

Abstract
The medicinal plant Xanthium strumarium L. (X. strumarium) is covered with glandular trichomes, which are the sites for synthesizing pharmacologically active terpenoids such as xanthatin. MicroRNAs (miRNAs) are a class of 21–24 nucleotide (nt) non-coding RNAs, most of which are identified as regulators of plant growth development. Identification of miRNAs involved in the biosynthesis of plant secondary metabolites remains limited. In this study, high-throughput Illumina sequencing, combined with target gene prediction, was performed to discover novel and conserved miRNAs with potential roles in regulating terpenoid biosynthesis in X. strumarium glandular trichomes. Two small RNA libraries from leaves and glandular trichomes of X. strumarium were established. In total, 1,185 conserved miRNAs and 37 novel miRNAs were identified, with 494 conserved miRNAs and 18 novel miRNAs being differentially expressed between the two tissue sources. Based on the X. strumarium transcriptome data that we recently constructed, 3,307 annotated mRNA transcripts were identified as putative targets of the differentially expressed miRNAs. KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis suggested that some of the differentially expressed miRNAs, including miR6435, miR5021 and miR1134, might be involved in terpenoid biosynthesis in the X. strumarium glandular trichomes. This study provides the first comprehensive analysis of miRNAs in X. strumarium, which forms the basis for further understanding of miRNA-based regulation on terpenoid biosynthesis.
Introduction
MicroRNAs (miRNAs) are small non-coding, endogenous RNAs consisting of ~22 nt in average, and are generated from large stem-loop precursors transcribed from non-protein-coding genes, introns or coding regions of the host genome[1, 2]. They interact with mRNAs through perfect or non-perfect complementarity to degrade mRNAs or repress translation, thus negatively regulating gene expression post-transcriptionally [3–7]. Plant miRNAs have been reported to be involved in various biological processes, including plant growth development, signal transduction, and stress responses against biotic or abiotic factors [8–12]. They also target genes with functions in metabolite biosynthesis [13,14]. With the aid of a high-throughput sequencing technology, there are increasing miRNAs identified and characterized from a number of medicinal plant species, e.g. Panax ginseng [15], Opium poppy [16], Taxus [17], and Catharanthus roseus [18], and their roles in regulating the production of secondary metabolites of interests were also suggested by the bioinformatics analysis. In the model plant Arabidopsis thaliana, there was direct evidence that miRNAs regulated the biosynthesis of secondary metabolites by modulating their expression in vivo. For instance, the overexpression of miRNA393 in Arabidopsis thaliana altered the levels of glucosinolate and camalexin via perturbing the auxin signaling pathway [19]. Other studies showed that the flavonoid biosynthesis of Arabidopsis thaliana was regulated by the expression of miRNA156 while the modulation of miRNA163 expression level changed the profiles of secondary metabolites [20, 21].
Xanthium strumarium L. (X. strumarium), an annual growth herb, belongs to the compositae family [22]. The whole plant, especially its leaf, root and fruit, has been used in traditional medicine for the treatment of rhinitis, malaria, rheumatism, tuberculosis, cancer, and ulcers [23–26]. Previous studies indicated that plants of the Asteraceae family are characteristically rich in sesquiterpene lactones, an important class of terpenoids, and the Xanthiums species are rich in such medicinal ingredients [27–31]. The pharmacological properties of X. strumarium are largely attributed to the presence of xanthanolides (a class of sesquiterpene lactones), which have been reported to possess antifungal, antibacterial, and cytotoxic activities, and exhibit a growth inhibitory activity against insects [30, 32–37]. Despite their multiple bioactivities, especially their anti-tumor and anti-cancer activities [38, 39], the knowledge on how xanthanolides are biosynthesized and how the pathway is regulated remains largely unknown. Answering this scientific question is one of the long-term aims in our laboratory. Previously, we discovered that xanthanolides were highly biosynthesized and accumulated in the glandular trichomes of the X. strumarium tissues, especially on its leaves at early stage [40]. To identify genes encoding enzymes involved in trichome-dependent biosynthesis of xanthanolides in X. strumarium, the transcriptome dataset from two related tissue sources—glandular cells isolated from young leaves and intact young leaves was recently analyzed by our group. To study the regulatory mechanisms of xanthanolides biosynthesis, we focused our attentions on miRNA-based regulations as there are increasingly published literatures reporting their roles in plant secondary metabolic activities [15–19]. To date, reports on miRNAs in X. strumarium remain lacking. In this study, X. strumarium miRNAs were firstly identified using high-throughput sequencing technology and the differentially expressed miRNAs between the isolated glandular cells and intact young leaves were discovered. Combined with the analysis of the X. strumarium transcriptome, the targets of those differentially expressed miRNAs were predicted and their functions were annotated, which suggested that some of the differentially expressed miRNAs might play roles in regulating terpenoid biosynthesis in X. strumarium glandular cells.
Materials and Methods
Plant materials
Young leaves (the first and second leaves from the top) were randomly collected from different individual X. strumarium plants grown at the Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, China (Aug. 10th, 2013). The age of the X. strumarium was three month-old. X. strumarium glandular trichomes were isolated from 20g of intact young leaves according to protocols described previously by Chen et al. with some modifications [40]. The young leaves were abraded in a cell disrupter (Bead-Beater, BIOSPEC, USA) using glass beads in an isolation buffer (25 mM MOPSO, pH 6.6, 200 mM sorbitol, 10 mM sucrose, 5 mM thiourea, 2 mM dithiothreitol, 5 mM MgCl2, 0.5 mM sodium phosphate, 0.6% (w/v) methylcellulose and 1% (w/v) polyvinylpyrrolidone). The disrupted extracts were filtered through a 425 μm nylon mesh, and the filtrate was then consecutively passed through 125, 80 and 42 μm nylon meshes with a resin buffer (25 mM MOPSO, pH 6.6, 200 mM sorbitol, 10 mM sucrose, 5 mM thiourea, 2 mM dithiothreitol, 5 mM MgCl2 and 0.5 mM sodium phosphate). The isolated glandular trichomes were retained on the 42 μm mesh. Each sample was flash frozen in liquid nitrogen and then stored at −80°C for RNA isolation.
Small RNA library construction and high-throughput sequencing
Total RNA was extracted from fresh young leaves and the isolated glandular trichomes with Trizol reagent (Ambion). The quantity and quality of RNA samples were measured by Eppendorf BioPhotometer plus to ensure that the OD260/OD280 values were between 1.8 and 2.2. The RNA integrity was examined by agarose gel electrophoresis. Small RNA sequencing was performed using an Illumina Genome Analyzer at the Beijing Genomics Institute (BGI, Shenzhen, China). Small RNA fractions with the length range from18 to 30 nt were purified and then ligated to a 5' and 3' adaptor. After the reverse transcription followed by 11 cycles of polymerase chain reactions, approximately 20 μg of the amplified products were used for sequencing.
Analysis of the sequenced data of the small RNAs
Small RNA reads with a length of 49 nt were produced by Illumina. Then data processing analysis was conducted as follows: (1) Removal of low-quality reads (more than four bases with sQ values below 10, and more than six bases with sQ values less than 13); (2) Removal of reads with 5′ adaptor contaminants; (3) Removal of reads without 3′ primer; (4) Removal of reads without an insert tag; (5) Removal of reads with poly A; (6) Removal of reads shorter than 18 nt; and (7) A summary of the length distribution of the clean reads. The remaining clean reads were mapped to X. strumarium transcriptome with less than two mismatches to analyze the expression and distribution of the small RNAs using SOAP software[41].To annotate the small RNAs, the sequences were aligned to the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) and Rfam (http://rfam.sanger.ac.uk/) 10.1 databases by a BLAST search[42, 43]. The matched tags, including rRNA, scRNA, snoRNA, snRNA, and tRNA were eliminated. The remaining tags were used to detect conserved and novel miRNAs. The transcriptome databases of the X. strumarium small RNAs and mRNAs were deposited at the sequence read archive (SRA) of National Center for Biotechnology Information (NCBI) under the accession numbers of SRP056720 and SRP056511, respectively.
Identification of the conserved miRNAs
There is no miRNA information for X. strumarium in miRBase. To identify the conserved miRNAs, the following strategy was used: first, considering the differences between species, clean data was aligned to mature miRNAs or miRNA precursors of all plants in miRBase 20.0 (http://www.mirbase.org)[44] allowing two mismatches using tag2miRNA software (developed by BGI); second, we chose the most abundant miRNA from each mature miRNA family to construct a temporary miRNA database; third, we aligned the clean data to the above temporary miRNA database and the expression of miRNA was generated by summing the count of all tags which were aligned to the temporary miRNA database within two mismatches. The small RNAs that were unaligned to any databases were defined as unannotated sequences.
Prediction of the novel miRNAs
The unannotated sequences ranging from 18 to 25 nt were used to identify novel miRNAs by Mireap software based on the following main criteria described by chen et al. [45]: (1) The tags which could be used to predict novel miRNAs came from the set of unannotated tags which were matched to the transcriptome of X. strumarium; (2) Those tags whose sequences and structures satisfied the two criteria: hairpin precursors could fold into secondary structures and the sequences were present in one arm of the hairpin precursors, will be considered as candidate novel miRNAs; (3) Hairpin precursors lack large internal loops or bulges; (4) The secondary structures of the hairpins are steady, with the free energy of hybridization lower than or equal to -18 kcal/mol; (5) The number of mature miRNAs with predicted hairpin precursors must be at least five in the alignment results.
Target gene prediction of the conserved and novel miRNAs
To obtain putative target genes, we matched the identified miRNAs to the X. strumarium transcriptome according to the rules published by Allen et al.[3] and Schwab et al.[7]. The criteria were (1) the number of mismatches between small RNAs and their targets should be less than four (G–U pairs count as half mismatch); (2) no more than two adjacent mismatches in the miRNA/target duplex; (3) no adjacent mismatches in positions 2 to 12 of the miRNA/target duplex from the 5′ miRNA end; (4) no mismatches in positions 10 to 11 of the miRNA/target duplex; (5) no more than 2.5 mismatches in positions 1 to 12 of the miRNA/target duplex from the 5′ miRNA end; and (6) the minimum free energy (MFE) of the miRNA/target duplex should be ≥ 75% of the MFE of the miRNA with its perfect complement.
Differential expression analysis of miRNAs between the leaves and glandular trichomes
To ensure the significance of the difference in miRNA expression, we normalized the expression of miRNAs in the two tissue sources (leaves and glandular trichomes) as transcript per million (TPM). Then those miRNAs with a P-value<0.05 (adjusted to a corrected P-value (q-value) lower than 0.05) and an absolute value of log2Ratio>1 were selected as the differentially expressed miRNAs. Target gene prediction of the differentially expressed miRNAs was also conducted to better understand the regulatory roles of the miRNAs. Alignments of the miRNAs to the corresponding target sites are shown in S1 Table.
GO (Gene Ontology) functional classification and KEGG pathway analysis for the potential targets of the differentially expressed miRNAs
GO is a classification system for gene function, which supplies a set of dynamically updated and controlled vocabulary to comprehensively describe the property of genes and gene products. There are 3 ontologies in GO: molecular function, cellular component and biological process. The basic unit of GO is GO-term, each of which belongs to one type of ontology. Therefore, to classify the function distribution of the potential targets of the differentially expressed miRNAs genes, the Blast2GO program was used to obtain their GO annotations [46] and the WEGO software to obtain their GO functional classifications [47]. The GO enrichment analysis of the targets was conducted and GO terms with a corrected P-value ≤ 0.05 were defined as significantly enriched terms. KEGG is a public database regarding metabolic pathways [48]. The target genes were mapped to the KEGG database to identify what pathways in which those targets of the differentially expressed miRNAs are involved.
Real-time quantitative PCR (RT-qPCR)
Stem-loop RT-qPCR was employed to validate the gene expression data from the Illumina sequencing according to the method previously described by Chen et al. [49]. The primers used for this part of the experiment were listed in S2 Table. First-strand cDNA synthesis was performed using RevertAid Reverse Transcriptase (Thermo Scientific). The reaction was carried out at 42°C for 60 min followed by incubation at 70°C for 10 min, and then held at 4°C thereafter. RT-qPCR was conducted using the FastStart Universal SYBR Green Master (Roche) and ABI 7500 Real-Time PCR System according to the manufacturer’s instructions. The reactions were undertaken at 95°C for 10 min for one cycle; at 95°C for 15s, then at 62°C for 1 min for 40 cycles. All reactions were performed in three independent biological samples with three technical repeats. The melting curve was generated to test the specificity of PCR products and avoid the amplicons only from primers themselves. The actin gene of X. strumarium (GenBank accession no.JF434698) was used as an internal standard to normalize the variation in each sample manipulation and the results were analyzed using the comparative 2-ΔΔCt method to quantify the relative expression [50].
Results
High-throughput sequencing analysis of small RNAs
In total, 12,325,132 raw reads for the leaves and 9,076,601 raw reads for the glandular trichomes were initially generated. After data preprocessing, 12,152,212 and 8,988,274 clean reads for the leaves and glandular trichomes remained for the analysis, generating 7,261,121 and 4,842,894 total unique sequences for the leaves and glandular trichomes, respectively. 6,193,697 and 3,775,470 unique sequences (85.3% and 77.96% of the total unique sequences) were specific to the leaves and glandular trichomes (Table 1). This was indicative of the diversity of small RNA sequences in each tissue source. Little difference was found in the length distribution of the sequences from both tissue sources: the most abundant was the 24 nt small RNAs, accounting for more than 60% of the total reads, followed by 21 nt small RNAs, and small RNAs with a length of 23 nt (Fig 1). In addition, 220,115 (3.03%) and 247,453 (5.11%) unique sequences for the leaves and glandular trichomes matched to the X. strumarium transcriptome data. After annotating and removing the non-coding RNAs, including rRNAs, tRNAs, snRNAs, and snoRNAs, 37,490 (0.52%) and 33,664 (0.7%) reads for the leaves and glandular trichomes were left for the identification of conserved miRNAs, and 7,138,288 (98.31%) and 4,735,851 (97.79%) unannotated reads for the leaves and glandular trichomes were used to predict novel miRNAs (Table 2).
Table 1. Statistics of small RNA sequencing.
| Type | Leaves | Glandular trichomes | ||
|---|---|---|---|---|
| count | % | count | % | |
| total raw reads | 12,325,132 | — | 9,076,601 | — |
| high quality reads | 12,195,632 | 100 | 9,021,559 | 100 |
| 3'adapter null reads | 16,018 | 0.13 | 14,806 | 0.16 |
| insert null reads | 881 | 0.01 | 638 | 0.01 |
| 5'adapter contaminants | 17,105 | 0.14 | 9797 | 0.11 |
| smaller than 18nt reads | 7103 | 0.06 | 5576 | 0.06 |
| polyA reads | 2313 | 0.02 | 2468 | 0.03 |
| clean reads | 12,152,212 | 99.64 | 8,988,274 | 99.63 |
| total unique reads | 7,261,121 | — | 4,842,894 | — |
| tissue_specific unique reads | 6,193,697 | 85.30 a | 3,775,470 | 77.96 a |
aThe percentage of the tissue_specific unique reads for the respective tissue source.
Fig 1. Size distribution of the miRNAs from the leaves and glandular cells.

Table 2. Distribution of small RNAs among different categories in leaves and glandular trichomes of X. strumarium.
| Type | Unique small RNAs | Total small RNAs | ||
|---|---|---|---|---|
| Leaves | Glandular trichomes | Leaves | Glandular trichomes | |
| total reads | 7,261,121(100%) | 4,842,894(100%) | 12,152,212(100%) | 8,988,274(100%) |
| matched reads a | 220,115(3.03%) | 247,453(5.11%) | 1,474,980(12.14%) | 1,819,939(20.25%) |
| miRNA | 37,490(0.52%) | 33,664(0.70%) | 719,520(5.92%) | 521,259(5.80%) |
| rRNA | 75,056(1.03%) | 60,161(1.24%) | 388,404(3.20%) | 405,948(4.52%) |
| snRNA | 1003(0.01%) | 1239(0.03%) | 1493(0.01%) | 2317(0.03%) |
| snoRNA | 347(0%) | 414(0.01%) | 478(0%) | 608(0.01%) |
| tRNA | 8937(0.12%) | 11,565(0.24%) | 38287(0.32%) | 339,486(3.78%) |
| unannotated | 7,138,288(98.31%) | 4,735,851(97.79%) | 11,004,030(90.55%) | 7,718,656(85.87%) |
aThe reads that were matched to the X. strumarium transcriptome.
Identifying conserved miRNAs in both tissue sources
In X. strumarium, no miRNA has been reported at the time of drafting this manuscript. We identified 978 conserved miRNA families with 745,146 total reads in the leaves and 894 miRNA families with a total of 550,246 reads in the glandular trichomes (S3 Table). There were 687 conserved miRNA families expressed in both tissue sources (Fig 2A), of which miR5565 was the most abundant miRNA family with 338,261 reads in the leaves and 249,096 in the glandular trichomes. The expression levels of a few other miRNAs, such as miR166, miR167, miR172, miR398, and miR156 were also very high in both samples, while some miRNAs, including miR2084, miR2670, miR2875 and miR2950, were expressed in extremely low abundance with only less than five reads.
Fig 2. Proportion of identified miRNAs in the leaves and glandular trichomes presented in a Venn diagram.
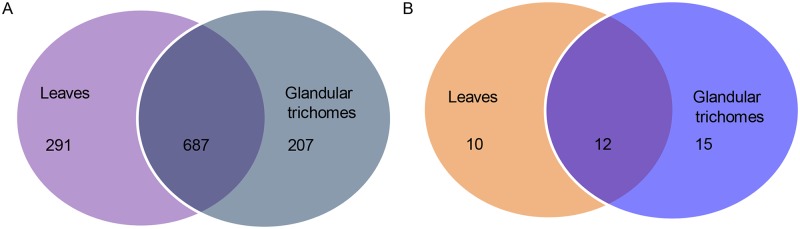
The miRNAs in the diagram consist of three portions: miRNAs that are exclusively present in leaves, miRNAs that are exclusively present in glandular trichomes, and miRNAs present in both tissue sources. (A) conserved miRNAs; (B) novel miRNAs.
Identifying potential novel miRNAs in X. strumarium
Based on the criteria described in the section of Materials and Methods, 22 potential novel miRNAs for the leaves and 27 for the glandular trichomes were identified in both tissue sources with at least five reads (S4 Table). Of these, only 12 novel miRNAs appeared in both samples (Fig 2B), suggesting that the expression profiling of novel miRNAs was different between the leaves and glandular trichomes.
The identified novel miRNAs ranged from 20 to 23 nt, with 21nt being the most abundant (59.46%) (Fig 3A). The length of the predicted precursors for the novel miRNAs were 66 to 323 nt, with that the majority was between 50 and 150 nt (54.06%) (Fig 3C). The folding energy of these hairpin structures for the precursors of novel miRNAs was -19.7 to -101.8 kcal/mol, which most values within the range of -40 to -80 kcal/mol (54.05%) (Fig 3B). These results were similar to those observed in Chinese cabbage, Arabidopsis thaliana, Oryza sativa and Arachis hypogaea [51–53]. The nucleotide bias analysis showed that novel miRNAs from both tissue sources had the similar tendency on the nucleotide bias at certain key positions, for example, a strong preference for adenosine (A) at the tenth position and for uridine (U) at the first position(Fig 4), which are the typical features of miRNAs[54, 55].
Fig 3. Summary of potential novel miRNAs identified in X. strumarium.

(A) Length frequency for the identified novel miRNAs. (B) Folding energy frequency of precursors for the potential novel miRNAs. (C) Length frequency of precursors for the potential novel miRNAs.
Fig 4. Nucleotide preference at each position of novel miRNAs.

(A) miRNA nucleotide bias of novel miRNAs in leaves; (B) miRNA nucleotide bias of novel miRNAs in glandular trichomes.
Target prediction of conserved and novel miRNAs in X. strumarium
Target genes for the conserved and novel miRNAs were predicted to better understand the biological functions of the identified miRNAs in X. strumarium. In total, we found 4,071 target genes for 544 conserved miRNAs and 116 target genes for 26 novel miRNAs in X. strumarium, with an average of 7.48 and 4.46 targets per conserved and novel miRNA (S5 Table). To annotate these potential targets, a BlastX search against the NCBI protein database with an E value lower than 10−5 was performed. Some targets were annotated as transcription factors, including WRKY, Basic helix–loop–helix (bHLH), SQUAMOSA Promoter Binding Protein-Like (SPL) and basic leucine zipper motif (bZIP) proteins. Other target genes included those involved in signal transduction, metabolism, stress response and those with unknown functions. The expression levels of these targets between the leaves and glandular trichomes were also compared (S5 Table).
The identification of the miRNAs differentially expressed between the two tissue sources
The total of 512 miRNAs, including 494 conserved and 18 novel miRNAs, were found to be differentially expressed between the two tissue sources. Among them, 262 conserved and 13 novel miRNAs were up-regulated, and 232 conserved and five novel miRNAs were down-regulated in the glandular trichomes (S6 Table). To validate the miRNA expression data from the sequencing, the expression levels of 13 differentially expressed miRNAs, including eight novel miRNAs and five conserved miRNAs, were measured using RT-qPCRs. As was shown in Fig 5, the expression trend of most of the miRNAs, except for miR1134, was consistent with the Illumina sequencing results, meaning that the gene expression data of miRNAs by the sequencing technique was credible.
Fig 5. RT-qPCR data for the transcript abundance of some miRNAs in the leaves and glandular trichomes.

The miRNA levels were normalized to an internal control (actin) and expressed relative to the values of leaves (control), which were given an arbitrary value of 1. Error bars indicate the standard deviation of three biological replicates.
Target prediction of the differentially expressed miRNAs
Based on the X. strumarium transcriptome, a total of 3,307gene targets were identified for those differentially expressed miRNAs (S6 Table). Among these targets, some encode transcription factors such as v-myb avian myeloblastosis viral oncogene homolog (MYB), WRKY, bHLH, APETALA2/ethylene-responsive factor (AP2/ERF), bZIP and SPL proteins. For instance, the unique genes, including CL6103.Contig1 targeted by miR5072, CL1989.Contig2_All targeted by miR7539 and Unigene12046_All targeted by miR1850, displayed high similarities to WRKY proteins. WRKY transcription factors have been reported to play roles in regulating the biosynthesis of terpenoids [56–58]. By mapping those targets to the KEGG pathway database, we were able to find that some targets seemed to encode putative enzymes in terpenoid biosynthesisin X. strumarium (the ones highlighted by yellow color in S7 Table), especially in sesquiterpene biosynthesis (Table 3). For example, the upstream enzymes in the pathways of terpenoid biosynthesis, including 1-deoxy-D-xylulose 5-phosphate synthase (DXS), 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR), isopentenyl diphosphate (IPP)/dimethylallyl diphosphate (DMAPP) synthase (IDS), and isopenteyl diphosphate isomerase (IDI), were predicted to be targeted by miR7539, miR5021 and miR1134. DXS, HMGR, IDS, and IDI are the enzymes involved in the biosynthesis of IPP and DMAPP, the common precursors of all the terpenoids [59]. In particular, HMGR is a rate-limiting enzyme of the pathway to synthesize IPP and DMAPP [60]. The target by miR6435 is homologous to germacrene A oxidase (GAO), the first key enzyme in the pathway to the biosynthesis of xanthanolides [61]. Interestingly, xanthanolides have been considered to be the major active compounds in X. strumarium [62]. In addition, some targets are homologs to the enzymes in the biosynthesis of di-, or tri-terpenoids. For example, the beta-amyrin synthase targed by miR5491 and ent-kaurene synthase targeted by miR6449 are key enzymes that catalyze the formation of the most common triterpene β-Amyrin and diterpene ent-kaurene [63, 64].
Table 3. Target genes for differentially expressed miRNAs involved in terpenoids biosynthesis.
| microRNAs | Target gene candidates | Annotation | Biosynthetic pathway |
|---|---|---|---|
| miR6435 | Unigene22477_All | Germacrene A oxidase | sesquiterpenoid[61] |
| miR5255 | Unigene26141_All | Squalene epoxidase | triterpenoid[65, 66] |
| miR5255 | Unigene26143_All | Squalene epoxidase | triterpenoid |
| miR5255 | Unigene26144_All | Squalene epoxidase | triterpenoid |
| miR5255 | Unigene26145_All | Squalene epoxidase | triterpenoid |
| miR5255 | Unigene26146_All | Squalene epoxidase | triterpenoid |
| miR5491 | CL1191.Contig1_All | beta-amyrin synthase | triterpenoid[64] |
| miR5491 | CL1191.Contig2_All | beta-amyrin synthase | triterpenoid |
| miR5491 | CL1191.Contig3_All | beta-amyrin synthase | triterpenoid |
| miR5491 | CL1191.Contig5_All | beta-amyrin synthase | triterpenoid |
| miR5491 | Unigene18850_All | beta-amyrin synthase | triterpenoid |
| miR5021 | CL12255.Contig3_All | HMGR | terpenoid backbone [59] |
| miR1134 | CL12255.Contig3_Al | HMGR | terpenoid backbone |
| miR5021 | CL3919.Contig4_All | IDS | terpenoid backbone [59] |
| miR5021 | Unigene24678_All | IDI | terpenoid backbone [59] |
| miR5021 | Unigene23634_All | IDI | terpenoid backbone |
| miR7539 | CL4414.Contig1_All | DXS | terpenoid backbone [59] |
| miR7540 | CL2999.Contig1_All | R-linalool synthase | monoterpenoid[67] |
| miR5183 | CL5257.Contig2_All | gibberellin 3-oxidase | diterpenoid[68] |
| miR6449 | CL5429.Contig1_All | ent-kaurene synthase | diterpenoid[63] |
| miR6449 | CL5429.Contig7_All | ent-kaurene synthase | diterpenoid |
| miR6449 | CL5429.Contig8_All | ent-kaurene synthase | diterpenoid |
GO analysis showed that these targets could be summarized into three main categories and classified into 47 functional groups (Fig 6). Based on the biological process category, the majority of the targets were involved in “cellular process”, “metabolic process” and “single-organism process”. In the case of molecular functions, a large number of genes were grouped into “binding” and “catalytic activity”. While in the cellular component, the genes were mostly related to “cell”, “cell part” and “organelle”. The GO enrichment analysis showed that the terms “mitochondrial respiratory chain complex IV” and “respiratory chain complex IV” are overrepresented in the cellular component. For the molecular function, the majority of genes were found to be involved in the “oxidoreductase activity” and “cytochrome-c oxidase activity”. The GO terms “heme a metabolic process” and “heme a biosynthetic process” account for a large proportion in the biological process, which indicated that the miRNAs might play roles in modulating plant metabolic processes (S8 Table).
Fig 6. GO functional classification for the predicted targets by the differentially expressed miRNAs.

X-axis, the three main GO categories and 47 GO terms assigned for the differentially expressed miRNA targets; Y-axis, the gene numbers corresponding to the GO terms.
Discussion
Plant miRNAs have been reported to be involved in a variety of important processes, including development, signal transduction, and responses to environmental stresses [12]. There was also evidence to show that miRNAs function in regulating secondary metabolic activities. For example, the molecule miRNA156 is involved in the regulation of flavonoid biosynthesis in Arabidopsis thaliana [21]. Glandular trichomes are one type of specialized structure in synthesizing a wide range of plant secondary metabolites [69], in X. strumarium, they are also the primary sites for accumulating xanthanolides, the compounds with multiple bioactivities [40]. We hypothesized that miRNA expression in glandular cells might play roles in regulating the biosynthesis of secondary metabolites in X. strumarium such as xanthanolides. However, to the best of our knowledge, no any information is available for the miRNAs from glandular trichomes of any plant species. As the beginning to address this hypothesis, glandular trichomes were physically isolated from the young leaves of X. strumarium in this study and large sets of miRNAs in this particular structure were identified using a high-throughput sequencing technology. A database for miRNAs from its intact young leaves was also constructed and used as a comparison. A total of 894 conserved miRNAs and 27 novel miRNAs were successfully identified from the glandular trichomes. The expression levels of more than 50% of these miRNAs seem to be up- or down-regulated in the glandular trichomes compared to intact leaves. The reliability of the gene expression data was confirmed by the Q-RT-PCR analysis of the five conserved and eight novel miRNAs that were randomly selected. The expression of, novel-mir-14, novel-mir-17, and novel-mir-34 is very high in glandular trichomes and more than 1000 folds to those in intact leaves, indicating that they may have physiological functions in this specialized structure. With respects to the miRNAs with the highest abundance in the glandular cells, they may be glandular trichome-specifically expressed or transported into the trichomes from the other parts of the leaves.
To understand the regulatory roles of miRNAs, it is essential to predict and annotate its target mRNAs. Based on the feature that plant miRNAs are perfectly complementary to their targets, miRNA target genes can be predicted by a bioinformatics approach [3, 7]. Using the bioinformatics tool, we were able to identify that the targets by the glandular trichomes conserved miRNAs included transcription factors and non-transcriptional factor proteins (S5 Table). Several transcription factors were predicted to be targeted by the same conserved miRNA molecule, for example, miR7539 targets MYB, bHLH, WAKY, zinc finger, DNA Binding With One Finger(DOF), SPL, and bZIP transcription factors and miR5658 targets MYB, bHLH, zinc finger, and bZIP transcription factors, suggesting that these miRNAs may play multiple roles in diverse physiological processes. Non-transcriptional factor proteins, such as DXS, HMGR, IDS and IDI, were predicted to be targets of miR7539, miR5021 and miR1134, respectively. These targets are essential enzymes in upstream isoprenoid pathway to produce IPP and DMAPP, the common precursors for all the downstream end terpenoids. In particular, HMGR is a key regulatory enzyme that controls the amounts of isoprenoids [60]. These data suggested that miR7539, miR5021 and miR1134 might be involved in regulating terpenoid biosynthesis by targeting upstream terpenoid pathway genes. Some miRNAs target putative downstream enzymes in the biosynthesis of mono-, sesqui-, di-, and tri-terpenoids. They were R-linalool synthase, gibberellin 3-oxidase, ent-kaurene synthase, squalene epoxidase, beta-amyrin synthase, and germacreneA oxidase, which were targeted by miR7540, miR5183, miR6449, miR5255, miR5491, and miR6435, respectively (Table 3). Most interestingly, germacrene A oxidase (GAO) targeted by miR6435 is a key P450 involved in the biosynthesis of xanthanolides[61], which was previously reported to be active molecules to contribute to the pharmacological property of X. strumarium [30, 34, 70]. Gene expression data from the miRNA-sequencing showed that the molecule miR6435 is glandular trichome-specifically expressed (S6 Table), which is also consistent with the feature that glandular trichomes are the primary sites to synthesize xanthanolides. The data allowed us to hypothesize that miR6435 might play a role in the regulation of xanthanolide biosynthesis in X. strumarium glandular trichomes. Identification of miRNAs which can perfectly bind to their mRNA targets may also provide alternate approach to isolate pathway genes, especially for those pathways that are not elucidated well. The discovery of X. strumarium glandular trichome miRNAs of this study may help to identify xanthanolide biosynthesis genes. The biosynthetic pathway of xanthanolides is not elucidated yet, especially for its downstream pathway in which cytochrome P450 enzymes are presumably involved. Here, we have tried to identify miRNAs whose targets were P450 mRNAs. In addition to GAO targeted by miR6435, we also have found that the targets of other miRNAs, including miR1512, miR3447, miR5678, miR6283, and miR7539, encode P450s, in particular, of these P450 mRNA sequences, the target mRNA by miR6283 seemed to be trichome-specifically expressed (S5 Table). Thus, it will be of interest to further experimentally perform functional analysis of miR6283 and its target. In contrast to the conserved miRNA targets, none of the targets by novel miRNAs presented in this research were transcription factors and many of them encoded cytochrome c oxidase, ABC transporter, and protein kinases, suggesting their roles in oxidation-reduction processes, transport, and signal transduction.
Conclusions
In conclusion, this is the first comprehensive identification of miRNAs from the plant glandular trichomes, the specialized structure to synthesize a wide range of medicinal molecules. We have been able to identify miRNAs and their mRNA targets that are trichome-specifically expressed. The data of this study provide the starting point to further investigation to elucidate the miRNAs regulatory mechanism underlying the biosynthesis of secondary metabolites, especially terpenoids, in X. strumarium glandular cells.
Supporting Information
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank Jia Li for the excellent technical assistance, Xin Wang for useful discussions with the data of this manuscript, and Junbo Gou for his help with collecting the plant materials.
Data Availability
All the relevant data described in this study are within the paper and its Supporting Information files. In addition, the transcriptome databases for the small RNAs and mRNAs of X. strumarium have been deposited at the sequence read archive (SRA) of NCBI under the accession numbers of SRP056720 and SRP056511, respectively.
Funding Statement
The funding sources are from the National Natural Science Foundation of China (grant number: 31370339) and the Grant of One Hundred Talent Program of the CAS (grant number: 29Y129441R0104444) to YZ. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. [DOI] [PubMed] [Google Scholar]
- 2. Ha M, Kim VN. Regulation of microRNA biogenesis.Nat Rev Mol Cell Biol. 2014;15(8):509–24. 10.1038/nrm3838 [DOI] [PubMed] [Google Scholar]
- 3. Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121(2):207–21. 10.1016/j.cell.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 4. Aukerman MJ, Sakai H. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell. 2003;15(11):2730–41. 10.1105/tpc.016238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009;136(2):215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004;303(5666):2022–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D. Specific effects of microRNAs on the plant transcriptome. Dev Cell. 2005;8(4):517–27. [DOI] [PubMed] [Google Scholar]
- 8. Frazier T, Zhang B. Identification of Plant microRNAs Using Expressed Sequence Tag Analysis In: Pereira A, editor. Plant Reverse Genetics. Methods in Molecular Biology. New York: Humana Press; 2011. pp. 13–25. 10.1007/978-1-60761-682-5_2 [DOI] [PubMed] [Google Scholar]
- 9. Jagadeeswaran G, Saini A, Sunkar R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis . Planta. 2009;229(4):1009–14. 10.1007/s00425-009-0889-3 [DOI] [PubMed] [Google Scholar]
- 10. Phillips JR, Dalmay T, Bartels D. The role of small RNAs in abiotic stress. FEBS Lett. 2007;581(19):3592–7. 10.1016/j.febslet.2007.04.007 [DOI] [PubMed] [Google Scholar]
- 11. Yang T, Xue L, An L. Functional diversity of miRNA in plants. Plant Sci. 2007;172(3):423–32. 10.1016/j.plantsci.2006.10.009 [DOI] [Google Scholar]
- 12. Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: a small regulatory molecule with big impact. Dev Biol. 2006;289(1):3–16. 10.1016/j.ydbio.2005.10.036 [DOI] [PubMed] [Google Scholar]
- 13. Robert-Seilaniantz A, Bari R, Jones JD. A biotic or abiotic stress? In:Pareek A, Sopory SK, Bohnert HJ, Govindjee, editors. Abiotic Stress Adaptation in Plants. Dordrecht: Springer; 2010. pp. 103–122. [Google Scholar]
- 14. Schommer C, Palatnik JF, Aggarwal P, Chételat A, Cubas P, Farmer EE, et al. Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol. 2008;6(9):e230 10.1371/journal.pbio.0060230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu B, Wang M, Ma Y, Yuan L, Lu S. High-throughput sequencing and characterization of the small RNA transcriptome reveal features of novel and conserved microRNAs in Panax ginseng. PLoS One. 2012;7(9):e44385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boke H, Ozhuner E, Turktas M, Parmaksiz I, Ozcan S, Unver T. Regulation of the alkaloid biosynthesis by miRNA in opium poppy. PlantBiotechnol J. 2015;13(3):409–20. 10.1111/pbi.12346 [DOI] [PubMed] [Google Scholar]
- 17. Hao DC, Yang L, Xiao PG, Liu M. Identification of Taxus microRNAs and their targets with high‐throughput sequencing and degradome analysis. Physiol Plant. 2012;146(4):388–403. 10.1111/j.1399-3054.2012.01668.x22708792 [DOI] [PubMed] [Google Scholar]
- 18. Prakash P, Ghosliya D, Gupta V. Identification of conserved and novel microRNAs in Catharanthusroseus by deep sequencing and computational prediction of their potential targets. Gene. 2015;554(2):181–95. 10.1016/j.gene.2014.10.046 [DOI] [PubMed] [Google Scholar]
- 19. Robert-Seilaniantz A, MacLean D, Jikumaru Y, Hill L, Yamaguchi S, Kamiya Y, et al. The microRNA miR393 re-directs secondary metabolite biosynthesis away from camalexin and towards glucosinolates. Plant J. 2011;67(2):218–31. 10.1111/j.1365-313X.2011.04591.x [DOI] [PubMed] [Google Scholar]
- 20. Ng DW, Zhang C, Miller M, Palmer G, Whiteley M, Tholl D, et al. cis-and trans-Regulation of miR163 and target genes confers natural variation of secondary metabolites in two Arabidopsis species and their allopolyploids. Plant Cell. 2011;23(5):1729–40. 10.1105/tpc.111.083915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gou J-Y, Felippes FF, Liu C-J, Weigel D, Wang J-W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor.Plant Cell. 2011;23(4):1512–22. 10.1105/tpc.111.084525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hammerschmidt D. Xanthium strumarium. J Lab Clin Med. 1998;132(1):86 [DOI] [PubMed] [Google Scholar]
- 23. Chandel S, Bagai U, Vashishat N. Antiplasmodial activity of Xanthium strumarium against Plasmodium berghei-infected BALB/c mice. Parasitol Res. 2012;110(3):1179–83. 10.1007/s00436-011-2611-1 [DOI] [PubMed] [Google Scholar]
- 24. Gautam R, Saklani A, Jachak SM. Indian medicinal plants as a source of antimycobacterial agents. J Ethnopharmacol. 2007;110(2):200–34. 10.1016/j.jep.2006.12.031 [DOI] [PubMed] [Google Scholar]
- 25. Ma Y-T, Huang M-C, Hsu F-L, Chang H-F. Thiazinedione from Xanthium strumarium .Phytochemistry. 1998;48(6):1083–5. 10.1016/S0031-9422(98)00084-3 16556487 [DOI] [Google Scholar]
- 26. Yin MH, Kang DG, Choi DH, Kwon TO, Lee HS. Screening of vasorelaxant activity of some medicinal plants used in Oriental medicines. J Ethnopharmacol. 2005;99(1):113–7. 10.1016/j.jep.2005.02.013 [DOI] [PubMed] [Google Scholar]
- 27. Abdei-Mogib M, Dawidar A, Metwally M, Abou-Elzahab M. Xanthanolides from Xanthium spinosum . Phytochemistry. 1991;30(10):3461–2. 10.1016/0031-9422(91)83230-I [DOI] [Google Scholar]
- 28. Ahmed AA, Jakupovic J, Bohlmann F, Regaila H, Ahmed A. Sesquiterpene lactones from Xanthium pungens . Phytochemistry. 1990;29(7):2211–5. 10.1016/0031-9422(90)83040-8 [DOI] [Google Scholar]
- 29. Mahmoud AA. Xanthanolides and Xanthane Epoxide Derivatives from Xanthium strumarium . Planta Med. 1998;64(08):724–7. [DOI] [PubMed] [Google Scholar]
- 30. Rodriguez E, Towers G, Mitchell J. Biological activities of sesquiterpene lactones. Phytochemistry. 1976;15(11):1573–80. 10.1016/S0031-9422(00)97430-2 [DOI] [Google Scholar]
- 31. Saxena V, Mondal S. A xanthanolide from Xanthium strumarium .Phytochemistry. 1994;35(4):1080–2. 10.1016/S0031-9422(00)90678-2 [DOI] [Google Scholar]
- 32. Harada A, Sakata K, Ina H, Ina K. Isolation and Identification of Xanthatin as an Anti-Attaching Repellent against Blue Mussel. Agric Biol Chem. 1985;49(6):1887–8. WOS:A1985AKZ5200054 [Google Scholar]
- 33. Kawazu K, Nakajima S, Ariwa M. Xanthumin and 8-epi-xanthatin as insect development inhibitors from Xanthium canadense Mill. Experientia. 1979;35(10):1294–5. [DOI] [PubMed] [Google Scholar]
- 34. Kim H, Lee I, Yeo S, Seong L, Yu T. Isolation and Characterization of Antitumor Agents from Xanthium strumarium L. Korean J Biotechnol Bioeng. 2003;18:324–8. [Google Scholar]
- 35. Lavault M, Landreau A, Larcher G, Bouchara J-P, Pagniez F, Le Pape P, et al. Antileishmanial and antifungal activities of xanthanolides isolated from Xanthium macrocarpum . Fitoterapia. 2005;76(3):363–6. 10.1016/j.fitote.2005.03.019 [DOI] [PubMed] [Google Scholar]
- 36. Roussakis C, Chinou I, Vayas C, Harvala C, Verbist J. Cytotoxic activity of xanthatin and the crude extracts of Xanthium strumarium . Planta Med. 1994;60(5):473 [DOI] [PubMed] [Google Scholar]
- 37. Sato Y, Oketani H, Yamada T, Singyouchi KI, Ohtsubo T, Kihara M, et al. A Xanthanolide with Potent Antibacterial Activity against Methicillin-resistant Staphylococcus aureus .J Pharm Pharmacol. 1997;49(10):1042–4. 10.1111/j.2042-7158.1997.tb06038.x [DOI] [PubMed] [Google Scholar]
- 38. Nibret E, Youns M, Krauth-Siegel RL, Wink M. Biological activities of xanthatin from Xanthium strumarium leaves. Phytother Res. 2011;25(12):1883–90. 10.1002/ptr.3651 [DOI] [PubMed] [Google Scholar]
- 39. Ramírez-Erosa I, Huang Y, Hickie RA, Sutherland RG, Barl B. Xanthatin and xanthinosin from the burs of Xanthium strumarium L. as potential anticancer agents. Can J PhysiolPharmacol. 2007;85(11):1160–72. . [DOI] [PubMed] [Google Scholar]
- 40. Chen F, Hao F, Li C, Gou J, Lu D, Gong F, et al. Identifying three ecological chemotypes of Xanthium strumarium glandular trichomes using a combined NMR and LC-MS method. PLoS One. 2013;8(10):e76621 10.1371/journal.pone.0076621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li R, Yu C, Li Y, Lam T- W, Yiu S-M, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7. 10.1093/bioinformatics/btp336 [DOI] [PubMed] [Google Scholar]
- 42. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. GenBank. Nucleic Acids Res. 2013;41(D1):D36–D42. 10.1093/nar/gks1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31(1):439–41. 10.1093/nar/gkg006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(suppl1):D140–D4. 10.1093/nar/gkj112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen M, Zhang X, Liu J, Storey KB. High-throughput sequencing reveals differential expression of miRNAs in intestine from sea cucumber during aestivation. PLoS One. 2013;8(10): e76120 10.1371/journal.pone.0076120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6. 10.1093/bioinformatics/bti610 [DOI] [PubMed] [Google Scholar]
- 47. Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(suppl2):W293–W7. 10.1093/nar/gkl031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(suppl 1):D480–D4. 10.1093/nar/gkm882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, et al. Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res. 2005;33(20):e179–e. 10.1093/nar/gni178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 51. Chi X, Yang Q, Chen X, Wang J, Pan L, Chen M, et al. Identification and characterization of microRNAs from peanut (Arachishypogaea L.) by high-throughput sequencing. PLoS One. 2011;6(11):e27530 10.1371/journal.pone.00275322110666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang F, Li H, Zhang Y, Li J, Li L, Liu L, et al. MicroRNA expression analysis of rosette and folding leaves in Chinese cabbage using high-throughput Solexa sequencing. Gene. 2013;532(2):222–9. 10.1016/j.gene.2013.09.039 [DOI] [PubMed] [Google Scholar]
- 53. Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA. Conservation and divergence of plant microRNA genes. Plant J. 2006;46(2):243–59. 10.1111/j.1365-313X.2006.02697.x [DOI] [PubMed] [Google Scholar]
- 54. Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5’ terminal nucleotide. Cell. 2008;133(1):116–27. 10.1016/j.cell.2008.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA.Mol Cell. 2004;14(6):787–99. 10.1016/j.molcel.2004.05.027 [DOI] [PubMed] [Google Scholar]
- 56. Eulgem T, Rushton PJ, Robatzek S, Somssich IE. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5(5):199–206. 10.1016/S1360-1385(00)01600-9 [DOI] [PubMed] [Google Scholar]
- 57. Ma D, Pu G, Lei C, Ma L, Wang H, Guo Y, et al. Isolation and characterization of AaWRKY1, an Artemisia annua transcription factor that regulates the amorpha-4, 11-diene synthase gene, a key gene of artemisinin biosynthesis. Plant Cell Physiol. 2009;50(12):2146–61. 10.1093/pcp/pcp149 [DOI] [PubMed] [Google Scholar]
- 58. Yang CQ, Fang X, Wu XM, Mao YB, Wang LJ, Chen XY. Transcriptional regulation of plant secondary metabolism. J Integr Plant Biol. 2012;54(10):703–12. 10.1111/j.1744-7909.2012.01161.x [DOI] [PubMed] [Google Scholar]
- 59. Ma Y, Yuan L, Wu B, Li X, Chen S, Lu S. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza . J Exp Bot. 2012;63(7):2809–23. 10.1093/jxb/err466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Choi D, Ward BL, Bostock RM. Differential induction and suppression of potato 3-hydroxy-3-methylglutaryl coenzyme A reductase genes in response to Phytophthorainfestans and to its elicitor arachidonic acid. Plant Cell. 1992;4(10):1333–44. 10.1105/tpc.4.10.1333 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Eljounaidi K, Cankar K, Comino C, Moglia A, Hehn A, Bourgaud F, et al. Cytochrome P450s from Cynara cardunculus L. CYP71AV9 and CYP71BL5, catalyze distinct hydroxylations in the sesquiterpene lactone biosynthetic pathway. Plant Sci. 2014;223:59–68. 10.1016/j.plantsci.2014.03.007 [DOI] [PubMed] [Google Scholar]
- 62. Kamboj A, Saluja AK. Phytopharmacological review of Xanthium strumarium L.(Cocklebur). Int J Green Pharm. 2010;4(3):129. [Google Scholar]
- 63. Hayashi K-i, Kawaide H, Notomi M, Sakigi Y, Matsuo A, Nozaki H. Identification and functional analysis of bifunctionalent-kaurene synthase from the moss Physcomitrella patens . FEBS Lett. 2006;580(26):6175–81. 10.1016/j.febslet.2006.10.018 [DOI] [PubMed] [Google Scholar]
- 64. Kushiro T, Shibuya M, Ebizuka Y. β-Amyrin synthase. Eur J Biochem. 1998;256(1):238–44. [DOI] [PubMed] [Google Scholar]
- 65. Jung JD, Park HW, Hahn Y, Hur CG, In DS, Chung HJ, et al. Discovery of genes for ginsenoside biosynthesis by analysis of ginseng expressed sequence tags. Plant Cell Rep. 2003;22(3):224–30. 10.1007/s00299-003-0678-6 [DOI] [PubMed] [Google Scholar]
- 66. Suzuki H, Achnine L, Xu R, Matsuda SP, Dixon RA. A genomics approach to the early stages of triterpene saponin biosynthesis in Medicago truncatula . Plant J. 2002;32(6):1033–48. 10.1046/j.1365-313X.2002.01497.x [DOI] [PubMed] [Google Scholar]
- 67. Landmann C, Fink B, Festner M, Dregus M, Engel K-H, Schwab W. Cloning and functional characterization of three terpene synthases from lavender (Lavandulaangustifolia). Arch BiochemBiophys. 2007; 465(2):417–29. 10.1016/j.abb.2007.06.011 [DOI] [PubMed] [Google Scholar]
- 68. Chen Y, Hou M, Liu L, Wu S, Shen Y, Ishiyama K, et al. The maize DWARF1 encodes a gibberellin 3-oxidase and is dual localized to the nucleus and cytosol. Plant Physiol. 2014;166(4):2028–39. 10.1104/pp.114.247486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Duke SO. Glandular trichomes-a focal point of chemical and structural interactions.Int JPlantSci. 1994;155(6):617–20. [Google Scholar]
- 70. Kupchan SM, Eakin M, Thomas A. Tumor inhibitors. 69. Structure-cytotoxicity relations among the sesquiterpene lactones. J Med Chem. 1971; 14(12):1147–52. 10.1021/jm00294a001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All the relevant data described in this study are within the paper and its Supporting Information files. In addition, the transcriptome databases for the small RNAs and mRNAs of X. strumarium have been deposited at the sequence read archive (SRA) of NCBI under the accession numbers of SRP056720 and SRP056511, respectively.