associated with excessive dopamine transmission.41 Drugs that either indirectly (PCP) or directly (amphetamine) stimulate the dopaminergic system have been previously used to investigate aspects of schizophrenia.42 In the open-field test, PCP increased the total distance traveled in both groups of mice. However, at low doses (1 and 2.5 mg kg−1) PCP elicited locomotor hyperactivity in control mice only, without affecting activity levels in Pv-mG5 −/− mice (Figure 5a). Pv-mG5−/− mice also displayed a blunted sensitivity to PCP-induced PPI deficits. A dose-dependent decrease in PPI was observed in both groups when mice were presented with the lower prepulse (pp) intensities (pp4 and pp8; Supplementary Figure 10). However, although control mice were susceptible to PCP-induced decrease in PPI at the highest prepulse intensity (pp12), this effect was not apparent in Pv-mG5−/− mice (Figure 5b). These findings suggest that a fully developed Pv-inhibitory circuit, or that mGluR5–NMDAR interaction on Pv+ cells, may have an important role in mediating behavioral disruptions after NMDA receptor antagonism.
Figure 5.

Pv-mG5−/− mice displayed altered responsiveness to phencyclidine (PCP) and amphetamine. (a) Pv-mG5−/− mice exhibited a diminished response to PCP-induced hyperlocomotion compared with controls (genotype × dose: F(5,191) = 2.41, P <0.05). PCP (1–10 mg kg−1) increased the distance traveled in control mice (P<0.05), but low-dose PCP (1 and 2.5 mg kg −1) had no effect in Pv-mG5 −/− mice. Locomotor activity was increased in Pv-mG5−/− mice as PCP dose increased (5–15 mg kg−1; P<0.05). (b) PCP-induced PPI impairments were absent in Pv-mG5−/− mice when presented with pp12 dB (prepulse × dose × genotype: F(8,372) = 2.62, P<0.01). PCP (5 and 10 mg/kg) impaired PPI (P<0.05) in control mice but not Pv-mG5−/− mice. PPI was lower in controls than in Pv-mG5−/− mice when PCP (2.5–10 mg/kg) was administered (P< 0.001). Males displayed higher PPI than females (sex: F(1,186) = 6.65, P <0.05; data not shown). (c) Pv-mG5−/− mice were more sensitive to amphetamine in the open-field test (genotype × dose: F(3,124) = 4.57, P <0.01) when administered 2 mg kg−1 amphetamine (P<0.001). (d) Amphetamine impaired PPI in both genotypes (dose: F(3,128) = 6.99, P <0.001) compared with saline-treated animals when 2 and 4 mg kg −1 were administered (P<0.05). PPI was increased in Pv-mG5−/− mice in all treatment groups (genotype: F(1,128) = 9.01, P <0.01). Data were analyzed with repeated-measures analysis of variance followed, when appropriate, by Fisher’s LSD post hoc test. Light bars reflect control mice, and dark bars reflect Pv-mG5−/− mice. Asterisk denotes difference between control and Pv-mG5−/− mice (***P<0.001). Plus denotes difference between treatment group and respective saline-treated animals (+P<0.05, ++P<0.01, +++P<0.001). Bar graphs depict means ± s.e.m.
Increased sensitivity to amphetamine-induced dopamine release is evident in schizophrenia patients.41 Amphetamine-induced hyperlocomotion is used to assess this feature in rodents. Amphetamine increased locomotor activity in all groups of mice. However, the response was greater in Pv-mG5−/− mice after amphetamine treatment (2 mg kg−1) compared with amphetamine-treated control animals (Figure 5c and Supplementary Figures 11a–d). We next sought to identify whether Pv-mG5 −/− mice exhibited increased sensitivity to amphetamine-induced PPI disruptions. As previously observed (Figures 4a and 5b), PPI was increased in mutant mice compared with controls (Figure 5d). In contrast to the open-field test, control and Pv-mG5−/− displayed equal sensitivity to amphetamine-induced sensorimotor gating disruptions, as 2 and 4 mg kg−1 impaired PPI in both groups (Figure 5d).
DISCUSSION
Conditional ablation of mGluR5 restricted to Pv neurons induced a reduction in the overall number of Pv+ cells and a reduction of synaptic terminals of the remaining Pv neurons. Consequently, these defects were accompanied by impaired inhibitory function. In addition, in vivo electrocorticography recordings revealed that Pv-mG5 −/− animals displayed altered auditory ERPs, with reductions of the 40 ms component, as observed in many animal models of mental disorders. The alterations in auditory ERPs were accompanied by increased evoked and induced theta and gamma oscillatory activity. In addition to altered neuronal network functions in the cortex of Pv-mG5−/− mice, these animals exhibited robust cognitive and behavioral impairments that are commonly observed in many neurodevelopmental disorders, including decreased recognition memory, increased compulsive-like repetitive behaviors, increased PPI and altered sensitivity to agents that perturb the dopamine system. Abnormal GABAergic function underlies many neurodevelopmental disorders1 and these alterations may derive from abnormal glutamate transmission during the functional development of the GABAergic network. Our findings indicate that postnatal mGluR5 activation on Pv+ interneurons is necessary for the correct E/I balance in different brain areas. Indeed, we observed GABAergic alterations in the PFC, hippocampus and striatum that resemble the altered E/I balance evident in several disorders, including schizophrenia,43,44 autism45 and Tourette syndrome.46
Multiple regions throughout the cortex and striatum express the genes coding for both Pv and mGluR5.47 It would be expected that Pv neurons in any of these regions will be altered by conditional mGluR5 ablation, thus contributing to the physiological alterations observed in our Pv-mG5−/− mice. Regions expressing high levels of Pv, such as the reticular nucleus of the thalamus and the cerebellum, show almost undetectable levels of mGluR547 and are not expected to contribute to the phenotypes exhibited by our Pv-mG5−/− mice.
Disruption of Pv-mGluR5 signaling is consistent with previous findings that suggest disrupted NMDAR activity may underlie inhibitory deficits in such disorders. For example, ablation of NMDAR from corticolimbic GABAergic interneurons decreased GAD67 and Pv expression13 led to profound alterations in inhibitory function.12–14 Similarly, disruption of NMDAR-mediated transmission during early neurodevelopment by, for example, administration of PCP or ketamine, also produces alterations in E/I balance when animals reach adulthood.42,48 Our results obtained after conditional ablation of mGluR5 in Pv+ neurons, in addition to studies targeting the NMDAR on GABAergic neurons, indicate that postnatal excitatory transmission, mediated by both ionotropic-and metabotropic glutamate receptors, is necessary for normal Pv+ neuron development and function. Therefore, disrupted mGluR5 signaling, in addition to ionotropic glutamate receptors, may contribute to the alterations in E/I balance observed in several neurodevelopmental disorders.
Our results, in addition to previous findings, indicate that disrupting glutamatergic signaling can result in abnormal oscillatory patterns that depend on the receptor class and neuronal population targeted. Indeed, although ERPs in Pv-mGluR5-deficient mice were accompanied by increased evoked and induced theta and gamma oscillatory activity, Pv-NMDAR ablation was associated with excitatory disinhibition13 and increased spontaneous gamma oscillations.11,13 The ablation of NMDAR from pyramidal neurons also increased pyramidal cell excitability and baseline gamma power and reduced gamma-evoked power.49 Deviations from normal oscillatory activity in specific frequency bands are thought to contribute to the cognitive deficits and psychopathological symptoms of schizophrenia. In particular, an attenuation in induced theta and gamma oscillations is frequently observed in schizophrenia patients,50 but an increased evoked51 and induced52 gamma power during specific stages of cognitive testing has been reported also. Increased beta and gamma oscillations have also been implicated in psychotic symptoms of schizophrenia.53–55 Importantly, alterations in gamma oscillations are observed also in other neurodevelopmental disorders, autism in particular.56–58
The oscillatory alterations in Pv-mG5−/− mice were accompanied by a sequelae of behavioral abnormalities, including pronounced deficits in social and object recognition, but not in spatial memory. Consistently, there was no evidence that hippocampal synaptic plasticity was compromised in Pv-mG5−/− mice. It is well known that spatial memory is largely hippocampal dependent,59 and that both recognition and spatial memory deficits are observed after disruption of ionotropic glutamatergic signaling on GABAergic cells.13–15,60,61 Here, we found that the disruption of mGluR5 function in Pv+ cells specifically affects recognition memory. This memory domain has been suggested to be less dependent on the hippocampus than spatial memory, requiring additional brain structures, such as the PFC and the perirhinal cortex.62 Therefore, it is possible that inhibitory deficits in those brain regions contribute to the recognition memory deficits in Pv-mG5−/− mice. Interestingly, global mGluR5 disruption20,63 impaired spatial memory, indicating that generalized memory impairments probably result from a combined loss of mGluR5 function in pyramidal and inhibitory neurons. Our findings revealed domain-specific memory deficits that arise from mGluR5 disruption in a much more specific inhibitory neuronal population.
Pv+ interneurons regulate striatal output by providing feedforward inhibition to medium spiny neurons.64 Inhibitory deficits in the striatum can lead to several impairments reminiscent of those observed in Pv-mG5−/− mice, including increased repetitive behaviors that are relevant to human disorders.65 For example, Tourette syndrome, a disease characterized by repetitive tics and high overlapping comorbidity with obsessive compulsive disorder, is also associated with a reduction of striatal Pv+ cells.46 Similarly, sapap3 mutant mice, used to model obsessive compulsive disorder, display reduced striatal Pv expression and excessive grooming.66 Repetitive behaviors similar to those exhibited by Pv-mG5−/− mice are also common in mouse models of autism67 and Rett syndrome.68 These commonalities strongly suggest that disruption of postnatal mGluR5 function in Pv+ cells, and its ensuing deficits in E/I in the striatum, are likely responsible for the phenotype observed in Pv-mG5−/− mice. Moreover, Pv+ cell expression in the striatum was only significantly reduced, compared with controls, in male Pv-mG5−/− mice. Male Pv-mG5−/− mice were also more sensitive to repetitive-like behaviors than female Pv-mG5−/− mice, further implicating inhibitory deficits within the striatum in the expression of repetitive behaviors. Intriguingly, autism has a higher incidence rate in males than females.69 Dysfunction of mGluR5 activity on Pv+ cells may therefore contribute to the development of repetitive behaviors in male patients with autism. Interestingly, deletion of mGluR5 from pyramidal neurons was shown to decrease, not increase, repetitive behavior.35 Therefore, in addition to domain-specific memory deficits, the mGluR5-dependent manifestation of compulsive-like behavior is specifically caused by loss of mGluR5 function in Pv+ cells. In light of recent evidence suggesting that mGluR5 antagonism could be an effective therapeutic agent in the treatment of Fragile X syndrome,70 our findings suggest that blockade of mGluR5 during critical developmental stages may have unintended consequences, potentially exacerbating features of those disorders.
Sensorimotor gating was disrupted in Pv-mG5−/− mice and mutants exhibited increased PPI. This finding was unexpected, as global mGluR5 deletion results in profound PPI deficits,19 and mGluR5 deletion from cortical excitatory neurons does not affect PPI.35 However, increased PPI has been observed in mouse models of Fragile X71 and Rett syndrome68 and both models also exhibit strong defects in inhibitory neurons.68,72 Intriguingly, this model of Rett syndrome displayed dysfunctional GABA signaling in the cortex and striatum68 and was also associated with increased PPI. Although deficient sensorimotor gating has been reported in autistic patients,40 increased PPI has also been observed in autistic patients when presented with low-intensity prepulse stimuli.38 This phenomenon was attributed to a heightened sensitivity in detecting stimuli of low salience, which is consistent with the hypothesis that increased gamma oscillations heighten the salience of information transfer.73 Thus, increased gamma oscillations observed in Pv-mG5−/− mice may result in abnormal processing of incoming sensory information. A hypersensitivity to incoming sensory information in turn may contribute to increased PPI in Pv-mG5−/− mice, as similarly suggested for affected PPI38 and gamma oscillations56 in autism.
PCP and amphetamine have been previously used to study aspects of schizophrenia. Here we found a reduced sensitivity to PCP-induced hyperactivity in the open-field and an attenuation of PCP-induced PPI deficits in Pv-mG5−/− mice. These findings are consistent with GABAergic-NMDA receptor ablation and reduced MK-801-induced hyperactivity.12,13 NMDA antagonists are hypothesized to exert their effects via GABAergic neurons.74 Thus, diminished postnatal glutamate transmission onto Pv+ cells through NMDARs or, similarly as shown here by mGluR5, results in impaired neuronal inhibition, which likely affects the treatment with NMDAR antagonist in Pv-mG5−/− mice. On the other hand, amphetamine-induced locomotor hyperactivity in rodents involves increased sub-cortical dopaminergic activity.75 Pv-mG5−/− mice showed heightened sensitivity to amphetamine in the open field. This effect has been observed after several neurodevelopmental manipulations relevant to schizophrenia, including neonatal NMDAR antagonism, social isolation and gestational methylazoxymethanol acetate exposure.42 A direct connection has been established between the loss of Pv+ interneurons, the associated hyperactivity of the hippocampus, and increases in the population activity of dopaminergic neurons, which can underlie the hyper-responsivity to amphetamine of rodents to amphetamine-induced increases in locomotor activity.76 A similar hyperactivation of the hippocampus and dopaminergic system is believed to underlie psychotic outbreaks in schizophrenia.77 Impaired postnatal development of the Pv+ cells, the neuronal circuits that they influence and the reduced inhibitory control of the hippocampus that is evident in Pv-mG5−/− mice may, therefore, cooperatively mediate the increased sensitivity to amphetamine-induced hyperactivity. Thus, disrupted mGluR5 signaling in Pv+ inhibitory neurons during neurodevelopment may be one of the neuromechanisms that contributes to psychotic outbreaks in schizophrenia.
In summary, our results demonstrated that postnatal ablation of mGluR5 from Pv+ fast-spiking interneurons resulted in long-term neurochemical, functional and behavioral alterations that are relevant to several neurodevelopmental disorders. These findings highlight the fundamental role of tightly controlled neuronal inhibition required for balanced neuronal network activity and normal behavior.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01MH91407 and R01MH94670 to MMB, R01MH62527 to A Markou and HHMI to TJS. EAM was supported by NIH/NINDS grant K99NS080911. AP-D was a recipient of a Calouste Gulbenkian Foundation Fellowship. We thank Dr Stephen F Heinemann and Jian Xu for providing us the mGluR5-LoxP mouse line, Joseph Chambers for technical assistance with animal breeding, Dr William F Loomis, Dr James Kesby and Dr Kyongmi Um for their insightful comments and Michael Arends for editorial assistance.
Footnotes
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
CONFLICT OF INTEREST
During the last 3 years, A Markou has received research contract support from Astra-Zeneca, Bristol-Myers-Squibb and Forest Laboratories, and honoraria from AbbVie, Germany.
References
- 1.Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- 2.Huang ZJ. Activity-dependent development of inhibitory synapses and innervation pattern: role of GABA signalling and beyond. J Physiology. 2009;587:1881–1888. doi: 10.1113/jphysiol.2008.168211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue M, Atallah BV, Scanziani M. Equalizing excitation-inhibition ratios across visual cortical neurons. Nature. 2014;511:596–600. doi: 10.1038/nature13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- 5.Uhlhaas PJ, Roux F, Rodriguez E, Rotarska-Jagiela A, Singer W. Neural synchrony and the development of cortical networks. Trends Cogn Sci. 2010;14:72–80. doi: 10.1016/j.tics.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Doischer D, Hosp JA, Yanagawa Y, Obata K, Jonas P, Vida I, et al. Postnatal differentiation of basket cells from slow to fast signaling devices. J Neurosci. 2008;28:12956–12968. doi: 10.1523/JNEUROSCI.2890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okaty BW, Miller MN, Sugino K, Hempel CM, Nelson SB. Transcriptional and electrophysiological maturation of neocortical fast-spiking GABAergic inter-neurons. J Neurosci. 2009;29:7040–7052. doi: 10.1523/JNEUROSCI.0105-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauer JF, Bartos M. Recruitment of early postnatal parvalbumin-positive hippocampal interneurons by GABAergic excitation. J Neurosc. 2010;30:110–115. doi: 10.1523/JNEUROSCI.4125-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- 10.Wang HX, Gao WJ. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology. 2009;34:2028–2040. doi: 10.1038/npp.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Sun QQ. Development of NMDA NR2 subunits and their roles in critical period maturation of neocortical GABAergic interneurons. Dev Neurobiol. 2011;71:221–245. doi: 10.1002/dneu.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 16.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Hooft JA, Giuffrida R, Blatow M, Monyer H. Differential expression of group I metabotropic glutamate receptors in functionally distinct hippocampal inter-neurons. J Neurosci. 2000;20:3544–3551. doi: 10.1523/JNEUROSCI.20-10-03544.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarihi A, Jiang B, Komaki A, Sohya K, Yanagawa Y, Tsumoto T. Metabotropic glutamate receptor type 5-dependent long-term potentiation of excitatory synapses on fast-spiking GABAergic neurons in mouse visual cortex. J Neurosci. 2008;28:1224–1235. doi: 10.1523/JNEUROSCI.4928-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brody SA, Dulawa SC, Conquet F, Geyer MA. Assessment of a prepulse inhibition deficit in a mutant mouse lacking mGlu5 receptors. Mol Psychiatry. 2004;9:35–41. doi: 10.1038/sj.mp.4001404. [DOI] [PubMed] [Google Scholar]
- 20.Lu Y-M, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, et al. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, Zhu Y, Contractor A, Heinemann SF. mGluR5 has a critical role in inhibitory learning. J Neurosci. 2009;29:3676–3684. doi: 10.1523/JNEUROSCI.5716-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dugan LL, Ali SS, Shekhtman G, Roberts AJ, Lucero J, Quick KL, et al. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS One. 2009;4:e5518. doi: 10.1371/journal.pone.0005518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- 25.Zembrzycki A, Chou SJ, Ashery-Padan R, Stoykova A, O’Leary DD. Sensory cortex limits cortical maps and drives top-down plasticity in thalamocortical circuits. Nat Neurosci. 2013;16:1060–1067. doi: 10.1038/nn.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 28.Nadler JJ, Moy SS, Dold G, Simmons N, Perez A, Young NB, et al. Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 2004;3:303–314. doi: 10.1111/j.1601-183X.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 29.Thomas A, Burant A, Bui N, Graham D, Yuva-Paylor LA, Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology (Berl) 2009;204:361–373. doi: 10.1007/s00213-009-1466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semenova S, Geyer MA, Sutcliffe JG, Markou A, Hedlund PB. Inactivation of the 5-HT(7) receptor partially blocks phencyclidine-induced disruption of prepulse inhibition. Biol Psychiatry. 2008;63:98–105. doi: 10.1016/j.biopsych.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moy SS, Perez A, Koller BH, Duncan GE. Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Res. 2006;1089:186–194. doi: 10.1016/j.brainres.2006.03.073. [DOI] [PubMed] [Google Scholar]
- 32.Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, et al. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolber BJ, Montana MC, Carrasquillo Y, Xu J, Heinemann SF, Muglia LJ, et al. Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain-like behavior. J Neurosci. 2010;30:8203–8213. doi: 10.1523/JNEUROSCI.1216-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballester-Rosado CJ, Albright MJ, Wu CS, Liao CC, Zhu J, Xu J, et al. mGluR5 in cortical excitatory neurons exerts both cell-autonomous and -nonautonomous influences on cortical somatosensory circuit formation. J Neurosci. 2010;30:16896–16909. doi: 10.1523/JNEUROSCI.2462-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jew CP, Wu C-S, Sun H, Zhu J, Huang J-Y, Yu D, et al. mGluR5 ablation in cortical glutamatergic neurons increases novelty-induced locomotion. PLoS One. 2013;8:e70415. doi: 10.1371/journal.pone.0070415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Tuathaigh CMP, Babovic D, O’Sullivan GJ, Clifford JJ, Tighe O, Croke DT, et al. Phenotypic characterization of spatial cognition and social behavior in mice with ‘knockout’ of the schizophrenia risk gene neuregulin 1. Neuroscience. 2007;147:18–27. doi: 10.1016/j.neuroscience.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 37.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madsen GF, Bilenberg N, Cantio C, Oranje B. Increased prepulse inhibition and sensitization of the startle reflex in autistic children. Autism Res. 2013;7:94–103. doi: 10.1002/aur.1337. [DOI] [PubMed] [Google Scholar]
- 39.Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 40.Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry. 2007;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 41.Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72. doi: 10.1016/s0006-3223(99)00067-0. [DOI] [PubMed] [Google Scholar]
- 42.Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br J Pharmacol. 2011;164:1162–1194. doi: 10.1111/j.1476-5381.2011.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woo TU, Whitehead RE, Melchitzky DS, Lewis DA. A subclass of prefrontal γ-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA. 1998;95:5341–5346. doi: 10.1073/pnas.95.9.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002;55:1–10. doi: 10.1016/s0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]
- 45.Gogolla N, LeBlanc J, Quast K, Südhof T, Fagiolini M, Hensch T. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodevelop Disord. 2009;1:172–181. doi: 10.1007/s11689-009-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kataoka Y, Kalanithi PS, Grantz H, Schwartz ML, Saper C, Leckman JF, et al. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol. 2010;518:277–291. doi: 10.1002/cne.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 48.Powell SB, Sejnowski TJ, Behrens MM. Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology. 2012;62:1322–1331. doi: 10.1016/j.neuropharm.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tatard-Leitman VM, Jutzeler CR, Suh J, Saunders JA, Billingslea EN, Morita S, et al. Pyramidal cell selective ablation of N-methyl-D-aspartate receptor 1 causes increase in cellular and network excitability. Biol Psychiatry. 2015;77:556–568. doi: 10.1016/j.biopsych.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–113. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- 51.Barr MS, Farzan F, Tran LC, Chen R, Fitzgerald PB, Daskalakis ZJ. Evidence for excessive frontal evoked gamma oscillatory activity in schizophrenia during working memory. Schizophr Res. 2010;121:146–152. doi: 10.1016/j.schres.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 52.Haenschel C, Bittner RA, Waltz J, Haertling F, Wibral M, Singer W, et al. Cortical oscillatory activity is critical for working memory as revealed by deficits in early-onset schizophrenia. J Neurosci. 2009;29:9481–9489. doi: 10.1523/JNEUROSCI.1428-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee SH, Wynn JK, Green MF, Kim H, Lee KJ, Nam M, et al. Quantitative EEG and low resolution electromagnetic tomography (LORETA) imaging of patients with persistent auditory hallucinations. Schizophr Res. 2006;83:111–119. doi: 10.1016/j.schres.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 54.Mulert C, Kirsch V, Pascual-Marqui R, McCarley RW, Spencer KM. Long-range synchrony of gamma oscillations and auditory hallucination symptoms in schizophrenia. Int J Psychophysiol. 2011;79:55–63. doi: 10.1016/j.ijpsycho.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spencer KM, Niznikiewicz MA, Nestor PG, Shenton ME, McCarley RW. Left auditory cortex gamma synchronization and auditory hallucination symptoms in schizophrenia. BMC Neurosci. 2009;10:85. doi: 10.1186/1471-2202-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orekhova EV, Stroganova TA, Nygren G, Tsetlin MM, Posikera IN, Gillberg C, et al. Excess of high frequency electroencephalogram oscillations in boys with autism. Biol Psychiatry. 2007;62:1022–1029. doi: 10.1016/j.biopsych.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 57.Rojas DC, Maharajh K, Teale P, Rogers SJ. Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism. BMC Psychiatry. 2008;8:66. doi: 10.1186/1471-244X-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cornew L, Roberts TL, Blaskey L, Edgar JC. Resting-state oscillatory activity in autism spectrum disorders. J Autism Dev Disord. 2012;42:1884–1894. doi: 10.1007/s10803-011-1431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Broadbent NJ, Squire LR, Clark RE. Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci USA. 2004;101:14515–14520. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andersen JD, Pouzet B. Spatial Memory deficits induced by perinatal treatment of rats with PCP and reversal effect of D-serine. Neuropsychopharmacology. 2004;29:1080–1090. doi: 10.1038/sj.npp.1300394. [DOI] [PubMed] [Google Scholar]
- 61.Harich S, Gross G, Bespalov A. Stimulation of the metabotropic glutamate 2/3 receptor attenuates social novelty discrimination deficits induced by neonatal phencyclidine treatment. Psychopharmacology. 2007;192:511–519. doi: 10.1007/s00213-007-0742-y. [DOI] [PubMed] [Google Scholar]
- 62.Brown MW, Warburton EC, Aggleton JP. Recognition memory: Material, processes, and substrates. Hippocampus. 2010;20:1228–1244. doi: 10.1002/hipo.20858. [DOI] [PubMed] [Google Scholar]
- 63.Christoffersen GR, Simonyi A, Schachtman TR, Clausen B, Clement D, Bjerre VK, et al. MGlu5 antagonism impairs exploration and memory of spatial and non-spatial stimuli in rats. Behav Brain Res. 2008;191:235–245. doi: 10.1016/j.bbr.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 64.Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends Neurosci. 2012;35:557–564. doi: 10.1016/j.tins.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci. 2013;14:278–291. doi: 10.1038/nrn3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burguière E, Monteiro P, Feng G, Graybiel AM. Optogenetic Stimulation of Lateral Orbitofronto-Striatal Pathway Suppresses Compulsive Behaviors. Science. 2013;340:1243–1246. doi: 10.1126/science.1232380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moy SS, Nadler JJ, Poe MD, Nonneman RJ, Young NB, Koller BH, et al. Development of a mouse test for repetitive, restricted behaviors: Relevance to autism. Behav Brain Res. 2008;188:178–194. doi: 10.1016/j.bbr.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chao HT. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol. 2013;26:146–153. doi: 10.1097/WCO.0b013e32835ee548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pop AS, Gomez-Mancilla B, Neri G, Willemsen R, Gasparini F. Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology (Berl) 2014;231:1217–1226. doi: 10.1007/s00213-013-3330-3. [DOI] [PubMed] [Google Scholar]
- 71.Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- 72.Selby L, Zhang C, Sun Q-Q. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett. 2007;412:227–232. doi: 10.1016/j.neulet.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sohal VS. Insights into cortical oscillations arising from optogenetic studies. Biol Psychiatry. 2012;71:1039–1045. doi: 10.1016/j.biopsych.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2012;62:1574–1583. doi: 10.1016/j.neuropharm.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Auclair A, Cotecchia S, Glowinski J, Tassin JP. D-amphetamine fails to increase extracellular dopamine levels in mice lacking alpha 1b-adrenergic receptors: relationship between functional and nonfunctional dopamine release. J Neurosci. 2002;22:9150–9154. doi: 10.1523/JNEUROSCI.22-21-09150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lodge DJ, Grace AA. Hippocampal dysfunction and disruption of dopamine system regulation in an animal model of schizophrenia. Neurotox Res. 2008;14:97–104. doi: 10.1007/BF03033801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lisman JE, Pi HJ, Zhang Y, Otmakhova NA. A thalamo-hippocampal-ventral tegmental area loop may produce the positive feedback that underlies the psychotic break in schizophrenia. Biol Psychiatry. 2010;68:17–24. doi: 10.1016/j.biopsych.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.