

Abstract
IGF-binding protein-3 (IGFBP-3) has been shown to induce apoptosis in an insulin-like growth factor (IGF)-independent manner in various cell systems, however, the underlying molecular mechanisms remain unknown. In the present study, we showed that IGFBP-3 significantly enhanced interleukin-24 (IL-24)-induced cell death in prostate cancer (PC) cell lines in vitro. Both the addition of IGFBP-3 to cell medium or the enforced expression of IGFBP-3 in the PC cell line inhibited activation of mammalian target of rapamycin (mTOR). Downregulation of mTOR/S6K reduced Mcl-1 protein expression and consequently promoted sensitization to IL-24 treatment. Overexpression of Mcl-1 reduced the level of cleaved poly(ADP-ribose) polymerase (PARP) induced by IL-24 and IGFBP-3, suggesting that the IL-24-induced apop-tosis is realized by way of Mcl-1. We then showed that the combination of IL-24 and IGFBP-3 significantly suppressed PC tumor growth in vivo. We propose that the IGFBP-3 and IL-24 non-toxic mTOR inhibitors can be used as an adjuvant in the treatment of PC.
Keywords: prostate cancer, IGF-binding protein-3, mammalian target of rapamycin
Introduction
Insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) is one of the six characterized binding partners of IGF-I, and is the most abundant IGFBP found in circulation (1). However, growing evidence points to IGF-independent effects of IGFBP-3 on cell growth (2). IGFBP-3 has been shown to act as a primary growth inhibitor and can be induced by other growth-inhibitory (and apoptosis-inducing) agents such as transforming growth factor-β1 (TGF-β1), retinoic acid, tumor necrosis factor-α (TNF-α) and the tumor-suppressor gene p53 (3). Growing evidence also suggests that IGFBP-3 is involved in induction of apoptosis (4). Notably, IGFBP-3 promotes ceramide-induced apoptosis in a breast cancer cell line (5) and enhances both p53-dependent (6,7) and p53-independent (8) apoptosis. However, IGFBP-3 alone shows little effect on cell apoptosis, indicating that other proteins are necessary in this process.
Interleukin (IL)-24 was originally identified as melanoma differentiation-associated gene-7 (mda-7), which is a cytokine tumor suppressor located in a cluster on chromosome 1q32 encoding IL-10, -19 and -20 (9). Interleukin-24 (IL-24) protein expression is lost during the progression of melanoma tumors; virtually all metastatic melanomas lack IL-24 expression (10). IL-24 functions as a pro-Th1 cytokine in human peripheral blood mononuclear cells and induces secretion of IL-6, interferon (IFN)-γ and TNF-α (11). Numerous studies demonstrate potent antitumor functions of IL-24 in a broad spectrum of human cancers, with no effects seen on normal cells (12).
This tumor cell-specific growth-inhibitory effect has also been observed in multiple animal models in vivo and in human clinical trials. IL-24 exerts its antitumor function mainly through inhibition of the epidermal growth factor receptor (EGFR) and phosphatidylinositol 3-kinase (PI3K) signaling pathways, and the induction of expression of double-stranded RNA-activated protein kinase (PKR) in human non-small cell lung cancer (NSCLC) and breast cancer (13,14). The tumor suppressor activity of IL-24 is independent of the status of other tumor suppressor genes, such as p53, Rb, p16 or Ras (15,16). IL-24 regulates many proliferative control mechanisms in tumor cells and downregulates anti-apoptotic proteins (Bcl-2/Bcl-xL) and upregulate pro-apoptotic proteins (Bax and Bak); this effect was not seen in normal cells (17,18). IL-24 has been established as a promising therapeutic candidate with potent antitumor, antiangiogenic and cytokine activities. However, the precise molecular mechanisms and signaling pathways of IL-24 in melanoma suppression remain largely unknown.
The mammalian target of rapamycin (mTOR) is a highly conserved serine/threonine kinase that regulates cell growth, cell cycle progression and metabolism. The PI3K/AKT signaling pathway activates mTOR, which in turn directly phosphorylates ribosome protein S6 kinase 1 (S6K) and eIF4E-binding protein 1, both of which are important in control of protein translation initiation (19,20). S6K phosphorylates S6, which regulates the translation of 50 terminal oligopyrimi-dine mRNAs that encode ribosomal proteins and translational factors. 4EBP1 binds to and inhibits eIF4E, initiating cap-dependent translation. mTOR is constitutively activated in the development of various types of human cancers, including ovarian, pancreatic and lung carcinomas (21). Thus, mTOR signaling networks have emerged as attractive targets for novel anticancer therapies.
IGFBP-3 is differentially expressed across normal prostate tissue types (6) and may be important in the regulation of prostate cell survival. However, though IGFBP-3 is both antiproliferative and pro-apoptotic, the molecular mechanisms behind its actions have not been elucidated. In the present study, we found that IGFBP-3 selectively enhances IL-24-induced cytotoxicity in prostate cancer (PC) cells, yet has no effect on the survival of adenoma-derived cells, which are resistant to IL-24-induced cell death. This result combined with previous findings leads us to hypothesize that IGFBP-3 promotes apoptosis through regulation of survival pathways activated in response to the induction of apoptosis. This hypothesis potentially explains why IGFBP-3 does not cause apoptosis when added directly to cell cultures (6,8). Our present results show, for the first time, that IGFBP-3 inhibits mTOR activation in response to IL-24-induced apoptosis. Inhibition of mTOR activation is important in improving efficacy of both chemotherapy and radiotherapy (22). Therefore, we propose that through inhibition of the mTOR pro-survival pathway, IGFBP-3 coupled with IL-24 may be a potent adjuvant in a number of cancer treatment regimens.
Materials and methods
Reagents, cell lines and cell culture
LNCap, PC-3, P69 and HEK293 cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in the recommended growth medium (Invitrogen, Carlsbad, CA, USA) at 5% CO2, 37°C. HEK293 cells transfected with, and stably expressing the pcDNA3 expression vector containing IGFBP-3 (IGFBP-3 cDNA was constructed by our laboratory). HEK293/IGFBP-3, and a vector control (HEK293/pcDNA3-control) were grown in 10% fetal bovine serum (FBS) Dulbecco's modified Eagle's medium (DMEM) supplemented with 200 mg/ml G418 for selection. Soluble recombinant human IGFBP-3 and IL-24 were purchased from PeproTech, USA.
Treatment with IL-24
Cells were seeded in triplicate flasks and grown under standard conditions until ~70% confluency. Cells were grown for 24 h in a serum-free medium (SFM) to remove IGFBP-3 from the serum and then grown for up to 24 h in SFM supplemented with or without IL-24 (0.2, 0.4 and 0.8 µM) to induce apoptosis.
Measurement of cell viability
Cell viability was measured by mitochondrial conversion of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) to a colored product. Following treatment with drugs, MTT was added. Cells were then solubilized in dimethylsulphoxide (DMSO). The amount of converted MTT was determined by measuring absorbance at 570 nm.
Plasmids and siRNAs
Full-length Mcl-1 or IGFBP-3 cDNA was cloned into the BamHI/XbaI sites of pcDNA3.1(+) (Invitrogen). S6K cDNA containing an N-terminal Myc (plasmid 26610) was purchased from Addgene (Cambridge, MA, USA). All constructs were verified by DNA sequencing. Mcl-1 (sc-35877), adenosine monophosphate-activated protein kinase (AMPK)α1/2 (sc-45312) and control (no. 1, sc-37007) siRNAs were purchased from Santa Cruz Biotechnology (Santa Cruz CA, USA). S6K (Hs_RPS6KB1_5) and control (1022076) siRNAs were purchased from Qiagen (Valencia, CA, USA), and mTOR (6381) and control (6568) siRNAs were purchased from Cell Signaling Technology (Beverly, MA, USA). Transfection experiments with plasmids and siRNAs were performed using Lipofectamine Plus and Lipofectamine 2000 (Invitrogen, UK) according to the manufacturer's instructions.
Colony formation assay
LNCap cells were transfected with an IGFBP-3 overexpression vector or Mcl-1 siRNAs for 24 h. The cells were reseeded in 60-mm plates and colony formation was monitored over the subsequent 10 days.
Treatment with IL-24 or IGFBP-3
Cells were seeded in duplicate flasks and grown under standard conditions until ~70% confluency, and then grown for 24 h in SFM to remove IGFBP-3 from the serum. The cells were then grown for up to 24 h in SFM supplemented with IL-24 (0.2–0.8 µM). The secreted proteins were harvested from human embryonic kidney HEK-(293) cells transfected with the IGFBP-3 pcDNA3 expression vector or from mock-transfected HEK293 cells. Upon reaching 70% confluency, cells were transfected using Lipofectamine 2000. After 6 h, the medium was replaced by SFM and the cells were harvested 48 h later. The IGFBP-3 content was adjusted to ~100 ng/ml by western blot analysis compared to IGFBP-3 protein standards (Upstate Biotechnology, USA).
Treatment with IL-24/IGF-IR antibody or anti-IGFBP-3 antibodies
Cells were first incubated in SFM for 24 h, and then in SFM plus an anti-IGF-IR antibody (Santa Cruz Biotechnology) or an anti-IGFBP-3 antiserum at a content level of 5 µg/ml for 24 h, followed by 0.8 µM IL-24 in the presence or absence of IGFBP-3 (GroPep, AUS). The cell yield and floating cell population were counted using a Neubauer counting chamber (VWR, UK).
Assessment of IGFBP-3
Subconfluent monolayers were grown in SFM for 24 h. The cells were then grown in SFM for 24 h, with or without IL-24. The media were then harvested, centrifuged to remove floating cells and stored at −70°C. The number of attached cells was quantified using a haemocytometer. Proteins from conditioned medium were concentrated using 'Microsep' 10 K centrifuge columns (Gelman Laboratory). The levels of IGFBP-3 were assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) immunoblotting and the IGFBP-3 protein was detected by anti-IGFBP-3 antiserum (polyclonal from Diagnostic System Laboratory, USA) using an enhanced chemiluminescence (ECL) system (KPL, USA).
Western blotting
Cell lysates and immunoprecipitates were separated by SDS-PAGE and transferred to nitrocellulose membranes, followed by immunoblotting with the specified primary antibodies and horseradish peroxidase-conjugated secondary antibodies. Immunoreactive bands were visualized with SuperSignal West Pico Chemiluminescent Substrates (Thermo Scientific Pierce, Rockford, IL, USA).
Generation of recombinant adenoviruses
Recombinant adenoviruses (pAd/CMV/IGFBP-3 or pAd/CMV/IL-24) contained IGFBP-3 or IL-24 between the cytomegalovirus (CMV) promoter and the polyadenylation signal (SV40pA). These adenoviral vectors were propagated in HEK293 cells (Invitrogen) using a Stratagene MBS mammalian transfection kit (La Jolla, CA, USA) with a modified calcium phosphate transfection protocol. The transfected cells were incubated at 37°C for 7 days, harvested and subjected to 4 cycles of freezing (dry ice and alcohol)/thawing in a 37°C water bath. The cell lysates were centrifuged at 12,000 × g and 4°C for 10 min; then the supernatant (primary virus stock) was transferred to a fresh screw-cap mini-centrifuge tube and stored at −80°C. Recombinant adenoviruses were further amplified using the same procedure; the cell lysates were centrifuged with cesium chloride step gradients at 60,000 × g and 4°C for 2 h, allowing the viruses to separate from the defective particles and empty capsids. The recovered virus bands were dialyzed 3 times against phosphate-buffered saline (PBS). The viruses Ad5.IGFBP-3 or Ad5.IL-24 were aliquoted in a buffer containing 10 mmol/l Tris-HCl (pH 8.1), 10 mmol/l MgCl2 and 10% v/v glycerol and then stored at −80°C.
Studies in vivo
Four-week-old athymic nude mice were obtained from the Animal Research Center, Shanghai Laboratory Animal Research Center, China. All procedures were performed in accordance with an animal protocol approved by the First Affiliated Hospital of Medical School, Xi'an Jiaotong University, and by the local Experimental Ethics Committee. All efforts were made to minimize animal suffering and reduce the number of animals used. Tumors were established by subcutaneous injection of 5×106 LNcap cells into the right flank of the mice. When a tumor grew to the size of ~50 mm3, the host mouse was randomly assigned to one of the 3 groups (each n=6): control (DMSO dissolved in 50 µl PBS and administered i.p. every other day), Ad5.IGFBP-3 (5×108 PFU in 50 µl of PBS, administered i.p. every other day) and Ad5.IGFBP-3+Ad5.IL-24 (5×108 PFU in 50 µl of PBS, administered i.p. once daily). Tumor size was measured every other day using calipers, and tumor volume (mm3) was estimated as length × width2/2. The mice were closely monitored for 24 days prior to euthanization and removal of tumors. Each tumor was stored at −80°C for further analysis.
Statistical analysis
All data represent the mean of 3 separate experiments. Each experiment was carried out in duplicate or triplicate parallel flasks. Each experiment was repeated 3 times, and results are presented as the mean of the 3 separate experiments. Statistical analysis was carried out using SPSS for Windows statistical software (release 10.0.5; SPSS, Inc., Chicago, IL, USA). Analysis of variance (ANOVA) was used to determine differences among means. Pair-wise comparisons were made using Tukey's post hoc test for multiple comparisons.
Results
LNCap and PC-3 lines are sensitive to IL-24-induced proliferation inhibition, yet not the P69 cell line
Differential sensitivity to IL-24 was observed in PC cell lines. Whereas the LNCap and PC-3 cell lines were sensitive to the inhibition of cell proliferation, the P69 cell line was not sensitive to the drug treatment. However, as experiments on the effects of IGFBP-3 were carried out in SFM, we initially sought to verify that the differential sensitivity to IL-24-induced cell proliferation inhibition was retained under serum-free conditions. Therefore, both LNCap, PC-3 and P69 cells were treated with IL-24 (0.2, 0.4 and 0.8 µM) for 24 h in SFM (Fig. 1A). In the 3 investigated tumorigenic cell lines, there was a significant decrease in attached cell yield and an associated increase in cell death [confirmed as apoptotic by poly(ADP-ribose) polymerase (PARP) cleavage, Fig. 1B and C] in the LNCap and PC-3 cell lines. In contrast, P69 cell line remained relatively resistant to IL-24-induced cell proliferation inhibition.
Figure 1.

Prostate cancer cell lines are sensitive to IL-24-induced apoptosis. (A) Cell proliferation 24 h after treatment with IL-24 (0.2, 0.4 or 0.8 µM). The results are expressed as mean ± SD (n=3). (B) Western blotting shows PARP and GAPDH (loading control) expression 24 h after treatment with IL-24 (0.8 µM). (C) Densitometry analysis from 3 independent western blot analyses of PARP (mean ± SD; n=3; **P<0.01, Student's t-test). IL-24, interleukin-24; PARP, poly(ADP-ribose) polymerase.
IGFBP-3 potentiates IL-24-induced cell proliferation inhibition in LNCap and PC-3 cells whereas not in the P69 cell line
As the pro-apoptotic IGFBP-3 protein improves the sensitivity of cancer cell lines to the induction of apoptosis (6,8), we investigated whether IGFBP-3 affected the response of cells to IL-24-induced cell proliferation inhibition. Cells were treated with 100 ng/ml IGFBP-3, which is a physiologically relevant dose (6) and equal to ~50 ng/106 cells. We investigated the exogenous IGFBP-3 protein to control in vitro. Protein was collected from the conditioned medium of HEK293 cells, which were transiently transfected to express IGFBP-3 protein. The secreted protein was harvested from the culture medium (referred to as IGFBP-3 medium, IGFBP3-M). The resulting concentrated conditioned medium contained ~100 ng/ml IGFBP-3 protein assessed by ELISA. These experiments were controlled by treatment of parallel cultures with concentrated media from mock-transfected HEK293 cells (referred to as mock media; MM). The results are shown in Fig. 2. Cancer cells were treated with IL-24 at a dose of 0.8 µM to induce moderate cell proliferation inhibition. For the PC cell lines, increasing the amount of IGFBP-3 in the media significantly enhanced cellular sensitivity to IL-24-induced cell proliferation inhibition (Fig. 2). However, the addition of IGFBP3-M to P69 had no effect on cell proliferation.
Figure 2.
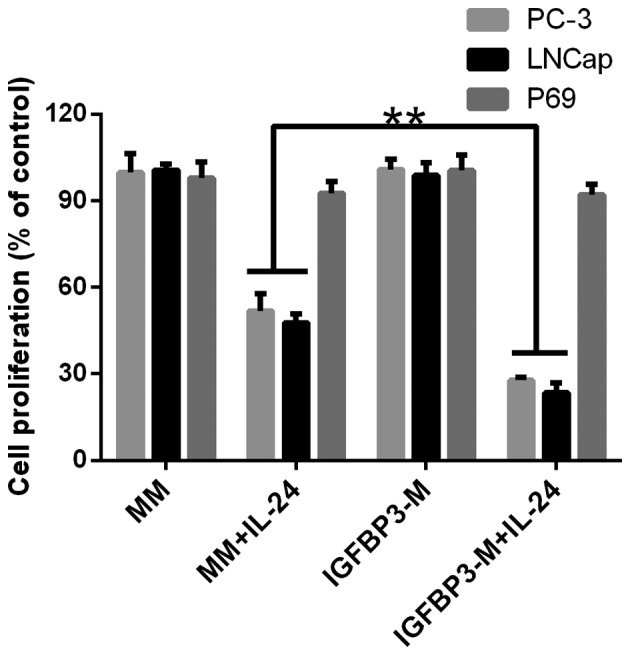
IGFBP-3 promotes IL-24-induced apoptosis in human prostate cancer cell lines. LNCap, PC-3 and P69 cells treated with 0.8 µM IL-24. Statistical difference determined by two-way ANOVA, **P<0.01. IGFBP-3, IGF-binding protein-3; IL-24, interleukin-24.
The IGF-1R blocking antibody αIR3 (5 µg/ml) failed to block IGFBP-3 potentiation of IL-24-induced cell proliferation inhibition in LNCap cell line, indicating that the cell proliferation decreasing observed was not due to the inhibition of IGF-dependent signaling pathways (Fig. 3). Furthermore, the potentiation of IGFBP-3 in IL-24-induced cell proliferation inhibition was completely blocked by the addition of a neutralizing IGFBP-3 antibody, confirming that IGFBP-3 promotes IL-24-induced cell proliferation inhibition.
Figure 3.
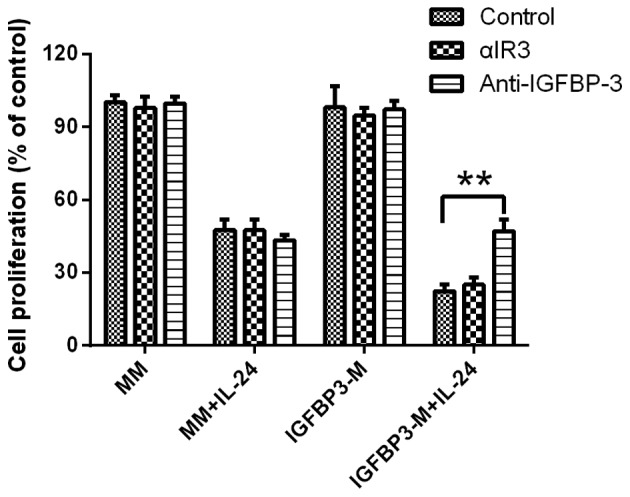
IGFBP-3 promotes IL-24-induced apoptosis in the presence of an IGF-1 receptor blocking antibody or IGFBP-3 antibody. Results shown represent the mean ± SD (n=3). Significant differences are denoted by **P<0.01. IGFBP-3, IGF-binding protein-3; IL-24, interleukin-24.
IGFBP-3 overexpression leads to inactivity of mTOR, which plays an important role in Mcl-1 downregulation
A recent study shows that PC cell lines express high levels of Mcl-1, and that suppression of Mcl-1 induces significant cell death (23). As expected, the downregulation of Mcl-1 with siRNA led to increased cell death and PARP protein cleavage in our experiments (Fig. 4A). Colony formation was also diminished in the presence of Mcl-1 siRNA compared with the control siRNA (Fig. 4B). To further clarify whether Mcl-1 plays an essential role in the survival of PC cells, we transfected Mcl-1 plasmids into cells treated with IGFBP3-M and IL-24 protein. The level of IGFBP3-M and IL-24-induced PARP cleavage were reduced upon overexpression of Mcl-1 (Fig. 4C and D). Our results indicate that downregulation of Mcl-1 is critical for the sensitizing effect of IGFBP-3 on IL-24-induced cell death.
Figure 4.

Mcl-1 is essential for survival of prostate cancer cells. (A) Survival of LNCap and PC-3 cells 24 (b) or 48 h (a) after transfection with siRNA targeting Mcl-1 or a control siRNA. (B) LNCap cells were transfected with siCon or Mcl-1 siRNA for 24 h. (C) LNCap cells were transfected with empty vector control (pcDNA3), Mcl-1 (pcDNA3/Mcl-1) or IGFBP3-M for 24 h, followed by treatment with 0.8 µM IL-24 for 24 h. (D) Densitometry analysis from 3 independent western blot analyses (mean ± SD; n=3; **P<0.01, Student's t-test). IGFBP-3, IGF-binding protein-3; IL-24, interleukin-24; PARP, poly(ADP-ribose) polymerase.
In the present study, we have shown that IGFBP-3 potentiates IL-24-induced apoptosis in PC cell lines. Adding to the former findings that IGFBP-3 enhanced apoptosis by suppressing certain survival signalling pathway. We then investigated the mechanism by which IGFBP-3 overexpression suppresses Mcl-1 protein. Initially, mTOR activity was assessed in cells treated with IGFBP3-M. IGFBP3-M suppressed mTOR activity, assessed on basis of weakened phosphorylation of mTOR and its effectors (S6K and S6) in LNCap cells (Fig. 5A). Since mTOR promotes cell survival through translational control of Mcl-1 (24), we determined the level of Mcl-1 protein in cells treated with mTOR siRNA. As expected, suppression of mTOR resulted in decreased expression of Mcl-1 protein and enhanced LNCap cell sensitivity to IL-24 (Fig. 5B and C). S6K as a downstream target of mTOR plays important roles in cell proliferation, protein translation and cell survival and is thus an effective target in cancer therapy (25). To ascertain whether S6K is involved in the regulation of Mcl-1 expression and consequent cell survival, we transfected the cells with S6K siRNA and then treated them with IL-24. S6K siRNA induced a decrease in Mcl-1 protein levels compared to the control siRNA (Fig. 6A). Moreover, knockdown of S6K enhanced IL-24-induced cell death (Fig. 6B and C). Overexpression of S6K alleviated IGFBP3-M-induced PARP cleavage (Fig. 6D). Based on these results, we propose that mTOR activity is suppressed upon IGFBP-3 overexpression and is critical for Mcl-1 downregulation.
Figure 5.

Overexpression of IGFBP-3 inhibits mTOR-mediated Mcl-1 downregulation. (A) LNCap cells were transfected with IGFBP-3 plasmid or control vector for 30 h. (B) LNCap cells were transfected with control or mTOR siRNA for 30 h. (C) LNCap cells were transfected with control or mTOR siRNA for 20 h, followed by treatment with 0.8 µM IL-24 for 24 h. Cell viability was measured using an MTT assay. The viability of the control siRNA (siCon)-treated group was set at 100%, and survival was calculated relative to the siCon. Cell viability data are shown as the mean of triplicates, and error bars reflect SD. *P<0.05 vs. the siCon/IL-24-treated group or simTOR-treated group. (D) The indicated protein levels were measured using western blot analysis. IGFBP-3, IGF-binding protein-3; IL-24, interleukin-24.
Figure 6.

The Mcl-1 decrease, induced by IGFBP-3 overexpression, is mediated by S6K downregulation. (A) LNCap cells were transfected with control or S6K siRNA for 30 h. (B and C) LNCap cells were transfected with control or S6K siRNA for 20 h, followed by treatment with 0.8 µM IL-24 for 24 h. The cell viability of the control siRNA (siCon)-treated group was set at 100%, and survival relative to the siCon is shown (B). (D) LNCap cells were transfected with increasing concentrations of Myc-tagged S6K, along with IGFBP-3 for 48 h. The indicated protein levels were measured using western blot analysis (A, C and D) Data shown are representative of at least two independent experiments. Cell viability data are shown as the mean of each group and error bars reflect SD. *P<0.05. IGFBP-3, IGF-binding protein-3; IL-24, interleukin-24; PARP, poly(ADP-ribose) polymerase.
Overexpression of IGFBP-3 leads to inhibition of mTOR through activation of AMPK
mTOR activity can be suppressed upon energy deprivation, as depleted ATP levels trigger AMPK activation (26). We further investigated whether AMPK is involved in the mTOR inhibition induced by IGFBP-3 overexpression. Treatment with IGFBP3-M stimulated the phosphorylation of AMPK (p-AMPK) and its substrate (acetyl-CoA carboxylase, p-ACC) in LNCap and PC-3 cells (Fig. 7A). Moreover, overexpression of IGFBP-3 led to a marked increase in the cellular ADP/ATP ratio, indicating a decrease in ATP content in both cell lines (Fig. 7B). To clarify the involvement of AMPK in IGFBP3-M-induced mTOR inactivity, cells overexpressing IGFBP-3 were transfected with AMPKα 1/2 siRNA. Downregulation of AMPK led to recovery of mTOR activity (Fig. 7C). These results suggest that IGFBP-3 overexpression is associated with ATP depletion, leading to AMPK-induced mTOR inhibition.
Figure 7.

IGFBP-3 overexpression leads to mTOR inhibition through AMPK. (A and B) LNCap cells were transfected with control, IGFBP-3 or Mcl-1 siRNA for 48 h. ADP/ATP ratios were determined using the EnzyLight ADP/ATP ratio assay kit. Data are expressed as the mean of 3 independent experiments and error bars reflect SD. (C) LNCap cells were transfected with control, IGFBP-3 or AMPKα1/2 siRNA for 48 h. The indicated protein levels were measured using western blot analysis (B and C) and blots shown are representative of 3 independent experiments. IGFBP-3, IGF-binding protein-3; IL-24, interleukin-24; AMPK, adenosine monophosphate-activated protein kinase.
IGFBP-3 and IL-24 significantly inhibit growth of human PC cells in vivo
To investigate the effects of IGFBP-3 and IL-24 on cell growth in vivo, LNcap cells were subcutaneously xenografted in nude mice. Tumor volume was reduced in the Ad5.IGFBP-3 group compared with the control group. After treatment with a combination of Ad5.IGFBP-3 and Ad5.IL-24, tumor size was significantly reduced compared with the control group (P<0.01) or the Ad5.IGFBP-3 group (P<0.01), indicating a higher suppression of tumor growth in vivo (Fig. 8A and B). Western blotting of tumor tissue lysates also revealed that the expression of PARP was significantly upregulated and mTOR was downregulated in the Ad5.IGFBP-3+Ad5.IL-24 group to a higher extent than in the Ad5.IGFBP-3 group (Fig. 8C). These results are consistent with our studies in vitro, providing further evidence that IL-24 potentiates the antitumor activity of IGBP-3 in vivo.
Figure 8.

Tumor growth and gene expression in vivo. (A) LNcap tumors were established subcutaneously in nude mice. A significant difference in tumor volume from the control is denoted by **P<0.01. (B) Representative animals and tumors are shown for each group. (C) Tumors from 3 groups were homogenized and subjected to western blot analysis to detect the expression of cleaved PARP and mTOR. GAPDH served as an internal control. PARP, poly(ADP-ribose) polymerase.
Discussion
IGF-binding protein-3 (IGFBP-3) is a pro-apoptotic protein previously shown to potentiate apoptotic cell death in a number of systems. In the present study, we showed that in human prostate cancer (PC) cells IGFBP-3 promotes IL-24-induced apoptosis. This important finding suggests that IGFBP-3 may be used to improve IL-24-induced apoptosis in cancer cells without affecting the survival of non-tumorigenic cells.
Apoptosis is induced by both extrinsic and intrinsic pathways initiated by the activation of death receptors and stress-inducing stimuli (27). Results from the present study contribute to the current literature demonstrating that IGFBP-3 enhances both extrinsic and intrinsic apoptotic pathways (4). Furthermore, IGFBP-3 is pro-apoptotic in several cell systems (breast, esophageal, colonic cancer cells), although its direct addition to cells will not induce apoptosis (7,8). These observations led us to hypothesize that instead of being an integral part of cell death signaling, IGFBP-3 instead modify the activity of pathways that modulate the cellular response to apoptosis. This hypothesis represents a more general mechanism by which IGFBP-3 is able to potentiate many death-inducing pathways.
The mTOR pathway plays multiple roles in cell growth, proliferation and survival. In the present study, we determined whether mTOR and its downstream factors are involved in IL-24 sensitization induced by IGFBP-3 overexpression. Unexpectedly, IGFBP-3 overexpression led to reduced levels of phosphorylated mTOR, as well as its downstream effector S6K. Downregulation of mTOR and S6K using siRNA made cells more sensitive to IL-24. Importantly, we showed that knockdown of either mTOR or S6K downregulates Mcl-1 protein levels. These results indicated that Mcl-1 is a downstream target of mTOR and S6K. Furthermore, Mcl-1 may be regulated at the translational level, as the main function of mTOR is to control translation via direct regulation of S6K and 4E-BP. This assumption is supported by several studies in the literature that Mcl-1 is a potential downstream target of eIF4E, which contains a unique G+C-rich 5′ untranslated region among the anti-apoptotic Bcl-2 members (24,28). In addition, inhibition of mTOR with rapamycin blocked the synthesis of Mcl-1 protein, but not that of Bcl-2 or Bcl-xL.
Surprisingly, overexpression of IGFBP-3 led to a marked increase in the ADP/ATP ratio within cells, indicating a decrease in ATP content. AMPK is a central cellular energy-sensing system that actively participates in the metabolism-cancer interaction via regulation of the mTOR pathway. AMPK activation directly phosphorylates and activates TSC2 by enhancing its GAP activity, leading to inhibition of mTOR signaling (29). Our findings confirm that overexpression of IGFBP-3 leads to AMPK activation and consequently mTOR inhibition. Downregulation of AMPK with AMPKα1/2 siRNA counteracted the inhibition of mTOR activity by IGFBP3-M. The present study, shows that overexpression of IGFBP-3 is associated with ATP depletion, leading to AMPK-induced mTOR inhibition. This finding highlights a novel underlying mechanism linking the IGFBP-3 and mTOR pathways. However, further research is necessary to clarify the exact mechanisms that link the overexpression of IGFBP-3 to ATP-depletion and subsequent activation of AMPK.
In summary, through inhibiting mTOR activation, IGFBP-3 increases the sensitivity of cancer cells to IL-24-induced apoptosis and may also be important in both the prevention of PC, and as a potential adjuvant for a number of other therapeutic regimens used in the treatment of metastatic prostate cancer.
Acknowledgments
The present study was supported by the National Science Foundation of China (grant nos. 30901500, 81072107 and 81372736), and the Science and Technology Program of Shaanxi Province (grant no. 2009JQ4002).
References
- 1.Gill ZP, Perks CM, Newcomb PV, Holly JM. Insulin-like growth factor-binding protein (IGFBP-3) predisposes breast cancer cells to programmed cell death in a non-IGF-dependent manner. J Biol Chem. 1997;272:25602–25607. doi: 10.1074/jbc.272.41.25602. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura M, Takakura M, Fujii R, Maida Y, Bono Y, Mizumoto Y, Zhang X, Kiyono T, Kyo S. The PRB-dependent FOXO1/IGFBP-1 axis is essential for progestin to inhibit endometrial epithelial growth. Cancer Lett. 2013;336:68–75. doi: 10.1016/j.canlet.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Hanafusa T, Shinji T, Shiraha H, Nouso K, Iwasaki Y, Yumoto E, Ono T, Koide N. Functional promoter upstream p53 regulatory sequence of IGFBP3 that is silenced by tumor specific methylation. BMC Cancer. 2005;5:9. doi: 10.1186/1471-2407-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baxter RC, Butt AJ, Schedlich LJ, Martin JL. Antiproliferative and pro-apoptotic activities of insulin-like growth factor-binding protein-3. Growth Horm IGF Res. 2000;10(10 Suppl A):S10–S11. doi: 10.1016/S1096-6374(00)90004-2. [DOI] [PubMed] [Google Scholar]
- 5.Ho L, Stojanovski A, Whetstone H, Wei QX, Mau E, Wunder JS, Alman B. Gli2 and p53 cooperate to regulate IGFBP-3-mediated chondrocyte apoptosis in the progression from benign to malignant cartilage tumors. Cancer Cell. 2009;16:126–136. doi: 10.1016/j.ccr.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Butt AJ, Williams AC. IGFBP-3 and apoptosis - a license to kill? Apoptosis. 2001;6:199–205. doi: 10.1023/A:1011388710719. [DOI] [PubMed] [Google Scholar]
- 7.Williams AC, Collard TJ, Perks CM, Newcomb P, Moorghen M, Holly JM, Paraskeva C. Increased p53-dependent apoptosis by the insulin-like growth factor binding protein IGFBP-3 in human colonic adenoma-derived cells. Cancer Res. 2000;60:22–27. [PubMed] [Google Scholar]
- 8.Hollowood AD, Lai T, Perks CM, Newcomb PV, Alderson D, Holly JM. IGFBP-3 prolongs the p53 response and enhances apoptosis following UV irradiation. Int J Cancer. 2000;88:336–341. doi: 10.1002/1097-0215(20001101)88:3<336::AID-IJC3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 9.Collard TJ, Guy M, Butt AJ, Perks CM, Holly JM, Paraskeva C, Williams AC. Transcriptional upregulation of the insulin-like growth factor binding protein IGFBP-3 by sodium butyrate increases IGF-independent apoptosis in human colonic adenoma-derived epithelial cells. Carcinogenesis. 2003;24:393–401. doi: 10.1093/carcin/24.3.393. [DOI] [PubMed] [Google Scholar]
- 10.Huang EY, Madireddi MT, Gopalkrishnan RV, Leszczyniecka M, Su Z, Lebedeva IV, Kang D, Jiang H, Lin JJ, Alexandre D, et al. Genomic structure, chromosomal localization and expression profile of a novel melanoma differentiation associated (mda-7) gene with cancer specific growth suppressing and apoptosis inducing properties. Oncogene. 2001;20:7051–7063. doi: 10.1038/sj.onc.1204897. [DOI] [PubMed] [Google Scholar]
- 11.Ellerhorst JA, Prieto VG, Ekmekcioglu S, Broemeling L, Yekell S, Chada S, Grimm EA. Loss of MDA-7 expression with progression of melanoma. J Clin Oncol. 2002;20:1069–1074. doi: 10.1200/JCO.20.4.1069. [DOI] [PubMed] [Google Scholar]
- 12.Chada S, Sutton RB, Ekmekcioglu S, Ellerhorst J, Mumm JB, Leitner WW, Yang HY, Sahin AA, Hunt KK, Fuson KL, et al. MDA-7/IL-24 is a unique cytokine - tumor suppressor in the IL-10 family. Int Immunopharmacol. 2004;4:649–667. doi: 10.1016/j.intimp.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Dent P, Yacoub A, Hamed HA, Park MA, Dash R, Bhutia SK, Sarkar D, Wang XY, Gupta P, Emdad L, et al. The development of MDA-7/IL-24 as a cancer therapeutic. Pharmacol Ther. 2010;128:375–384. doi: 10.1016/j.pharmthera.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu S, Oshima T, Imada T, Masuda M, Debnath B, Grande F, Garofalo A, Neamati N. Stabilization of MDA-7/IL-24 for colon cancer therapy. Cancer Lett. 2013;335:421–430. doi: 10.1016/j.canlet.2013.02.055. [DOI] [PubMed] [Google Scholar]
- 15.Gupta P, Emdad L, Lebedeva IV, Sarkar D, Dent P, Curiel DT, Settleman J, Fisher PB. Targeted combinatorial therapy of non-small cell lung carcinoma using a GST-fusion protein of full-length or truncated MDA-7/IL-24 with Tarceva. J Cell Physiol. 2008;215:827–836. doi: 10.1002/jcp.21369. [DOI] [PubMed] [Google Scholar]
- 16.Deng WG, Kwon J, Ekmekcioglu S, Poindexter NJ, Grimm EA. IL-24 gene transfer sensitizes melanoma cells to erlotinib through modulation of the Apaf-1 and Akt signaling pathways. Melanoma Res. 2010;21:44–56. doi: 10.1097/CMR.0b013e3283382155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauane M, Gopalkrishnan RV, Sarkar D, Su ZZ, Lebedeva IV, Dent P, Pestka S, Fisher PB. MDA-7/IL-24: Novel cancer growth suppressing and apoptosis inducing cytokine. Cytokine Growth Factor Rev. 2003;14:35–51. doi: 10.1016/S1359-6101(02)00074-6. [DOI] [PubMed] [Google Scholar]
- 18.Mhashilkar AM, Schrock RD, Hindi M, Liao J, Sieger K, Kourouma F, Zou-Yang XH, Onishi E, Takh O, Vedvick TS, et al. Melanoma differentiation associated gene-7 (mda-7): A novel anti-tumor gene for cancer gene therapy. Mol Med. 2001;7:271–282. [PMC free article] [PubMed] [Google Scholar]
- 19.Chada S, Mhashilkar AM, Liu Y, Nishikawa T, Bocangel D, Zheng M, Vorburger SA, Pataer A, Swisher SG, Ramesh R, et al. mda-7 gene transfer sensitizes breast carcinoma cells to chemotherapy, biologic therapies and radiotherapy: Correlation with expression of bcl-2 family members. Cancer Gene Ther. 2006;13:490–502. doi: 10.1038/sj.cgt.7700915. [DOI] [PubMed] [Google Scholar]
- 20.Lebedeva IV, Sarkar D, Su ZZ, Kitada S, Dent P, Stein CA, Reed JC, Fisher PB. Bcl-2 and Bcl-xL differentially protect human prostate cancer cells from induction of apoptosis by melanoma differentiation associated gene-7, mda-7/IL-24. Oncogene. 2003;22:8758–8773. doi: 10.1038/sj.onc.1206891. [DOI] [PubMed] [Google Scholar]
- 21.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 22.She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, Solit DB, Rosen N. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 24.Cusack JC., Jr Overcoming antiapoptotic responses to promote chemosensitivity in metastatic colorectal cancer to the liver. Ann Surg Oncol. 2003;10:852–862. doi: 10.1245/ASO.2003.07.518. [DOI] [PubMed] [Google Scholar]
- 25.Fabbri F, Brigliadori G, Carloni S, Ulivi P, Vannini I, Tesei A, Silvestrini R, Amadori D, Zoli W. Zoledronic acid increases docetaxel cytotoxicity through pMEK and Mcl-1 inhibition in a hormone-sensitive prostate carcinoma cell line. J Transl Med. 2008;6:43. doi: 10.1186/1479-5876-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci USA. 2008;105:10853–10858. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan S. Targeting the mammalian target of rapamycin (mTOR): A new approach to treating cancer. Br J Cancer. 2004;91:1420–1424. doi: 10.1038/sj.bjc.6602162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N. mTOR signaling: Implications for cancer and anticancer therapy. Br J Cancer. 2007;96(Suppl):R11–R15. [PubMed] [Google Scholar]
- 29.Adams JM. Ways of dying: Multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]