

Abstract
Background & Aims
Acute liver injury is a clinically important pathology and results in the release of Danger Associated Molecular Patterns, which initiate an immune response. Withdrawal of the injurious agent and curtailing any pathogenic secondary immune response may allow spontaneous resolution of injury. The role B cells and Immunoglobulin M (IgM) play in acute liver injury is largely unknown and it was proposed that B cells and/or IgM would play a significant role in its pathogenesis.
Methods
Tissue from 3 models of experimental liver injury (ischemia-reperfusion injury, concanavalin A hepatitis and paracetamol-induced liver injury) and patients transplanted following paracetamol overdose were stained for evidence of IgM deposition. Mice deficient in B cells (and IgM) were used to dissect out the role B cells and/or IgM played in the development or resolution of injury. Serum transfer into mice lacking IgM was used to establish the role IgM plays in injury.
Results
Significant deposition of IgM was seen in the explanted livers of patients transplanted following paracetamol overdose as well as in 3 experimental models of acute liver injury (ischemia-reperfusion injury, concanavalin A hepatitis and paracetamol-induced liver injury). Serum transfer into IgM-deficient mice failed to reconstitute injury (p = 0.66), despite successful engraftment of IgM. Mice deficient in both T and B cells (RAG1-/-) mice (p<0.001), but not B cell deficient (μMT) mice (p = 0.93), were significantly protected from injury. Further interrogation with T cell deficient (CD3εKO) mice confirmed that the T cell component is a key mediator of sterile liver injury. Mice deficient in B cells and IgM mice did not have a significant delay in resolution following acute liver injury.
Discussion
IgM deposition appears to be common feature of both human and murine sterile liver injury. However, neither IgM nor B cells, play a significant role in the development of or resolution from acute liver injury. T cells appear to be key mediators of injury. In conclusion, the therapeutic targeting of IgM or B cells (e.g. with Rituximab) would have limited benefit in protecting patients from acute liver injury.
Background
The term acute liver injury (ALI) encompasses a spectrum of sterile or infective hepatocellular insults characterised by acute inflammation within the liver. Injury results in the release of Danger Associated Molecular Patterns (DAMPs), which initiate an immune response. Withdrawal of the injurious agent and curtailing any pathogenic secondary immune response may allow spontaneous resolution of injury [1, 2].
ALI may progress to acute liver failure, which is associated with a mortality of up to 50% [3, 4]. In the developing world, infections (esp. Hepatitis A, B and E viruses) are the commonest aetiology, whereas in the developed world sterile causes predominate [3, 5]. Sterile triggers include drug toxicity (mainly paracetamol/acetaminophen toxicity), autoimmunity and ischemia (ischemia-reperfusion injury (IRI), hypoxic hepatitis). Survival is improving as a result of early diagnosis, improvements in critical care and the growing use of emergency liver transplantation [6]. However, there is still an unmet clinical need to understand how intervention targeting the secondary immune response can benefit patients at risk, or in the early phases, of ALI. One such scenario is ischaemia-reperfusion injury during liver resection or transplantation.
IRI results from the interruption then reinstatement of an organ’s blood supply. It limits access to donor organs and has been linked to early graft failure, as well as both acute and chronic rejection [7, 8]. IRI involves both ischemic and immune-mediated reperfusion phases of injury; numerous mediators and immune cells have been identified as being important in the evolution of this injury and common pathways appear to exist in the pathogenesis of IRI irrespective of the affected organ [9, 10]. Early elevation in pro-inflammatory cytokines in patients following liver resection surgery is linked to worse clinical outcome [11].
B cells are capable of shaping the nature of an immune response through their ability to present antigen and via their ability to produce both cytokines and antibodies. This may have a pro-inflammatory or regulatory influence on the resulting immune response [12]. B cells have been shown to have a pathogenic role in anti-CD40-induced liver injury [13] and in fibrotic liver disease [14]. Numata and colleagues have previously published that mice deficient in both B and T cells (RAG2-/-) had significantly reduced injury compared to wildtype controls 6 hours following administration of a toxic dose of paracetamol [15]. Similarly, mice deficient in both B and T cells (RAG1-/-) were also protected from hepatic IRI 24 hours post-operatively [16, 17]. B cells have a pathogenic role in the aetiology of renal IRI, with mice deficient in B cells (μMT) being protected injury [18]. The presence of B cells in the post-ischemic kidney was also associated with lower levels of IL-10 and slower resolution of injury [19].
Immunoglobulin M (IgM) is produced by B cells and is a normal component of serum. Natural IgM is thought to be produced as a response to self-antigen; it is polyreactive and has a role in the clearance of apoptotic cells [20]. IgM was shown to play a role in intestinal [21], skeletal muscle [22], renal [23] and cardiac [24] models of IRI; it was hypothesised that the IgM in this context was critical for activating complement via the lectin pathway [25].
The role IgM and B cells play in acute liver injury is largely unknown and it was proposed that IgM and/or B cells would play a significant role in its pathogenesis.
Methods
Ethical approval & Welfare
Animal
Following local ethical approval at the University of Edinburgh, animal work was carried out according to UK Home Office regulations (Animals Scientific Procedures Act 1986) under licenses 60/4045 and 60/2605. Mice were housed under specific pathogen-free conditions at the University of Edinburgh. General anaesthesia (GA) was induced via inhaled isoflurane and post-operatively subcutaneous opioid analgesia (buprenorphine) was administered. Animals were sacrificed under GA by terminal exsanguination by way of cardiac puncture.
Human
The Tissue Governance Unit (South East Scotland SAHSC BioResource) provided ethical approval for the human tissue used in this study.
Animals
All wild type (WT), knockout and transgenic mice were on a C57BL/6J background and matched for age and sex. RAG1-/- lack IgM and both mature B and T cells [26]. μMT mice lack mature B cells and IgM [27]. CD3εKO mice lack mature T cells [28].
Models of liver injury
In this model of warm partial hepatic IRI, an atraumatic clamp was applied to the vascular pedicle supplying the left lobe for 40 minutes; the liver was then allowed to reperfuse for between 1 hour and 7 days. Care was taken to maintain intraoperative core body temperature at 36°C to minimise the masking effects of hypothermia on liver IRI [29]. In serum reconstitution experiments, 500μl serum from either wildtype or RAG1-/- donors was injected i.p. 24 hours pre-operatively; successful engraftment was confirmed by the deposition of IgM within areas of liver injury.
Paracetamol-induced liver injury and concanavalin A (con A) hepatitis models were used as alternative models of sterile liver inflammation. Briefly, following a period of 18–24 hours of fasting, mice were given a 200mg/kg intraperitoneal injection of paracetamol (acetaminophen, Sigma-Aldrich, Poole, UK). Con A hepatitis is a model of immune-mediated hepatitis [30] and was induced by an intravenous injection of 10mg/kg con A (Sigma-Aldrich). In both models, the injury was assessed at 24 hours post-injection.
The extent of any liver injury was assessed in terms of the serum level of alanine aminotransfersase (ALT), a biochemical marker of liver injury [31]. ALT was measured on a Cobas Fara centrifugal analyser (Roche Diagnostics Ltd, Welwyn Garden City, UK) using a commercial kit (Alpha Laboratories Ltd, Eastleigh, UK). ALT was correlated with the histological evidence of injury seen on sections stained with haematoxylin and eosin (H&E).
Immunohistochemistry
The Tissue Governance Unit (South East Scotland SAHSC BioResource) kindly provided formalin-fixed sections from the explanted livers of patients requiring transplantation following paracetamol overdose; tissue blocks were specifically chosen by a consultant pathologist with a special interest in transplantation to include areas of both injured and uninjured liver. Antigen retrieval on these sections was performed by heating samples with Borg Decloaker [Biocare Medical, Birmingham, UK]. Tissue from the models of murine liver injury were fixed in methacarn and did not require a specific stage of antigen retrieval.
Sections were deparaffinised and rehydrated before endogenous peroxidase and avidin/biotin activity were quenched, prior to incubating with a goat anti-mouse IgM antibody (AI-2020, Vector Laboratories, Peterborough, UK) at a dilution of 1 in 5000 or goat anti-human IgM antibody (AI-3020, Vector Laboratories) at a dilution of 1 in 1000. Slides were subsequently incubated at room temperature with a polyclonal rabbit anti-goat biotinylated secondary antibody (E0466, DAKO, Ely, UK) at 1 in 400 dilution for 40 minutes. Sections were then developed with VectaStain RTU Elite (Vector Laboratories) followed by diaminobenzidine, before being counterstained.
IgM deposition was quantified from 6 randomly taken images at 200x magnification from each sample using ImageJ software (NIH, Bethesda, USA); it is expressed as percentage of total area.
Flow cytometry
Single cell preparations from the liver were generated by a combination of mechanical (GentleMACS, Miltenyi Biotec, Bisley, UK) and enzymatic (2mg/ml Collagenase D, Roche) digestion. This was then passed through a 70μm filter and centrifuged at 50g for 5 minutes to remove hepatocytes, clumps of cells and debris. Red cells were lysed (Red Cell Lysis Buffer, Sigma-Aldrich). Immune cells were isolated by positive selection using a CD45+ MicroBead AutoMACS separation (Miltenyi Biotec) and then stained with a fixable Live-Dead marker (Life Technologies, Paisley, UK) and a multi-colour panel of antibodies, including CD3, CD4, CD8, CD19 and NK1.1 (Biolegend, San Diego, USA). Samples were then run on a BD LSR II Fortessa (BD Biosciences, Oxford, UK) and analysed with FlowJo software (Tree Star, Ashland, USA). B cells were defined as CD3-CD19+ cells, within a forward-side scatter defined lymphocyte gate.
Statistical analysis
Groups were analysed with the aid of Prism 5 for Mac OSX [Graphpad Software, La Jolla, USA]; statistical methods are referred to specifically in the results section.
Results
IgM is deposited within areas of injured human and murine liver
To investigate whether IgM was deposited in areas of acute liver injury, sections from the explanted livers of patients transplanted for fulminant liver failure following paracetamol overdose were stained for IgM. Significant staining for IgM was seen in areas of injury (Fig 1A). This deposition of IgM was also found in 3 murine models of acute liver injury (Fig 1B & 1C): paracetamol induced liver injury, con A hepatitis and hepatic IRI. All 3 models demonstrated a similar zone 3 pattern of IgM deposition (and injury) to that seen in the explanted human liver. IgM was deposited rapidly in areas of liver injury and was found to disappear with macroscopic resolution of injury (defined as restoration of normal liver architecture) (Fig 1C & 1D).
Fig 1. IgM deposition in acute sterile liver injury.

Tissue sections of explanted livers from patients transplanted for fulminant liver failure secondary to paracetamol overdose were stained looking for evidence of IgM deposition; extensive deposition of IgM is demonstrated within areas of parenchymal injury (A). Tissue sections from male WT mice, with demonstrable sterile liver injury on sections stained with H&E following injection of 200mg/kg paracetamol or 10mg/kg con A also showed extensive IgM staining upon immunohistochemical staining (B). Male mice underwent 40 minutes left lobe ischemia and were then reperfused for 0 hours to 7 days (n = 2–6 per timepoint). The extent of IgM deposition was compared with the histological injury seen on slides stained with H&E; x25 magnification slides are shown at selected time points (C). IgM deposition was calculated as a percentage of total section area across the time-course (median + Standard Error of Mean (SEM)) (D); this deposition of IgM was rapid (seen within 1 hour) and disappeared with macroscopic resolution of injury.
IgM is not critical to the pathogenesis of injury
RAG1-/- are deficient in mature T cells, B cells and IgM [26]. RAG1-/- mice, which lack IgM and both mature B and T cells, were significantly protected from IRI (Fig 2A). In order to assess the importance of IgM in the pathogenesis of acute liver injury, serum from WT mice was transferred into RAG1-/- recipient mice. This failed to significantly reconstitute injury (Fig 2B), despite successful engraftment of IgM as evidenced by the deposition of IgM within visible areas of liver injury (Fig 2C). In spite of rapid IgM deposition within areas of liver injury (Fig 1), IgM does not appear to be critical to the development of significant ALI.
Fig 2. The pathogenicity of IgM in IRI.

WT and RAG1-/- (n = 6) mice underwent experimental warm partial hepatic IRI (40 minutes ischemia and 24 hours reperfusion) to elicit the role T cells, B cells and/or IgM play in the pathogenesis of injury. There was significant reduction in the serum biochemical marker of injury (ALT) observed in RAG1-/- mice compared to WT controls (Mann-Whitney p = 0.0014, median + SEM) (A). To assess whether this protection was a result in the deficiency of IgM, RAG1-/- were injected with serum (sTx) from WT (n = 6) or RAG1-/- (n = 6) donors 24 hours prior to IRI. RAG1-/- mice were injected with PBS (n = 3) as an additional control group. Following 40 minutes ischemia and 24 hours reperfusion, there was no reconstitution of injury (median WT injury calculated from n = 32 mice) or significant differences between the groups [Kruskal-Wallis p = 0.66] (B). Successful engraftment of IgM from WT donors was confirmed by the deposition of IgM into areas of injury (C).
B cells are not critical to injury
Given the protection seen in RAG1-/- mice was not attributable to deficiency in immunoglobulins normally present in serum (Fig 2), it was decided to investigate whether B cells themselves were a major contributor to injury.
Following reperfusion, there was a rapid accumulation of immune cells within the injured liver (Fig 3A); these were predominantly neutrophils (data not shown). This rapid influx of neutrophils fits with seen by other authors in the field [32]. Despite the increase in the number of immune cells, there was a progressive decrease in the number of live B cells following reperfusion (Fig 3B); this decrease is due to cell death (rather than an efflux) of B cells (Fig 3C).
Fig 3. B cells following hepatic IRI.

Following 40 minutes left lobe ischemia, WT mice were allowed to reperfuse for 3, 6 or 12 hours. Mice were then sacrificed and their ischemic left lobes digested. The isolated immune cells were analysed by flow cytometry. There was a significant influx of immune cells (defined as CD45+ cells) into the injured left lobe [Kruskall- Wallis p = 0.0080] (A). B cells were defined as CD3-CD19+ gated lymphocytes. The number of live B cells was found to decrease significantly with time following reperfusion [Kruskall- Wallis p = 0.0043] (B); this was predominantly due to cell death (as defined by positive staining for a live-dead marker) [Kruskall- Wallis p = 0.0060] (C).
B cell deficient (μMT) mice were subjected to hepatic IRI and no significant protection was seen compared to wild type controls (Fig 4). This is further evidence that IgM is not critical to injury, as these mice also lack IgM, yet still get significant injury. In contrast mice deficient in T cells (CD3εKO) mice, were significantly protected from injury (Fig 4). This points to the T cell compartment being a critical mediator of injury and explains the protection seen in RAG1-/- mice, given the apparent lack of pathogenicity of IgM or B cells in this setting.
Fig 4. The role of B and T cells in hepatic IRI.
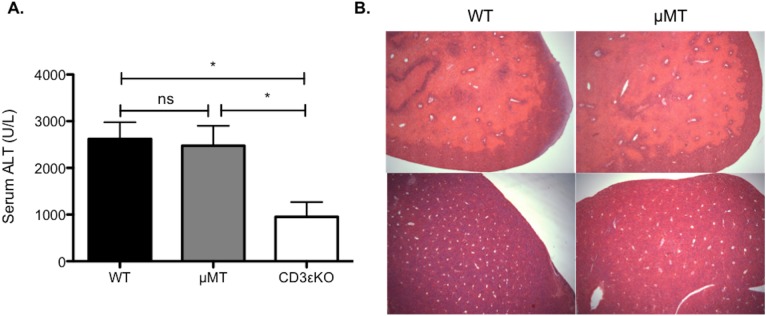
To investigate whether the protection seen in RAG1-/- mice resulted from B and/or T cells, WT controls and mice deficient respectively in mature B cells (μMT) and T cells (CD3εKO) underwent 40 minutes left hepatic lobe ischemia (n = 12/group). Following 24 hours, reperfusion a significant reduction (Kruskall-Wallis p = 0.0037) was seen in mice lacking T, but not B cells.
B cells and IgM are not critical to the resolution of injury
Deposition of IgM occurred early after injury and disappeared with injury resolution (Fig 1). To test the hypothesis that IgM and/or B cells could play an important role in the resolution of acute liver injury (e.g. by activating complement and promoting the opsonisation of cells for subsequent phagocytic clearance), mice underwent IRI and the livers were examined at 7 days post-injury to see if there was a significant delay in injury resolution in the absence of IgM and B cells. There was no significant delay in repair in μMT compared with wild type mice (Fig 5), with all mice at 7 days in both groups having <2% histological evidence of residual injury.
Fig 5. IgM and B cells are not critical to resolution of injury in the liver.
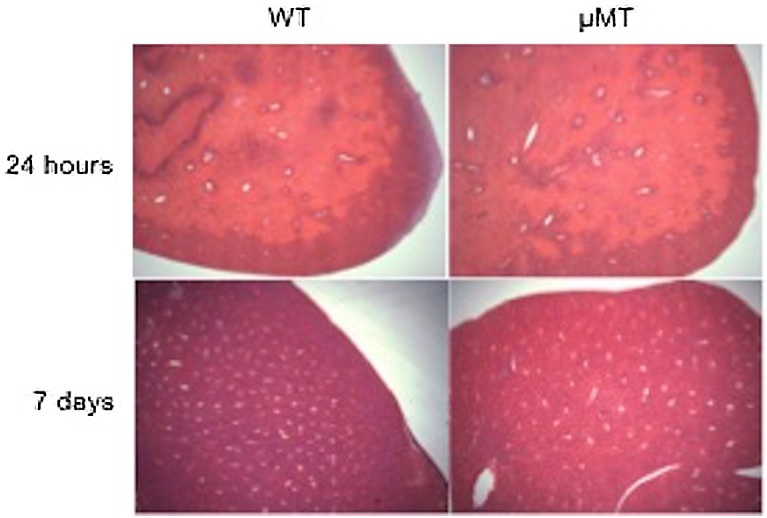
Given the apparent lack of pathogenicity of IgM or B cells in this model of hepatic IRI, it was decided to see if they had a significant role in the resolution from injury. The recovery from injury was compared between WT and μMT mice. Representative H&E sections (x25 magnification), demonstrate similar injury at 24 hours, which resolved almost completely by day 7 post-IRI in both WT and μMT mice (residual necrosis<2% all mice [WT (n = 3), μMT (n = 4)]).
This implies that neither B cells nor IgM were important in the resolution of tissue injury in the post-ischemic liver.
Discussion
Paracetomol toxicity and hypoxic liver injury typically cause zone 3 necrosis. In this study, the pattern of IgM staining in experimental models of these injuries, as well as in sections of liver from patients transplanted as a result of liver failure induced by paracetomol toxicity, mirrored this zone 3 distribution of injury (Fig 1). Deposition of IgM appears to be a good marker of the extent of liver injury, mapping closely the hepatocellular injury identified in serial H&E sections. Based on work with mouse strains deficient in IgM, including serum transfer experiments, this appears to be an epiphenomenon or at least that there is sufficient redundancy in the mechanisms underpinning sterile liver injury that IgM does not have a critical role in either the pathogenesis or subsequent resolution of acute liver injury (Figs 2 & 5). In other organ systems, IgM may have a more significant pathogenic role in the aetiology of IRI [21–24]. Although it should be noted that Lobo et al published conflicting data in which they found significantly worse renal IRI (and increased cardiac allograft rejection) in mice lacking IgM (IgMko), pointing to some of the important anti-inflammatory properties of IgM including inhibition of TLR-4–induced NF-κB nuclear translocation [33].
Given the time-frame in which this occurs (hours), the IgM “painting” injured cells is likely to represent polyreactive natural antibody (IgM) produced by B cells and found naturally in serum [20]. This “painting” of cells with IgM, has been described in other examples of tissue stress [34] and has been found to optimise complement mediated phagocytosis of apoptotic cells [35, 36]. Ciurana and colleagues found that serum IgM was essential for activating complement by necrotic cells [37]. The exact nature of the molecular target bound by IgM in this context remains unclear, but may result from the binding of natural antibody to “hidden” DAMPs exposed as a result of the sterile injury.
We also found that RAG1-/- mice, which also lack both B and T cells, were significantly protected from injury (Fig 2A); this correlates with work from other groups who observed similar degrees of hepatoprotection in these mice [16, 17]. We found that mice deficient in B cells (μMT), but with a functional T cell compartment, were not protected from injury and had no delay in their recovery (Figs 4 & 5). The role of B cells has not previously been reported in hepatic IRI. In work on renal IRI published by Burne-Taney and colleagues, they saw significant protection in the same strain of B cell deficient mice [18]. However, they were unable to recapitulate injury with adoptive transfer of B cells prior to injury, but did partially recreate injury by a serum transfer. A subsequent paper from the same group, demonstrated that the presence of B cells was associated with delayed repair in the post-ischemic kidney [19]. Others have not been able to reproduce this protection and conversely found that μMT mice sustained worse renal IRI compared to controls and postulated that this was due to the decreased levels of IL-10 they observed in these mice [23].
This is the first study, to look directly at the role T cells play in hepatic IRI with the use of a specific T cell knockout mice (CD3εKO). Work using TCR-/- mice has been performed in models of renal IRI, where T cells were also found to be key mediators of injury [38, 39]. The protection seen in T cell deficient (CD3εKO) mice, but not B cells and IgM deficient (μMT) mice, points to the protected phenotype seen in RAG1-/- mice by us and others [16, 17] arising from the T cell compartment, rather than from B cells or immunoglobulins (Fig 4). Data here adds directly to that from other labs, whose work has previously suggested a pathogenic role for T cells in hepatic IRI [40–42]. Conflictingly, Caldwell et al found significantly worse injury in CD4-/- mice, which could be attenuated with the adoptive transfer of CD4+ lymphocytes [43]. We have previously shown that (CD4+Foxp3+) regulatory T cells do not play a critical role in injury [44]. T cells in IRI have been variously found to be both antigen-dependent [41] and antigen independent [40]. CD8+ T cells have not been implicated in the aetiology of this injury across a number of models of IRI [10, 32]. While data from some groups suggest it is conventional (CD4+) T cells that are the key mediators of hepatic IRI [43, 45], others point to non-conventional (NKT, γδ) T cell populations [41, 42]. Further studies are needed to more fully understand the role T cells play in this injury.
Based on our data, therapeutic intervention targeting IgM or B cells (such as with Rituximab) would have no significant benefit to patients or impact on their recovery following liver injury.
Acknowledgments
We would like to thank Forbes Howie for processing the serum samples, Gary Borthwick for assistance with the animal work as well as Judi Allen and David Gray for kindly donating RAG1-/- and μMT/CD3εKO mice respectively for this study. We would also like to acknowledge the Tissue Governance Unit (South East Scotland SAHSC BioResource) for providing the human tissue used in this study.
Data Availability
All relevant data are within the paper.
Funding Statement
This study was made possible by the support of (JR): The Wellcome Trust (WT 092494/Z/10/Z), www.wellcome.ac.uk; The Royal College of Surgeons of Edinburgh (Maurice-Wohl Fellowship and Small Research Grant), www.rcsed.ac.uk; Tenovus Scotland, www.tenovus-scotland.org.uk; The Dowager Countess Eleanor Peel Trust, www.peeltrust.com. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Iredale JP. Cirrhosis: new research provides a basis for rational and targeted treatments. BMJ. 2003;327(7407):143–7. Epub 2003/07/19. 10.1136/bmj.327.7407.143 327/7407/143 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117(3):539–48. Epub 2007/03/03. 10.1172/JCI30542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369(26):2525–34. Epub 2013/12/27. 10.1056/NEJMra1208937 . [DOI] [PubMed] [Google Scholar]
- 4. Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459–63. Epub 2007/07/05. doi: AJG1388 [pii] 10.1111/j.1572-0241.2007.01388.x . [DOI] [PubMed] [Google Scholar]
- 5. Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137(12):947–54. Epub 2002/12/18. doi: 200212170–00007 [pii]. . [DOI] [PubMed] [Google Scholar]
- 6. Bernal W, Hyyrylainen A, Gera A, Audimoolam VK, McPhail MJ, Auzinger G, et al. Lessons from look-back in acute liver failure? A single centre experience of 3300 patients. J Hepatol. 2013;59(1):74–80. Epub 2013/02/27. 10.1016/j.jhep.2013.02.010 S0168-8278(13)00132-3 [pii]. . [DOI] [PubMed] [Google Scholar]
- 7. Devey LR, Friend PJ, Forsythe JL, Mumford LL, Wigmore SJ. The use of marginal heart beating donor livers for transplantation in the United kingdom. Transplantation. 2007;84(1):70–4. Epub 2007/07/14. 10.1097/01.tp.0000268072.04260.69 . [DOI] [PubMed] [Google Scholar]
- 8. Tilney NL, Guttmann RD. Effects of initial ischemia/reperfusion injury on the transplanted kidney. Transplantation. 1997;64(7):945–7. Epub 1997/11/05. . [DOI] [PubMed] [Google Scholar]
- 9. Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17(11):1391–401. Epub 2011/11/09. 10.1038/nm.2507 nm.2507 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Linfert D, Chowdhry T, Rabb H. Lymphocytes and ischemia-reperfusion injury. Transplantation Reviews. 2009;23(1):1–10. 10.1016/j.trre.2008.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strey CW, Marquez-Pinilla RM, Markiewski MM, Siegmund B, Oppermann E, Lambris JD, et al. Early post-operative measurement of cytokine plasma levels combined with pre-operative bilirubin levels identify high-risk patients after liver resection. Int J Mol Med. 2011;27(3):447–54. Epub 2011/01/06. 10.3892/ijmm.2010.592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–41. Epub 2012/01/10. 10.1146/annurev-immunol-020711-074934 . [DOI] [PubMed] [Google Scholar]
- 13. Kimura K, Moriwaki H, Nagaki M, Saio M, Nakamoto Y, Naito M, et al. Pathogenic role of B cells in anti-CD40-induced necroinflammatory liver disease. Am J Pathol. 2006;168(3):786–95. Epub 2006/03/02. doi: S0002-9440(10)62139-6 [pii] 10.2353/ajpath.2006.050314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. 2005;115(11):3072–82. Epub 2005/11/09. 10.1172/JCI24798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Numata K, Kubo M, Watanabe H, Takagi K, Mizuta H, Okada S, et al. Overexpression of suppressor of cytokine signaling-3 in T cells exacerbates acetaminophen-induced hepatotoxicity. J Immunol. 2007;178(6):3777–85. Epub 2007/03/07. doi: 178/6/3777 [pii]. . [DOI] [PubMed] [Google Scholar]
- 16. Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203(12):2639–48. Epub 2006/11/08. doi: jem.20061097 [pii] 10.1084/jem.20061097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng M, Li G, Qian X, Fan Y, Huang X, Zhang F, et al. IL-17A-producing NK cells were implicated in liver injury induced by ischemia and reperfusion. Int Immunopharmacol. 2012;13(2):135–40. Epub 2012/04/03. 10.1016/j.intimp.2012.03.007 S1567-5769(12)00073-2 [pii]. . [DOI] [PubMed] [Google Scholar]
- 18. Burne-Taney MJ, Ascon DB, Daniels F, Racusen L, Baldwin W, Rabb H. B cell deficiency confers protection from renal ischemia reperfusion injury. J Immunol. 2003;171(6):3210–5. Epub 2003/09/10. . [DOI] [PubMed] [Google Scholar]
- 19. Jang HR, Gandolfo MT, Ko GJ, Satpute SR, Racusen L, Rabb H. B cells limit repair after ischemic acute kidney injury. J Am Soc Nephrol. 2010;21(4):654–65. Epub 2010/03/06. 10.1681/ASN.2009020182 ASN.2009020182 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. 2010;10(11):778–86. Epub 2010/10/16. 10.1038/nri2849 nri2849 [pii]. . [DOI] [PubMed] [Google Scholar]
- 21. Williams JP, Pechet TT, Weiser MR, Reid R, Kobzik L, Moore FD Jr., et al. Intestinal reperfusion injury is mediated by IgM and complement. J Appl Physiol. 1999;86(3):938–42. Epub 1999/03/06. . [DOI] [PubMed] [Google Scholar]
- 22. Weiser MR, Williams JP, Moore FD Jr., Kobzik L, Ma M, Hechtman HB, et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183(5):2343–8. Epub 1996/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, et al. B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol. 2010;185(7):4393–400. Epub 2010/09/03. doi: jimmunol.0903239 [pii] 10.4049/jimmunol.0903239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haas MS, Alicot EM, Schuerpf F, Chiu I, Li J, Moore FD, et al. Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc Res. 2010;87(4):618–27. Epub 2010/05/14. doi: cvq141 [pii] 10.1093/cvr/cvq141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, et al. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006;177(7):4727–34. Epub 2006/09/20. doi: 177/7/4727 [pii]. . [DOI] [PubMed] [Google Scholar]
- 26. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–77. Epub 1992/03/06. doi: 0092-8674(92)90030-G [pii]. . [DOI] [PubMed] [Google Scholar]
- 27. Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350(6317):423–6. Epub 1991/04/04. 10.1038/350423a0 . [DOI] [PubMed] [Google Scholar]
- 28. Malissen M, Gillet A, Ardouin L, Bouvier G, Trucy J, Ferrier P, et al. Altered T cell development in mice with a targeted mutation of the CD3-epsilon gene. EMBO J. 1995;14(19):4641–53. Epub 1995/10/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Devey L, Festing MF, Wigmore SJ. Effect of temperature control upon a mouse model of partial hepatic ischaemia/reperfusion injury. Lab Anim. 2008;42(1):12–8. Epub 2008/03/20. 10.1258/la.2007.06009e . [DOI] [PubMed] [Google Scholar]
- 30. Wang HX, Liu M, Weng SY, Li JJ, Xie C, He HL, et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J Gastroenterol. 2012;18(2):119–25. Epub 2012/01/19. 10.3748/wjg.v18.i2.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Champe P, Harvey R, Ferrier D. Biochemistry. 3rd Edition ed. Baltimore: Lippincott Williams and Wilkins; 2005. [Google Scholar]
- 32. Zwacka RM, Zhang Y, Halldorson J, Schlossberg H, Dudus L, Engelhardt JF. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest. 1997;100(2):279–89. Epub 1997/07/15. 10.1172/jci119533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lobo PI, Bajwa A, Schlegel KH, Vengal J, Lee SJ, Huang L, et al. Natural IgM anti-leukocyte autoantibodies attenuate excess inflammation mediated by innate and adaptive immune mechanisms involving Th-17. J Immunol. 2012;188(4):1675–85. Epub 2012/01/21. 10.4049/jimmunol.1101762 jimmunol.1101762 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vollmers HP, Brandlein S. Natural IgM antibodies: from parias to parvenus. Histol Histopathol. 2006;21(12):1355–66. . [DOI] [PubMed] [Google Scholar]
- 35. Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity. 2005;38(4):259–64. . [DOI] [PubMed] [Google Scholar]
- 36. Chen Y, Park YB, Patel E, Silverman GJ. IgM antibodies to apoptosis-associated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells. J Immunol. 2009;182(10):6031–43. 10.4049/jimmunol.0804191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ciurana CL, Zwart B, van Mierlo G, Hack CE. Complement activation by necrotic cells in normal plasma environment compares to that by late apoptotic cells and involves predominantly IgM. Eur J Immunol. 2004;34(9):2609–19. 10.1002/eji.200425045 . [DOI] [PubMed] [Google Scholar]
- 38. Savransky V, Molls RR, Burne-Taney M, Chien CC, Racusen L, Rabb H. Role of the T-cell receptor in kidney ischemia-reperfusion injury. Kidney Int. 2006;69(2):233–8. Epub 2006/01/13. doi: 5000038 [pii] 10.1038/sj.ki.5000038 . [DOI] [PubMed] [Google Scholar]
- 39. Hochegger K, Schatz T, Eller P, Tagwerker A, Heininger D, Mayer G, et al. Role of alpha/beta and gamma/delta T cells in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2007;293(3):F741–7. Epub 2007/06/15. doi: 00486.2006 [pii] 10.1152/ajprenal.00486.2006 . [DOI] [PubMed] [Google Scholar]
- 40. Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T cells contribute to postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets. Hepatology. 2006;43(2):306–15. 10.1002/hep.21017 [DOI] [PubMed] [Google Scholar]
- 41. Kuboki S, Sakai N, Tschop J, Edwards MJ, Lentsch AB, Caldwell CC. Distinct contributions of CD4+ T cell subsets in hepatic ischemia/reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2009;296(5):G1054–9. Epub 2009/03/07. doi: 90464.2008 [pii] 10.1152/ajpgi.90464.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eggenhofer E, Rovira J, Sabet-Baktach M, Groell A, Scherer MN, Dahlke MH, et al. Unconventional RORgammat+ T Cells Drive Hepatic Ischemia Reperfusion Injury. J Immunol. 2013;191(1):480–7. Epub 2013/06/07. 10.4049/jimmunol.1202975 jimmunol.1202975 [pii]. . [DOI] [PubMed] [Google Scholar]
- 43. Caldwell CC, Okaya T, Martignoni A, Husted T, Schuster R, Lentsch AB. Divergent functions of CD4+ T lymphocytes in acute liver inflammation and injury after ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol. 2005;289(5):G969–76. Epub 2005/07/09. 10.1152/ajpgi.00223.2005 . [DOI] [PubMed] [Google Scholar]
- 44. Devey LR, Richards JA, O'Connor RA, Borthwick G, Clay S, Howie AF, et al. Ischemic preconditioning in the liver is independent of regulatory T cell activity. PLoS One. 2012;7(11):e49647 Epub 2012/11/28. 10.1371/journal.pone.0049647 PONE-D-12-26937 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shen X, Wang Y, Gao F, Ren F, Busuttil RW, Kupiec-Weglinski JW, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology. 2009;50(5):1537–46. Epub 2009/08/12. 10.1002/hep.23153 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.