

Abstract
Histone deacetylases (HDACs) are enzymes involved in transcriptional repression. We aimed to examine the significance of HDAC1 and HDAC2 gene expression in the prediction of recurrence and survival in 156 patients with hepatocellular carcinoma (HCC) among a South East Asian population who underwent curative surgical resection in Singapore. We found that HDAC1 and HDAC2 were upregulated in the majority of HCC tissues. The presence of HDAC1 in tumor tissues was correlated with poor tumor differentiation. Notably, HDAC1 expression in adjacent non-tumor hepatic tissues was correlated with the presence of satellite nodules and multiple lesions, suggesting that HDAC1 upregulation within the field of HCC may contribute to tumor spread. Using competing risk regression analysis, we found that increased cancer-specific mortality was significantly associated with HDAC2 expression. Mortality was also increased with high HDAC1 expression. In the liver cancer cell lines, HEP3B, HEPG2, PLC5, and a colorectal cancer cell line, HCT116, the combined knockdown of HDAC1 and HDAC2 increased cell death and reduced cell proliferation as well as colony formation. In contrast, knockdown of either HDAC1 or HDAC2 alone had minimal effects on cell death and proliferation. Taken together, our study suggests that both HDAC1 and HDAC2 exert pro-survival effects in HCC cells, and the combination of isoform-specific HDAC inhibitors against both HDACs may be effective in targeting HCC to reduce mortality.
Keywords: hepatocellular carcinoma, mortality, Southeast Asian population, histone deacetylases
Introduction
Histone deacetylases (HDACs) are enzymes involved in transcriptional repression mainly through removal of acetyl groups from the positively charged histone tails and allowing histones to wrap the DNA tightly. In addition to deacetylating histones, they also deacetylate non-histone proteins, resulting in protein degradation via the ubiquitin-proteasome pathway (1). HDACs exist as components in multiprotein complexes with transcriptional co-repressors, such as mSin3, N-CoR and SMRT (2). These complexes target specific genomic regions by interacting with DNA binding factors such as transcription factors, nuclear receptors, methyl-binding proteins, DNA methyl transferases and histone methyltransferases.
In mammals, there are 18 known HDAC homologs, subdivided into classes I, IIa, IIb, III and IV (3). HDAC1 was the first identified mammalian HDAC and is considered the prototype of the HDAC family. It belongs to class I of HDACs and shares 83% amino acid identity with another class I member, HDAC2 (4). Both HDAC1 and HDAC2 have almost identical genomic organization and this indicates that they have arisen from relatively recent gene duplication and probably share similar biological functions. They are ubiquitously expressed in many tissues and since they lack nuclear export signals, they are found exclusively in the nucleus (5). Despite having some overlapping biological functions, HDAC1 and HDAC2 also have distinct and even opposing biological functions. For example, the deletion of HDAC1 alone is lethal in mouse embryos (6). There is also evidence for distinct functions of HDACs in cancer cells. HDAC2, but not HDAC1, can inhibit proliferation and induce senescence in MCF7 breast cancer cells (7). In addition, cellular responses to growth factors appear to be specific to different HDACs - silencing of HDAC1 suppresses transforming growth factor (TGF) β1-induced apoptosis while silencing of HDAC2 increases spontaneous apoptosis and enhances TGF β1-induced apoptosis in AML12 murine hepatocytes due to opposing effects on extracellular-regulated kinase (ERK)1/2 (8).
Recent studies have shown elevated expression of class I HDACs in solid human tumors (9). HDAC1 and HDAC2 physically associate with an N-terminally truncated form of p63 (ΔNp63α) to repress the transcription of pro-apoptotic genes in squamous cell carcinoma (10). In this study, we demonstrate that HDAC1 and HDAC2 are upregulated in hepatocellular carcinoma (HCC) tissues compared to matched adjacent non-tumor controls. This is consistent with previous studies that show upregulation of HDAC1 and HDAC2 in hepatocellular cancer (11) and other cancers (12–14).
HCC is the most common form of liver cancer, accounting for 85 to 90% of all primary liver cancers (15) and liver cancer is the fifth most common cancer worldwide, with an alarming 748,300 new cases and 695,900 cancer deaths in 2008 (16). Current treatment for HCC includes surgical resection of the tumor, liver transplantation, and chemotherapy, but each has its own problems and limitations (17–20). The use of HDAC inhibitors to target solid tumors such as HCC is still at an early stage of development. HDAC inhibitors are thought to function by reversing the aberrant epigenetic states of cancer cells. Although the effects of pan-HDAC inhibitors have been widely studied, not much is known of the differing roles of the various HDAC homologs in cancer. In this study, we aimed to evaluate the prognostic significance of HDAC1 and HDAC2 gene expression in HCC by applying a competing risk approach.
Materials and methods
Patients and tissue samples
HCC tumor samples, matched adjacent non-tumor hepatic tissues and clinical data from 156 patients who underwent curative surgical resection between 1989 and 2009 were obtained from the Department of Pathology, National University Hospital of Singapore (NUH). This study was approved by the National University of Singapore (NUS)-Institutional Review Board (IRB 09-112).
Tissue microarray and immunohistochemistry
The tissues were deparaffinized and rehydrated before antigen retrieval was performed by heating the sections for 30 min in antigen unmasking solution (Vector Laboratories Inc., Burlingame, CA, USA) using the microwave oven. The sections were then treated with 3% hydrogen peroxide, washed in phosphate-buffered saline with Tween-20 (PBST) (1X PBS, 0.1% Tween-20), and incubated with primary antibodies overnight at 4°C. Rabbit polyclonal antibody against HDAC1 from Abcam Inc. (Cambridge, MA, USA) was used at a 1:8,000 dilution, while rabbit polyclonal antibody against HDAC2 from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) was used at a 1:200 dilution. Sections were then washed and incubated with a goat anti-rabbit IgG conjugated with avidin-biotinylated horseradish peroxidase (Dako, Glostrup, Denmark), before final washing and incubation for 1 min with DAB substrate. Counterstaining was conducted using hematoxylin solution (Sigma-Aldrich, St. Louis, MO, USA).
Scoring of the tissue microarray
Each sample on the tissue microarray slides was scored based on the intensity of HDAC staining in the nuclei of the hepatocytes, as the presence of nuclear HDACs is relevant to their transcription-modifying activities. A score of 0 indicated no staining while a score of 1, 2 and 3 represented low, moderate and intense staining respectively.
Statistical analysis
The deceased and living status of patients was determined using the in-house hospital database as well as the registry of births and deaths, while the recurrence status was determined using the in-house hospital database. Results are expressed as mean ± SD for continuous variables or percentages for dichotomous variables.
As for descriptive statistics, overall frequencies and distribution of demographic and clinicopathological parameters by HDAC1 and HDAC2 expression status were calculated by either 2 by 2 or 2 by K tables with Chi-squared test and Fisher's exact test with the frequency weights (fweights) option to take into account duplicated observations of tumors and adjacent non-tumor. And two sample mean comparison test between biomarkers positive and negative statuses were made using ANCOVA test with the frequency weights (fweights) option to take into account duplicated observations of tumors and adjacent non-tumor. The comparisons between HDAC1 and HDAC2 expression status between tumor and non-tumor tissue were made using McNemar test. The presence of HDAC1 expression in tumor and non-tumor tissue was identified using the cut-off score value of ≥1, and no expression of HDAC1 in tumor and non-tumor score was identified using '0' and treated as a reference group. In addition, the levels of HDAC1 expression in tumor and non-tumor tissue were identified as low level of expression with a score cut-off 0 to 1 point, and identified as high level of expression with a score cut-off >2 points in tumor and non-tumor tissues. Similar comparisons were applied to the HDAC2 gene expression.
Associations of HDAC1 and HDAC2 gene expression with disease-specific mortality and recurrence were assessed using standard univariate and multivariate Cox proportional hazard model with robust standard error estimates to allow for intragroup correlation for tumor and adjacent non-tumor tissue measurements in the same patient. Kaplan-Meier's survival curves with log-rank tests for comparisons of survival curves were also constructed. The duration of follow-up in this study was 10 years with median survival time of 6.96 (95% CI, 4.85–7.46).
Standard Cox proportional hazards model, which assumes that the hazard ratio is constant over time, was applied initially. The proportionality assumption of the Cox regression model was assessed graphically and with the use of Schoenfeld residuals (21–22) using 'estat phtest' STATA command after fitting a model with stcox. However, violations of the Cox proportional hazards assumption were observed for both HDAC1 and HDAC2 gene expression in tumor and non-tumor tissue to predict mortality and disease-free survival (DFS), as discussed in the Results section below. Hence, the competing risk regression model was used to estimate the impact of HDAC1 and HDAC2 expression (as main covariates of interest) on the probability of mortality due to HCC, in which disease recurrence was treated as a competing event. Similarly, death due to HCC was treated as a competing event when estimating the impact of HDAC1 and HDAC2 expression on the probability of recurrence due to HCC. The results (effect sizes) are expressed as sub-hazard ratios (SHR). In addition, we adjusted for various demographic, clinicopathological factors, including tumor staging. The crude cumulative probability of mortality while accounting for the dependence of the cumulative probability function (CPF) on the hazards of other competing event (i.e., disease recurrence) was calculated. We then compared the resulting curves between two groups based on the absence vs. presence of HDAC2 expression in tumor and non-tumor tissue. With the same approach, we also compared the resulting curves between two groups based on low expression (score of 0–1) vs. high expression (score of ≥2) of HDAC1 in tumor and non-tumor tissue.
All statistical analyses were performed using Stata statistical software (version 11.1; StataCorp LP, College Station, TX, USA).
Cell lines
The human colon cancer cell line, HCT116, and human hepatocarcinoma cell lines, HEP3B, HEPG2 and PLC5, were purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA). The HCT116 cells were cultured in McCoy's 5A (Sigma-Aldrich) medium supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA). The HEP3B, HEPG2 and PLC5 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (Gibco). The cells were maintained in an incubator at 37°C in a 5% CO2 humidified atmosphere.
Transfection
Twenty-four hours before transfection, the cells were counted and plated. Twenty picomoles of Invitrogen's Stealth Select siRNA (a pool of three different sequences targeting the same gene) or universal control siRNA with matching percentage GC content and Lipofectamine RNAiMax reagent (Invitrogen) was used according to the manufacturer's instructions. The siRNA sequences along the HDAC1 mRNA sequence (5′–3′) were: aacgaauugccugugaggaa gaguu (siRNA 1A), gcaugacucauaauuugcugcucaa (siRNA 1B), and caguauucgauggccuguuugaguuc (siRNA 1C). The siRNA sequences along the HDAC2 mRNA sequence (5′–3′) were: ucu aacagucaaaggucaugcuaaa (siRNA 2A), gaagauccagacaagagaa uuucua (siRNA 2B), and ggugauggagauguaucaaccuagu (siRNA 2C).
Cell cycle analysis
Cells were washed in 1X PBS, trypsinized, and collected. Both the live adherent and dead floating cells were collected and pelleted at 2,500 rpm for 5 min at 4°C. They were washed in 1X PBS, fixed in cold 70% ethanol and stored in −20°C for overnight. The fixed cells were centrifuged at 2,500 rpm for 5 min at 4°C and washed once in 1X PBS. They were then resuspended in 500 µl propidium iodide (PI) staining solution (0.1% Triton X-100, 0.2 mg/ml RNaseA, 0.02 mg/ml PI, in 1X PBS) and incubated for 15 min at 37°C in the dark. The PI-stained cells were filtered through a 40-µm filter before passing through a Beckman Coulter Epics Altra at the Flow Cytometry Unit at the National University Medical Institute. Ten thousand cells were analyzed for each sample to generate a cell cycle profile. Analysis was carried out using WinMDI software.
WST-1 cell proliferation assay
To measure cell proliferation, the colorimetric assay was performed using WST-1 reagent (Roche Applied Science, Indianapolis, IN, USA). Twelve hours post-transfection, the cells were trypsinized, counted and replated at 500 cells/well in a 96-well plate. Twenty-four hours after replating, 10 µl of WST-1 reagent was added to each well containing 100 µl of media. The plate was returned to the incubator for 4 h before the absorbance was read at 460 nm using a spectrophotometer. A 'blank' well with only the media and WST-1 reagent but without cells was used as negative control.
Colony formation assay
Between 24 to 48 h after transfection or drug treatment, cells were harvested, counted and plated in triplicate at 1,000 cells/well in a 6-well plate. After 10 to 14 days, the wells were washed in 1X PBS and the colonies were stained with crystal violet solution. The plates were then scanned and the images were analyzed using the ImageJ (NIH) software to measure the number of colonies formed in each well.
Results
Expression of HDAC1 and HDAC2 in HCC tissues and correlation with clinicopathological parameters
Immunohistochemistry was performed on a tissue microarray constructed from 156 pairs of HCC tissues and their matched adjacent non-tumor tissues to examine the expression of HDAC1 and HDAC2. Scoring was based on the HDAC staining intensity of the hepatocyte nuclei, where HDACs are known to function, with 0 being undetectable and 3 being the most intensely stained. An IgG-isotype antibody was used as the negative control. The overall cumulative mortality rate of patients is shown in Fig. 1. The scoring intensities for HDAC1 and HDAC2 in tumor tissues are shown in Fig. 2, respectively, and the scoring intensities for the respective non-tumor tissues are shown in Fig. 3. The overall frequencies and distribution of demographic and clinicopathological parameters by status of HDAC1 and HDAC2 expression status are shown in Table 1A and B.
Figure 1.

Overall cumulative mortality rate among patients with HCC. Duration of follow-up was 10 years; median survival time was 6.96 (95% CI, 4.85–7.46). HCC, hepatocellular carcinoma.
Figure 2.

Immunohistochemical analysis of the hepatocellular carcinoma tissue microarray. (A) Representative examples of scoring performed for HDAC1 staining on tumor tissue. A tissue section with negative staining was given a score of 0 while scores 1–3 indicate the intensity of the nuclear staining of the hepatocytes. (B) Representative examples of scoring performed for HDAC2 staining on tumor tissue. A tissue section with negative staining was given a score of 0 while scores 1–3 indicate the intensity of nuclear staining of the hepatocytes. Scale bar, 50 µm. HDAC, histone deacetylase.
Figure 3.
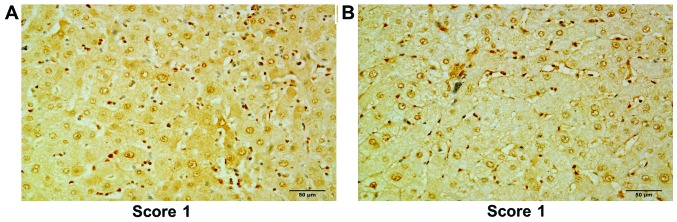
Immunohistochemical analysis of the hepatocellular carcinoma tissue microarray. (A) A representative example of scoring for HDAC1 staining on matched adjacent non-tumor tissue. (B) A representative example of scoring for HDAC2 staining on matched adjacent non-tumor tissue. Scale bar, 50 µm. HDAC, histone deacetylase.
Table I.
Overall frequencies and distribution of demographic and clinicopathological parameters by HDAC1 and HDAC2 expression status.
| A, HDAC1 status and patient parameters
| |||
|---|---|---|---|
| Variables | HDAC1(−) | HDAC1(+) | P-value |
| Age, mean (± SD) | 57.81 (±13.02) | 58.27 (±13.24) | 0.8219 |
| Gender, n (%) | |||
| Female | 4 (10.81) | 26 (21.85) | 0.137 |
| Male | 33 (89.19) | 93 (78.15) | |
| Race, n (%) | |||
| Non-Chinese | 29 (78.38) | 90 (76.27) | 0.791 |
| Chinese | 8 (21.62) | 28 (23.73) | |
| Smoking, n (%) | |||
| Non-smoker | 21 (58.33) | 60 (61.86) | 0.711 |
| Smoker | 15 (41.67) | 37 (38.14) | |
| Alcohol, n (%) | |||
| Non-drinker | 21 (58.33) | 62 (60.78) | 0.796 |
| Drinker | 15 (41.67) | 40 (39.22) | |
| HBV status, n (%) | |||
| Non-carrier | 13 (36.11) | 41 (35.04) | 0.907 |
| Carrier | 23 (63.89) | 76 (64.96) | |
| HCV status, n (%) | |||
| Non-carrier | 34 (91.89) | 112 (94.92) | 0.493 |
| Carrier | 3 (8.11) | 6 (5.08) | |
| Liver capsule, n (%) | |||
| Absence | 0 | 3 (4.55) | 0.570 |
| Presence | 22 (100) | 63 (95.45) | |
| Satellite nodules, n (%) | |||
| Absence | 35 (94.59) | 104 (87.39) | 0.364 |
| Presence | 2 (5.41) | 15 (12.61) | |
| Multiple lesions, n (%) | |||
| No | 29 (78.38) | 104 (87.39) | 0.177 |
| Yes | 8 (21.62) | 15 (12.61) | |
| Histological grade, n (%)(differentiation) | |||
| Well | 13 (38.24) | 18 (17.14) | 0.028 |
| Moderate | 16 (47.06) | 73 (69.52) | |
| Poor | 5 (14.71) | 14 (13.33) | |
| Lymphatic invasion, n (%) | |||
| No | 11 (91.67) | 57 (85.07) | 1.000 |
| Yes | 1 (8.33) | 10 (14.93) | |
| Vascular invasion, n (%) | |||
| No | 19 (65.52) | 67 (64.42) | 0.913 |
| Yes | 10 (34.48) | 37 (35.58) | |
| Cirrhosis, n (%) | |||
| No | 16 (43.24) | 63 (52.94) | 0.303 |
| Yes | 21 (56.76) | 56 (47.06) | |
| Tumor stage, n (%) | |||
| Stage 1 | 16 (44.44) | 60 (50.42) | 0.559 |
| Stage 2 | 14 (38.89) | 35 (29.41) | |
| Stage 3 and above | 6 (16.67) | 24 (20.17) | |
| AFP | 1,544.24 (±9,235.63) | 3,149.28 (±5,784.32) | 0.5184 |
| CEA | 3.60 (±2.88) | 2.75 (±2.54) | 0.2939 |
| B, HDAC2 status and patient parameters
| |||
|---|---|---|---|
| Variables | HDAC2(−) | HDAC2(+) | P-value |
| Age, mean (± SD) | 56.09 (±13.53) | 58.69 (±12.97) | 0.961 |
| Gender, n (%) | |||
| Female | 1 (10) | 29 (19.86) | 0.688 |
| Male | 9 (90) | 117 (80.14) | |
| Race, n (%) | |||
| Non-Chinese | 8 (80) | 111 (76.55) | 1.000 |
| Chinese | 2 (20) | 34 (23.45) | |
| Smoking, n (%) | |||
| Non-smoker | 6 (60) | 75 (60.98) | 1.000 |
| Smoker | 4 (40) | 48 (39.02) | |
| Alcohol, n (%) | |||
| Non-drinker | 7 (70) | 76 (59.38) | 0.740 |
| Drinker | 3 (30) | 52 (40.63) | |
| HBV status, n (%) | |||
| Non-carrier | 1 (10) | 53 (37.06) | 0.099 |
| Carrier | 9 (90) | 90 (62.94) | |
| HCV status, n (%) | |||
| Non-carrier | 10 (100) | 136 (93.79) | 1.000 |
| Carrier | 0 | 9 (6.21) | |
| Liver capsule, n (%) | |||
| Absence | 1 (33.33) | 2 (2.35) | 0.100 |
| Presence | 2 (66.67) | 83 (97.65) | |
| Satellite nodules, n (%) | |||
| Absence | 10 (100) | 129 (88.36) | 0.602 |
| Presence | 0 | 17 (11.64) | |
| Multiple lesions, n (%) | |||
| No | 8 (80) | 125 (85.62) | 0.643 |
| Yes | 2 (20) | 21 (14.38) | |
| Histological grade, n (%)(differentiation) | |||
| Well | 3 (33.33) | 28 (21.54) | 0.222 |
| Moderate | 6 (66.67) | 83 (63.85) | |
| Poor | 0 | 19 (14.62) | |
| Lymphatic invasion,n (%) | |||
| No | 2 (100) | 66 (85.71) | 1.000 |
| Yes | 0 | 11 (14.29) | |
| Vascular invasion, n (%) | |||
| No | 4 (57.14) | 82 (65.08) | |
| Yes | 3 (42.86) | 44 (34.92) | |
| Cirrhosis, n (%) | |||
| No | 3 (30) | 76 (52.05) | 0.207 |
| Yes | 7 (70) | 70 (47.95) | |
| Tumor stage, n (%) | |||
| Stage 1 | 4 (40) | 72 (49.66) | 0.416 |
| Stage 2 | 5 (50) | 44 (30.34) | |
| Stage 3 and above | 1 (10) | 29 (20) | |
| AFP | 1,372.45 (±2,933.55) | 2,862 (±9,098.68) | 0.4536 |
| CEA | 2.03 (±1.24) | 3.42 (±3.44) | 0.4292 |
HDAC, histone deacetylase.
Overall, we observed that the levels of both HDAC1 and HDAC2 in tumor tissues were significantly higher than those noted in adjacent non-tumor tissues (P<0.0001, Table II). In addition, the levels of HDAC1 and HDAC2 expression (i.e., expression level ≥2) were significantly high in the tumor tissues when compared to the adjacent non-tumor tissues (P<0.0001 for both HDAC1 and HDAC2 by McNemar test) (Table II). HDAC1 expression in tumor tissues (but not HDAC2) was higher in the moderately and poorly differentiated tumors compared to that in the well-differentiated tumors (P=0.028).
Table II.
Distribution of biomarkers according to tumor and adjacent non-tumor tissues.
| Variables | HDAC1 | P-value | HDAC2 | P-value |
|---|---|---|---|---|
| Overall, n (%) | ||||
| No | 139 (44.55) | 0.0791 | 69 (22.12) | <0.001 |
| Yes | 173 (55.45) | 243 (77.88) | ||
| Expression status | ||||
| (+) in T | 119 (76.28) | <0.0001 | 146 (93.59) | <0.0001 |
| (+) in NT | 54 (34.62) | 97 (62.18) | ||
| Scores in T | ||||
| 0–1 | 88 (56.41) | <0.0001 | 60 (38.46) | <0.008 |
| ≥2 | 68 (43.59) | 96 (61.54) | ||
| Scores in NT | ||||
| 0–1 | 142 (91.03) | 136 (87.18) | ||
| ≥2 | 14 (8.97) | 20 (12.82) | ||
T, tumor tissues; NT, non-tumor tissues; HDAC, histone deacetylase.
Prognostic value of HDAC1 and HDAC2 and competing risk regression model
The competing risk regression method was used to estimate the impact of HDAC1 and HDAC2 expression (as main covariates of interest) on mortality and disease recurrence. The final fitted competing risk regression model showed that the presence of HDAC2 expression (score ≥1) in tumor tissues is a significant independent predictor of mortality after adjusting for other significant risk factors while taking into account disease recurrence as the other competing risk event (Table III and Fig. 4A). The final adjusted SHR estimate was 9.24 (95% CI, 1.76–48.43; P=0.009) (Table III). The final adjusted SHR in the final competing risk model was adjusted for other clinically important and statistically significant variables in the univariate testing. These adjusting variables were age at diagnosis, tumor stage, presence of cirrhosis, presence of multiple lesions, the presence of vascular invasion, and presence of HDAC1 expression in tumor and non-tumor tissues.
Table III.
Adjusted hazard ratio estimates and SHR estimates of HDAC1 and HDAC2 gene expression in the prediction of cancer-specific mortality among patients with HCC by final fitted Cox proportional hazard model and final fitted competing risk model.
| Variables | Multivariate Cox regression (final fitted model)
|
Multivariate competing risk regression (final fitted model)
|
||||
|---|---|---|---|---|---|---|
| Adjusted HR | 95% CI | P-value | Adjusted SHR | 95% CI | P-value | |
| Tumor stage | ||||||
| Stage I | 1.0 | – | – | – | – | – |
| Stage II | 2.15 | 0.83–5.60 | 0.117 | 0.52 | 0.14–1.91 | 0.328 |
| Stage ≥III | 5.67 | 1.77–18.13 | 0.003 | 0.37 | 0.04–3.45 | 0.379 |
| Cirrhosis | ||||||
| Absence | 1.0 | – | – | 1.0 | – | – |
| Presence | 2.05 | 1.10–3.79 | 0.023 | 3.84 | 1.14–13.01 | 0.030 |
| Multiple lesions | ||||||
| Absence | 1.0 | – | – | 1.0 | – | – |
| Presence | 0.74 | 0.23–2.37 | 0.615 | 2.00 | 0.30–13.46 | 0.478 |
| Vascular invasion | ||||||
| Absence | 1.0 | – | – | 1.0 | – | – |
| Presence | 1.69 | 0.72–3.95 | 0.227 | 1.83 | 0.47–7.08 | 0.380 |
| Age at diagnosis | 1.01 | 0.99–1.04 | 0.282 | 1.03 | 0.98–1.07 | 0.220 |
| HDAC1 gene expression status | ||||||
| HDAC1(−) (T and NT) | 1.0 | – | – | 1.0 | – | – |
| HDAC1(+) (NT) | 1.25 | 0.66–2.34 | 0.490 | 1.65 | 0.44–6.16 | 0.453 |
| HDAC1(+) (T) | 1.23 | 0.60–2.52 | 0.570 | 0.52 | 0.12–2.27 | 0.382 |
| HDAC2 gene expression status | ||||||
| HDAC2(−) (T and NT) | 1.0 | – | – | 1.0 | – | – |
| HDAC2(+) (NT) | 1.77 | 0.83–3.79 | 0.141 | 6.04 | 0.66–55.57 | 0.112 |
| HDAC2(+) (T) | 1.43 | 0.63–3.23 | 0.393 | 9.24 | 1.76–48.43 | 0.009 |
T, tumor tissues; NT, non-tumor tissues; SHR, sub-hazard ratio; HDAC, histone deacetylase; HCC, hepatocellular carcinoma.
Figure 4.

Cancer-specific mortality based on HDAC expression. (A) The cumulative incidence for cancer-specific mortality comparing the presence and absence of HDAC2 (H2) gene expression in tumor and non-tumor tissues based on the competing risks regression model. (B) The cumulative incidence for cancer-specific mortality comparing levels of HDAC1 (H1) gene expression in tumor and non-tumor tissues based on the competing risks regression model. T, tumor tissues; NT, non-tumor tissues; HDAC, histone deacetylase.
The prediction of mortality by levels of HDAC1 expression (i.e., score 0–1 vs. ≥2) as a main covariate of interest showed a tendency towards higher risk of mortality in patients with high HDAC1 expression in tumors (Table IV and Fig. 4B), although the presence of HDAC1 expression alone was not found to be a significant predictor of mortality. The SHR estimate for high HDAC1 expression levels (score ≥2) in the tumor tissues was 2.48 with 95% CI, 0.88–7.00. However, this value did not reach statistically significant levels. As for the prediction of disease recurrence using multivariate competing risk regression approach, neither the presence of HDAC1 or HDAC2 (i.e., score ≥1), nor the levels of HDAC1 or HDAC2 expression (i.e., score 0–1 vs. score ≥2) prognosticated HCC recurrences in our population (Tables V and VI).
Table IV.
Adjusted hazard ratio estimates and SHR estimates of the HDAC1 and HDAC2 genes with high level expression in the prediction of cancer-specific mortality among patients with HCC by final fitted Cox proportional hazard model and final fitted competing risk model.
| Variables | Multivariate Cox regression (final fitted model)
|
Multivariate competing risk regression (final fitted model)
|
||||
|---|---|---|---|---|---|---|
| Adjusted HR | 95% CI | P-value | Adjusted SHR | 95% CI | P-value | |
| Tumor stage | ||||||
| Stage I | 1.00 | – | – | 1.00 | – | – |
| Stage II | 1.83 | 0.76–4.40 | 0.174 | 0.61 | 0.16–2.32 | 0.467 |
| Stage ≥III | 5.85 | 2.04–16.77 | 0.001 | 0.54 | 0.08–3.70 | 0.531 |
| Cirrhosis | ||||||
| Absence | 1.00 | – | – | 1.00 | – | – |
| Presence | 1.99 | 1.09–3.65 | 0.025 | 3.46 | 1.10–10.81 | 0.033 |
| Multiple lesions | ||||||
| Absence | 1.00 | – | – | 1.00 | – | – |
| Presence | 0.86 | 0.32–2.35 | 0.772 | 2.31 | 0.41–12.89 | 0.339 |
| Vascular invasion | ||||||
| Absence | 1.00 | – | – | 1.00 | – | – |
| Presence | 1.55 | 0.73–3.28 | 0.256 | 1.38 | 0.43–4.43 | 0.588 |
| Age at diagnosis | 1.02 | 0.99–1.04 | 0.167 | 1.03 | 0.98–1.08 | 0.223 |
| HDAC1 gene expression | ||||||
| HDAC1 score 0–1 (T and NT) | 1.00 | – | – | 1.00 | – | – |
| HDAC1 score ≥2 (NT) | 0.97 | 0.27–3.45 | 0.964 | 1.08 | 0.07–17.64 | 0.959 |
| HDAC1 score ≥2 (T) | 1.62 | 0.92–2.85 | 0.092 | 2.48 | 0.88–7.00 | 0.085 |
| HDAC2 gene expression | ||||||
| HDAC2 score 0–1 (T and NT) | 1.00 | – | – | 1.00 | – | – |
| HDAC2 score ≥2 in NT | 0.10 | 0.01–0.97 | 0.047 | 0.75 | 0.04–14.70 | 0.850 |
| HDAC2 score ≥2 in T | 0.76 | 0.48–1.22 | 0.261 | 0.65 | 0.23–1.82 | 0.411 |
T, tumor tissues; NT, non-tumor tissues; SHR, sub-hazard ratio; HDAC, histone deacetylase; HCC, hepatocellular carcinoma.
Table V.
Adjusted hazard ratio estimates and SHR estimates of HDAC1 and HDAC2 gene expression in the predictions of recurrence of HCC among patients with HCC by final fitted Cox proportional hazard model and final fitted competing risk model.
| Variables | Multivariate Cox regression (final fitted model)
|
Multivariate competing risk regression (final fitted model)
|
||||
|---|---|---|---|---|---|---|
| Adjusted HR | 95% CI | P-value | Adjusted SHR | 95% CI | P-value | |
| Tumor stage | ||||||
| Stage I | 1.0 | – | – | 1.0 | – | – |
| Stage II | 0.69 | 0.28–1.68 | 0.413 | 1.59 | 0.85–2.98 | 0.146 |
| Stage ≥III | 1.12 | 0.40–3.17 | 0.828 | 2.98 | 1.34–6.61 | 0.007 |
| Cirrhosis | ||||||
| Absence | 1.0 | – | – | 1.0 | – | – |
| Presence | 1.40 | 0.83–2.34 | 0.203 | 0.97 | 0.62–1.51 | 0.897 |
| Multiple lesions | ||||||
| Absence | 1.0 | – | – | 1.0 | – | – |
| Presence | 2.79 | 0.80–9.78 | 0.109 | 0.64 | 0.24–1.73 | 0.378 |
| Vascular invasion | ||||||
| Absence | 1.0 | – | – | 1.0 | – | – |
| Presence | 3.03 | 1.40–6.59 | 0.005 | 1.48 | 0.81–2.70 | 0.206 |
| Age at diagnosis | 0.98 | 0.96–1.00 | 0.024 | 1.01 | 0.99–1.02 | 0.305 |
| HDAC1 gene expression | ||||||
| HDAC1 score 0–1 (T and NT) | 1.0 | – | – | 1.0 | – | – |
| HDAC1 score ≥2 (NT) | 1.35 | 0.75–2.42 | 0.323 | 0.98 | 0.60–1.59 | 0.921 |
| HDAC1 score ≥2 (T) | 1.35 | 0.76–2.37 | 0.302 | 1.24 | 0.74–2.08 | 0.404 |
| HDAC2 gene expression | ||||||
| HDAC2 score 0–1 (T and NT) | 1.0 | – | – | 1.0 | – | – |
| HDAC2 score ≥2 (NT) | 1.21 | 0.67–2.20 | 0.529 | 1.37 | 0.87–2.17 | 0.179 |
| HDAC2 score ≥2 (T) | 0.94 | 0.51–1.76 | 0.854 | 1.05 | 0.61–1.79 | 0.858 |
T, tumor tissues; NT, non-tumor tissues; SHR, sub-hazard ratio; HDAC, histone deacetylase; HCC, hepatocellular carcinoma.
Table VI.
Adjusted hazard ratio estimates and SHR estimates of HDAC1 and HDAC2 genes with high-level expression in the predictions of recurrence of HCC among patients with HCC by final fitted Cox proportional hazard model and final fitted competing risk model.
| Variables | Multivariate Cox regression (final fitted model)
|
Multivariate competing risk regression (final fitted model)
|
||||
|---|---|---|---|---|---|---|
| Adjusted HR | 95% CI | P-value | Adjusted SHR | 95% CI | P-value | |
| Tumor stage | ||||||
| Stage I | 1.00 | – | – | 1.00 | – | – |
| Stage II | 0.73 | 0.30–1.78 | 0.483 | 1.58 | 0.84–2.96 | 0.156 |
| Stage ≥III | 1.17 | 0.42–3.27 | 0.762 | 3.09 | 1.39–6.87 | 0.006 |
| Cirrhosis | ||||||
| Absence | 1.00 | – | – | 1.00 | – | – |
| Presence | 1.41 | 0.84–2.36 | 0.194 | 0.97 | 0.62–1.50 | 0.875 |
| Multiple lesion | ||||||
| Absence | 1.00 | – | – | 1.00 | – | – |
| Presence | 2.88 | 0.84–9.91 | 0.093 | 0.65 | 0.24–1.73 | 0.382 |
| Vascular invasion | ||||||
| Absence | 1.00 | – | – | 1.00 | – | – |
| Presence | 2.71 | 1.23–5.93 | 0.013 | 1.45 | 0.79–2.64 | 0.231 |
| Age at diagnosis | 0.98 | 0.96–1.00 | 0.060 | 1.01 | 0.99–1.03 | 0.277 |
| HDAC1 gene expression | ||||||
| HDAC1 score 0–1 (T and NT) | 1.00 | – | – | 1.00 | – | – |
| HDAC1 score ≥2 (NT) | 0.77 | 0.43–1.41 | 0.401 | 0.86 | 0.45–1.64 | 0.652 |
| HDAC1 score ≥2 (T) | 1.02 | 0.61–1.69 | 0.944 | 0.92 | 0.60–1.41 | 0.696 |
| HDAC2 gene expression | ||||||
| HDAC2 score 0–1 (T and NT) | 1.00 | – | – | 1.00 | – | – |
| HDAC2 score ≥2 (NT) | 0.71 | 0.43–1.18 | 0.185 | 1.01 | 0.56–1.82 | 0.982 |
| HDAC2 score ≥2 (T) | 0.87 | 0.58–1.32 | 0.516 | 0.96 | 0.69–1.33 | 0.795 |
T, tumor tissues; NT, non-tumor tissues; SHR, sub-hazard ratio; HDAC, histone deacetylase; HCC, hepatocellular carcinoma.
There was no significant interaction between HDAC1 and HDAC2 in prediction of both cancer recurrence and cancer-specific mortality in this study when HDAC1 and HDAC2 were treated as either presence or absence categories or different levels of expression in the competing risk model.
Combined knockdown of HDAC1 and HDAC2 increases cell death and reduces cell proliferation and colony formation
The expression of HDAC1 and HDAC2 was knocked down by siRNAs both individually and in combination in three liver cancer cell lines, HEP3B (Fig. 5A and B), HEPG2 (Fig. 6A), PLC5 (Fig. 6C) and a colorectal cancer cell line, HCT116 (Fig. 6E). To investigate the effects of HDAC1 and HDAC2 on cell proliferation, WST-1 assay was performed on HEP3B cells treated with HDAC1 and HDAC2 siRNAs in combination or individually, and compared with scrambled siRNA-treated (Scr) and untransfected control cells (Ctrl). Silencing of both HDAC1 and HDAC2 in HEP3B cells significantly reduced cell growth compared to the controls. However, knockdown of either HDAC1 or HDAC2 alone did not affect cell proliferation (Fig. 5C). In addition, colony formation was reduced when both HDAC1 and HDAC2 were knocked down in HEP3B cells, but not when HDAC1 and HDAC2 were silenced individually (Fig. 5D). Similarly, significant reduction in colony formation was only observed when combined knockdown of HDAC1 and HDAC2 was performed in the HEPG2 (Fig. 6B), PLC5 (Fig. 6D) and HCT116 (Fig. 6F) cell lines.
Figure 5.

Effects of siRNA knockdown of HDAC1 and/or HDAC2 on HDAC activity in HEP3B cells. Cells were transfected with HDAC1 siRNA (H1), or HDAC2 siRNA (H2), or both (H1+2), or non-silencing control siRNA (Scr) for 72 h. (A) Cells were harvested and RNA was extracted and used for quantitative real time RT-PCR, carried out in replicates. The sequences specific for HDAC1 (1A and 1B) resulted in reduced RNA expression of HDAC1 compared to the non-silencing control (Scr) but not HDAC2, and the sequences specific for HDAC2 (2A, 2B, and 2C) resulted in reduced RNA expression of HDAC2 compared to the non-silencing control (Scr) but not HDAC1. (B) Cells transfected with a cocktail of HDAC1 siRNAs and/or HDAC2 siRNAs were harvested for protein, and western blot analysis using HDAC1- and HDAC2-specific antibodies was performed. GAPDH was used as the loading control. (C) WST-1 assay showed that combined knockdown of HDAC1 and HDAC2 reduced the cell growth over time. HEP3B cells were transfected with HDAC1 siRNA (H1), or HDAC2 siRNA (H2), or both (H1+2), or non-silencing control siRNA (Scr) or untransfected (Ctrl). (D) Left panel: Quantification of colony formation. The average number of colonies in each of the triplicate wells was plotted against the treatment. Error bars indicate standard deviations. Right panel: Representative images of the crystal violet-stained colonies. The combined knockdown of both HDAC1 and HDAC2 reduced colony formation. Cells were plated at a low density for 10 days and colonies were stained with crystal violet before imaging. HDAC, histone deacetylase.
Figure 6.

HDAC1 and/or HDAC2 knockdown. Western blot analysis shows the knockdown of protein expression of HDAC1 and/or HDAC2 in (A) HEPG2, (C) PLC5 and (E) HCT116 cells. Cells were transfected with HDAC1 siRNA (H1) or HDAC2 siRNA (H2), or both (H1+2), or non-silencing control siRNA (Scr). After 72 h, cells were harvested for protein, and western blot analysis using HDAC1- and HDAC2-specific antibodies was carried out. GAPDH was used as a loading control. Quantification of colony formation in (B) HEPG2, (D) PLC5 and (F) HCT116 after HDAC1 and/or HDAC2 were knocked down in the cells. The average number of colonies in each of the triplicate wells was plotted against the treatment. Error bars showed standard deviations. The knockdown of both HDAC1 and HDAC2 together was required to reduce colony formation. HDAC, histone deacetylase.
The effects of HDAC1 and HDAC2 on cell death was assessed by PI staining of fixed HEP3B cells treated with HDAC1 and HDAC2 siRNAs and control siRNA as above. Flow cytometric analysis showed that combined knockdown of both HDAC1 and HDAC2 significantly increased the sub-G1 fraction by 22.5% at 96 h post-transfection, compared to ~2% in the scrambled siRNA and untreated controls, indicating increased cell death (Fig. 7A and B). In contrast, knockdown of either HDAC1 or HDAC2 alone did not significantly affect cell death. The expression of various proteins involved in apoptosis was investigated by western blotting (Fig. 7C). Consistent with the flow cytometric data on cell death, cleavage of caspase-3 as well as its substrate PARP were observed in cells in which both HDAC1 and HDAC2 were knocked down up to 120 h post-transfection, but not in cells treated with either HDAC1 or HDAC2 siRNA. Taken together, our data suggest that the combined targeting of HDAC1 and HDAC2 in hepatocellular and colon cancer cells is more effective in reducing their growth potential and inducing cell death than selective targeting of either HDAC.
Figure 7.

Analysis of cell death in HEP3B cells after treatment with HDAC1 and/or HDAC2 siRNA. (A) Cell cycle analysis at 96 h after siRNA treatment. The combined knockdown of both HDAC1 and HDAC2 increased the percentage of cells in the subG1 and reduced the percentage of cells in the G1 phase of the cell cycle, indicating increased cell death. Representative flow diagrams of PI-stained cells are shown. (B) The percentages of cells in each phase of the cell cycle were quantified at 96 h after siRNA treatment. The graph shown is representative of at least three experiments. (C) Increase in apoptotic proteins after HDAC1 and HDAC2 knockdown. Expression of apoptotic proteins in HEP3B was detected by western blot analysis at 96 and 120 h after transfection with siRNA against HDAC1 (H1) or HDAC2 (H2) or both (1+2) or non-silencing control (Scr) or untransfected control (ctrl). There was cleavage of caspase-3 (as indicated by the white arrow) as well as PARP (as indicated by the red arrow). β-actin was used as the loading control. HDAC, histone deacetylase; PI, propidium iodide.
Discussion
In the Singapore cohort of 156 patients, we found that the expression levels of HDAC1 and HDAC2 in the tumor tissues were significantly higher than those in matched adjacent non-tumor tissues, as reported in other studies (13,23–25). HDAC1 expression was also higher in moderately and poorly differentiated tumors compared to well-differentiated tumors. Our data are consistent with previous observations whereby high tumor HDAC1 was found to be associated with poorer differentiation and invasion into the portal vein (26). Multivariate analysis using the competing risk regression model was performed for our cohort. The standard Cox proportional hazard model was not used as it found to be inappropriate for our analysis - violation of the Cox proportional hazards assumption was noted when we applied the presence or absence of HDAC1 and HDAC2 gene expression in tumor and non-tumor tissues to predict mortality and DFS (Fig. 8A). A similar violation was found when we applied the high and low level of HDAC1 and HDAC2 gene expression (i.e., 0–1 vs. ≥2) in tumor and non-tumor tissues to predict mortality and DFS (Fig. 8B). The competing risk regression model revealed that high expression of HDAC1 in tumor tissues (score ≥2) independently predicted cancer-specific mortality. Our findings are consistent with Rikimaru et al, who showed that high HDAC1 expression was associated with poorer survival in HCC patients after hepatic resection (26).
Figure 8.

Kaplan-Meier cumulative incidence estimates of mortality by HDAC expression. (A) Comparison of Kaplan-Meier cumulative incidence estimates of mortality by HDAC2 (H2) expression status in tumor and non-tumor tissues via log-rank test. (B) Comparison of Kaplan-Meier cumulative incidence estimates of mortality by levels of HDAC1 (H1) expression in tumor and non-tumor tissues via log-rank test. T, tumor tissues; NT, non-tumor tissues; HDAC, histone deacetylase.
Recent evidence suggests that HDAC2 promotes malignant progression in HCC by activating Akt signaling and epithelial-mesenchymal transformation (23). Consistent with the roles of HDAC2 in cancer progression, we found that the presence of HDAC2 in tumor tissues (score ≥1) was an independent predictor of cancer-specific mortality. This is in agreement with other studies which demonstrate a correlation between high HDAC2 expression and shorter overall survival in several cancers including HCC (23,24), oral cancer (27), gastric cancer (28) and colorectal cancer (14). In addition, HDAC2 presence in adjacent non-tumor hepatic tissues was also identified by the competing risk regression model as a potential predictor of poorer survival, although this did not reach levels of significance (P=0.112).
Importantly, we also found a significant correlation between HDAC1 expression in adjacent non-tumor hepatic tissues with the presence of satellite nodules and multiple lesions, suggesting that HDAC1 activation in the adjacent tumor environment may contribute to tumor spread. With respect to HDAC2, its presence in adjacent non-tumor hepatic tissues was significantly associated with smoking. Previous observations have implicated smoking as a risk factor in the development of HCC (29), while HDAC inhibition was shown to reduce the risk of smoking-related cancers such as head and neck cancers (30). It is thus possible that HDAC2 upregulation occurs as an early event in smoking-related hepatocellular transformation.
Our in vitro studies showed that combined knockdown of HDAC1 and HDAC2 (but not knockdown of either alone) reduced cell proliferation and survival in HCC and colon cancer cell lines. The requirement for both HDAC1 and HDAC2 for cell proliferation was previously observed in B-cell development, whereby HDAC1 and HDAC2 act together to promote G1 to S phase progression by inhibiting the transcription of cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p57Kip2 (31). In HCC, HDAC2 was shown to be important for oncogenic potential and inhibits the transcription of p21WAF1/Cip1 by binding to Sp1-binding site enriched proximal region of the p21WAF1/Cip1 promoter (32). In addition, a positive feedback mechanism that upregulates HDAC2 expression in response to growth factors via the PI3 kinase/mTORC1/NF-κBp50 signaling was also identified in HCC cells (23). In these cells, increased HDAC2 activity in turn was found to be required for sustaining elevated PI3 kinase/Akt and mTOR signaling to promote cancer cell growth and survival (23). Notably, both HDAC1 and HDAC2 have been found together in association with transcriptional complexes that regulate oncogenic processes, such as the ZEB1/HDAC transcription complex which represses E-cadherin expression and promotes migration of pancreatic cancer cells (33). Our clinical and in vitro data suggest that HDAC1 and HDAC2 also have independent functions that contribute to cancer progression, hence targeting both HDACs could be a useful therapeutic consideration to reduce cancer spread and improve the mortality rate.
Acknowledgments
The authors thank Assistant Professor Yaw Chyn Lim for her help in taking the bright field images and Mrs. Tamilarasi Jegadeesan for her support in the laboratory logistics.
References
- 1.Ito K, Adcock IM. Histone acetylation and histone deacetylation. Mol Biotechnol. 2002;20:99–106. doi: 10.1385/MB:20:1:099. [DOI] [PubMed] [Google Scholar]
- 2.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 3.Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnstone RW. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 6.Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harms KL, Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67:3145–3152. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 8.Lei WW, Zhang KH, Pan XC, Wang DM, Hu Y, Yang YN, Song JG. Histone deacetylase 1 and 2 differentially regulate apoptosis by opposing effects on extracellular signal-regulated kinase 1/2. Cell Death Dis. 2010;1:e44. doi: 10.1038/cddis.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–176. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 10.Ramsey MR, He L, Forster N, Ory B, Ellisen LW. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 2011;71:4373–4379. doi: 10.1158/0008-5472.CAN-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buurman R, Gürlevik E, Schäffer V, Eilers M, Sandbothe M, Kreipe H, Wilkens L, Schlegelberger B, Kühnel F, Skawran B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology. 2012;143:811–820. e1–e15. doi: 10.1053/j.gastro.2012.05.033. [DOI] [PubMed] [Google Scholar]
- 12.Fritzsche FR, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8:381. doi: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98:604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weichert W, Röske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14:1669–1677. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 15.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 16.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 17.Lau WY. The history of liver surgery. J R Coll Surg Edinb. 1997;42:303–309. [PubMed] [Google Scholar]
- 18.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. SHARP Investigators Study Group: Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 19.Portolani N, Coniglio A, Ghidoni S, Giovanelli M, Benetti A, Tiberio GA, Giulini SM. Early and late recurrence after liver resection for hepatocellular carcinoma: Prognostic and therapeutic implications. Ann Surg. 2006;243:229–235. doi: 10.1097/01.sla.0000197706.21803.a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahbari NN, Koch M, Schmidt T, Motschall E, Bruckner T, Weidmann K, Mehrabi A, Büchler MW, Weitz J. Meta- analysis of the clamp-crushing technique for transection of the parenchyma in elective hepatic resection: Back to where we started? Ann Surg Oncol. 2009;16:630–639. doi: 10.1245/s10434-008-0266-7. [DOI] [PubMed] [Google Scholar]
- 21.Barlow WE, Prentice RL. Residuals for relative risk regression. Biometrika. 1988;75:65–74. doi: 10.1093/biomet/75.1.65. [DOI] [Google Scholar]
- 22.Fisher LD, Lin DY. Time-dependent covariates in the Cox proportional-hazards regression model. Annu Rev Public Health. 1999;20:145–157. doi: 10.1146/annurev.publhealth.20.1.145. [DOI] [PubMed] [Google Scholar]
- 23.Noh JH, Bae HJ, Eun JW, Shen Q, Park SJ, Kim HS, Nam B, Shin WC, Lee EK, Lee K, et al. HDAC2 provides a critical support to malignant progression of hepatocellular carcinoma through feedback control of mTORC1 and AKT. Cancer Res. 2014;74:1728–1738. doi: 10.1158/0008-5472.CAN-13-2109. [DOI] [PubMed] [Google Scholar]
- 24.Quint K, Agaimy A, Di Fazio P, Montalbano R, Steindorf C, Jung R, Hellerbrand C, Hartmann A, Sitter H, Neureiter D, et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011;459:129–139. doi: 10.1007/s00428-011-1103-0. [DOI] [PubMed] [Google Scholar]
- 25.Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY, et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS. 2005;113:264–268. doi: 10.1111/j.1600-0463.2005.apm_04.x. [DOI] [PubMed] [Google Scholar]
- 26.Rikimaru T, Taketomi A, Yamashita Y, Shirabe K, Hamatsu T, Shimada M, Maehara Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology. 2007;72:69–74. doi: 10.1159/000111106. [DOI] [PubMed] [Google Scholar]
- 27.Chang HH, Chiang CP, Hung HC, Lin CY, Deng YT, Kuo MY. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009;45:610–614. doi: 10.1016/j.oraloncology.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Weichert W, Röske A, Gekeler V, Beckers T, Ebert MP, Pross M, Dietel M, Denkert C, Röcken C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008;9:139–148. doi: 10.1016/S1470-2045(08)70004-4. [DOI] [PubMed] [Google Scholar]
- 29.Koh WP, Robien K, Wang R, Govindarajan S, Yuan JM, Yu MC. Smoking as an independent risk factor for hepatocellular carcinoma: The Singapore Chinese Health Study. Br J Cancer. 2011;105:1430–1435. doi: 10.1038/bjc.2011.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang H, Gillespie TW, Goodman M, Brodie SA, Brandes M, Ribeiro M, Ramalingam SS, Shin DM, Khuri FR, Brandes JC. Long-term use of valproic acid in US veterans is associated with a reduced risk of smoking-related cases of head and neck cancer. Cancer. 2014;120:1394–1400. doi: 10.1002/cncr.28479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C, Matthias P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010;24:455–469. doi: 10.1101/gad.552310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noh JH, Jung KH, Kim JK, Eun JW, Bae HJ, Xie HJ, Chang YG, Kim MG, Park WS, Lee JY, et al. Aberrant regulation of HDAC2 mediates proliferation of hepatocellular carcinoma cells by deregulating expression of G1/S cell cycle proteins. PLoS One. 2011;6:e28103. doi: 10.1371/journal.pone.0028103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, Friess H, Büchler M, Evert M, Lerch MM, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61:439–448. doi: 10.1136/gutjnl-2011-300060. [DOI] [PubMed] [Google Scholar]