

Abstract
We previously reported that podophyllotoxin acetate (PA) radiosensitizes NCI-H460 cells. Here, we confirmed that PA treatment also induces cell death among two other non-small cell lung cancer (NSCLC) cell lines: NCI-H1299 and A549 cells (IC50 values = 7.6 and 16.1 nM, respectively). Our experiments further showed that PA treatment was able to induce cell death via various mechanisms. First, PA dose-dependently induced cell cycle arrest at G2/M phase, as shown by accumulation of the mitosis-related proteins, p21, survivin and Aurora B. This G2/M phase arrest was due to the PA-induced inhibition of microtubule polymerization. Together, the decreased microtubule polymerization and increased cell cycle arrest induced DNA damage (reflected by accumulation of γ-H2AX) and triggered the induction of intrinsic and extrinsic apoptotic pathways, as shown by the time-dependent activations of caspase-3, -8 and -9. Second, PA time-dependently activated the pro-apoptotic ER stress pathway, as evidenced by increased expression levels of BiP, CHOP, IRE1-α, phospho-PERK, and phospho-JNK. Third, PA activated autophagy, as reflected by time-dependent increases in the expression levels of beclin-1, Atg3, Atg5 and Atg7, and the cleavage of LC3. Collectively, these results suggest a model wherein PA decreases microtubule polymerization and increases cell cycle arrest, thereby inducing apoptotic cell death via the activation of DNA damage, ER stress and autophagy.
Keywords: podophyllotoxin acetate, cell cycle arrest, apoptosis, ER stress, autophagy, lung cancer
Introduction
Since the prehistoric period, human beings have treated various diseases with natural products, from plants, marine materials, and animals. The earliest recorded medical text, which came from ancient Egypt, described several plant-derived medicinal substances. Today, plants are the main resource for modern pharmacological research and the development of new drugs, including anticancer agents. The first plant-derived anticancer drugs were the vinca alkaloids, vinblastine and vincristine. They come from the Madagascar periwinkle, Catharanthus roseus G. Don, which was traditionally used to treat diabetes (1).
Podophyllotoxin was isolated as an active ingredient of a plant used for the traditional treatment of skin cancer and warts. Podophyllotoxin acetate (PA), which is a naturally occurring derivative of podophyllotoxin, is obtained as an abundant lignan from podophyllin, which is a type of resin produced by Podophyllum peltatum Linnaeus. The lignans are a family of abundant natural products and secondary metabolites that are manufactured through the shikimic acid pathway, and consist of two bound phenylpropane units. Podophyllotoxin exhibits the aryltetralin structure of a cyclolignan, which is a lignin in which the two phenylpropane units are joined by a carbocycle that consists of two single carbon-carbon bonds that occur between the side chains (one at the β-β′ positions). In terms of biological effects, podophyllotoxin is known to have immunosuppressive activity and antiviral effects against herpes, measles, influenza and venereal warts (2).
It is also considered to be a candidate anticancer agent, as it reversibly binds tubulin and interrupts its polymerization, thereby preventing the formation of mitotic spindles to trigger cell cycle arrest and inhibit cell proliferation (2). Many investigators have synthesized various derivatives in an effort to improve the antitumor effects of podophyllotoxin. Three kinds of representative semi-synthetic epipodophyllotoxin derivatives have been developed: etoposide, teniposide and etopophos. These drugs do not inhibit microtubule polymerization due to the presence of a bulky glucoside moiety in their chemical structure. Instead, their anticancer activity arises from their ability to bind DNA topoisomerases, which are ubiquitous enzymes that control the topological state of DNA in cells. There are two forms of DNA topoisomerase: type I enzymes cleave a single strand of DNA, while type II enzymes cleave both strands. Together, they decide the topology of DNA in actively proliferating cancer cells. Thus, DNA topoisomerases are among the main targets of anticancer drug development. The three semi-synthetic epipodophyllotoxin derivatives act on type II DNA topoisomerases, preventing the re-ligation of DNA. Treatment of cells with these drugs leads to the formation of a DNA-drug-enzyme complex, the breakage of one or both of the DNA strands, and eventual cell death or apoptosis (3).
In a previous study, we isolated PA from a library of natural compounds and showed that it could induce radio-sensitization of NCI-H460 cells (one of NSCLC cell line) and inhibit their proliferation at a very low concentration (4). Here, we tested the effect of PA on various NSCLS cell lines and sought to detail the molecular mechanisms underlying PA-induced cell death.
Materials and methods
Cell culture and chemical reagents
The A549 and NCI-H1299 human NSCLC cell lines were purchased from the American Type Culture Collection (Rockville, MD, USA). All the cells were incubated at 37°C with 5% CO2 incubator. Propidium iodide (PI) was obtained from Sigma-Aldrich (St. Louis, MO, USA). The Natural Product Collection, which included PA, was obtained from MicroSource Discovery Systems, Inc. (Gaylordsville, CT, USA).
MTT assay and IC50 determination
A549 and NCI-H1299 cells (4×103 cells/well) were exposed to different concentrations of PA for 72 h, and then treated with 50 μl of 3-(4,5-dimethylthiazol-2-yle)-2,5-diphenyltetrazolium bromide (MTT) solution (2 mg/ml) for 2 h. All incubations were performed at 37°C. The formazan crystals generated in living cells were dissolved in 200 μl/well of dimethyl sulfoxide (DMSO), and the absorbance of individual wells was read at 545 nm using a microplate reader (Original Multiscan; Thermo Scientific Co., Waltham, MA, USA). The 50% inhibitory concentration (IC50) was calculated from a concentration-response analysis performed using Softmax Pro software (Molecular Devices, Sunnyvale, CA, USA).
Microtubule assembly assay
The microtubule assembly assay was performed as described in a previous study (5). Briefly, A549 and NCI-H1299 cells were seeded (1×106 cells per 100-mm culture dish) and treated with 20 or 10 nM PA, respectively, for 24–48 h. Cells were lyses, and the supernatant fractions were collected as containing soluble αβ-tubulin dimers, while the pellets were collected as containing polymerized microtubules.
Immunocytochemical staining
A549 and NCI-H1299 cells (1×104) were seeded in chamber slides and treated with 20 or 10 nM PA, respectively, for 48 h. The treated cells were fixed with 1% paraformaldehyde and then stained with an anti-α-tubulin antibody and DAPI. Images of stained cells were acquired with a 710 confocal microscope (Carl-Zeiss, Germany).
Immunoblot analysis
Immunoblot analyses were performed as described previously (6). Membranes were probed with antibodies against BiP, CHOP, phospho-PERK, IRE1-α, beclin-1, Atg3, Atg5, Atg7, LC 3, caspase-3, -8 and -9, phospho-JNK, Aurora B (all from Cell Signaling Technology, Beverly, MA, USA), α-tubulin, Cdc2, cyclin B1, p21, survivin (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA). An anti-β-actin antibody (Sigma-Aldrich) was used as the loading control. The relative band densities were determined by densitometry and normalized with respect to that of β-actin, using ImageJ software (NIH, Bethesda, MD, USA).
Cell cycle analysis
A549 and NCI-H1299 cells (3×105 cells per 60-mm dish) were incubated with or without 20 or 10 nM PA for 24 or 48 h. The cells were then trypsinized, washed twice with ice-cold phosphate-buffered saline (PBS), and fixed with ice-cold 70% ethanol. Fixed cells were incubated with 50 μg/ml PI and 40 μg/ml RNase at 37°C for 30 min, and then analyzed with a FACSort flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA). The percentages of cells in each phase of the cell cycle were determined using Cell Quest software (Becton-Dickinson).
PI-Annexin V staining
PI-Annexin V staining was performed using a FITC Annexin V Apoptosis Detection kit I (BD Pharmingen, San Jose, CA, USA). Briefly, A549 and NCI-H1299 cells (3×105 cells) were incubated with or without 15 or 7.5 nM PA, respectively, for 24 or 48 h. Treated cells were harvested by trypsinization, washed twice with cold PBS, and resuspended in 500 μl of a solution containing 1 μg/ml each of PI and Annexin V. The apoptotic fraction was evaluated using a FACSort flow cytometer (Becton-Dickinson) as described by the manufacturer.
Statistical analysis
Data were analyzed using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA), and the significance of differences between experimental groups was determined using the Student's t-test. P-values <0.05 were considered significant, and the individual P-values in the figures are denoted by asterisks (**P<0.01; ***P<0.001). The number above each point or bar represents the mean results from three independent experiments, and the error bars signify the standard deviations (SDs).
Results
PA induces cell death of NSCLCs
PA (Fig. 1A) was previously screened from a natural product library as having radiosensitizing effects against the NCI-H460 cell line in vitro and in vivo. Here, we tested whether PA could also induce cell death against two other NSCLC cell lines (A549 and NCI-H1299 cells), and calculated its IC50 values against these cells over 24 or 48 h. After 48 h of PA treatment, the IC50 values of PA against and A549 and NCI-H1299 cells were found to be 16.08 and 7.53 nM, respectively (Fig. 1B and C). We used these IC50 values as standard doses in the following experiments.
Figure 1.
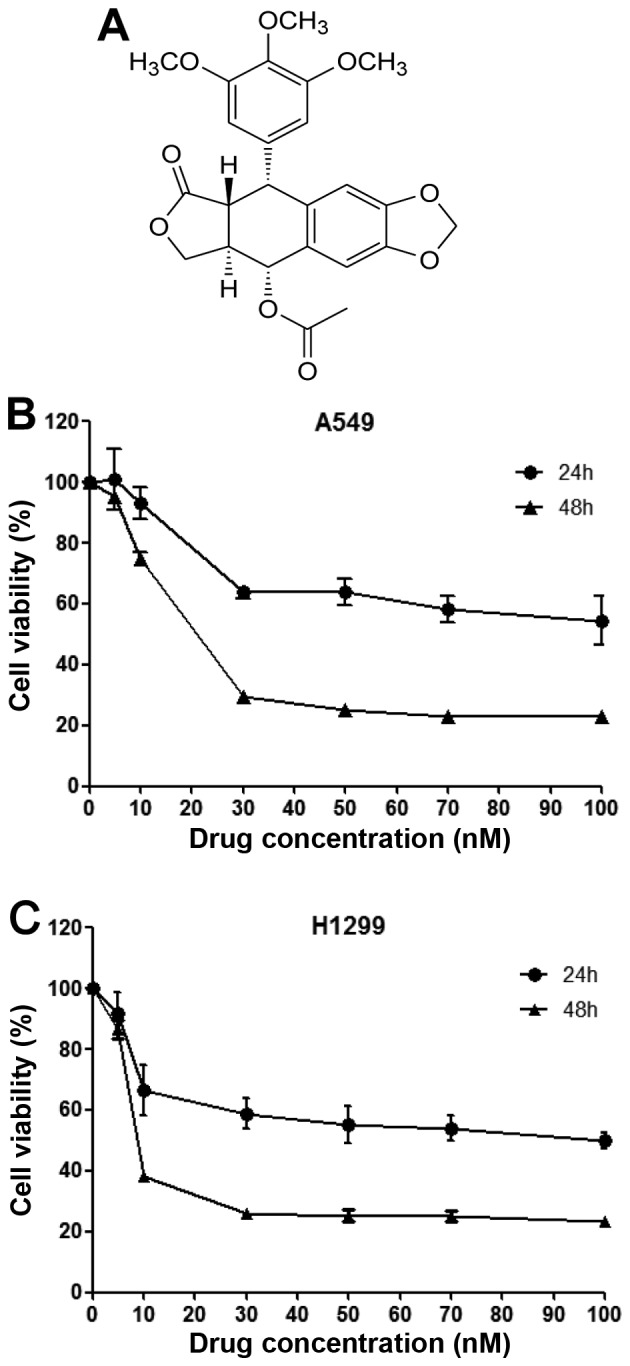
PA inhibits the proliferation of the NSCLC cell lines, A549 and NCI-H1299. (A) Molecular structure of PA. (B and C) Determination of IC50 values for PA against A549 and NCI-H1299 cells by MTT assays performed at 24 or 48 h post-treatment.
PA inhibits microtubule polymerization and induces cell cycle arrest at G2/M phase
One of the anticancer activities of podophyllotoxin, the precursor of PA, is the ability to interrupt of tubulin polymerization and thereby trigger cell cycle arrest. To test whether the anticancer potential of PA involves a similar mechanism, we examined microtubule organization in PA-treated cells. Our microtubule assembly assays and immunocytochemical staining using an anti-α-tubulin antibody revealed that PA treatment time-dependently decreased microtubule polymerization and disrupted the organization of microtubules in A549 and NCI-H1299 cells (Fig. 2). To examine whether the observed microtubule damage was followed by blockade of the cell cycle, as is seen with various microtubule-targeting anticancer reagents, we treated A549 cells with 5, 10 or 20 nM PA for 8 or 16 h, and treated NCI-H1299 cells with 5 or 10 nM PA for 8 or 16 h, and assessed their cell cycle distributions. Our results showed that PA-treated cells exhibited time- and dose-dependent increases inG2/M phase arrest (Fig. 3A and B, and Table I). The G2/M arrest of A549 cells was maximal following the 20 nM/16-h treatment, while that of NCI-H1299 cells was maximal following the 10 nM/8-h treatment dose- and time-dependently. Because intact cell cycle patterns of NCI-H1299 cells were not detected after 16 h of PA treatment (data not shown), cell cycle analysis of NCI-H1299 were performed until 16 h. We also observed G2/M phase arrest-related molecules in A549 or NCI-H1299 cells treated with 20 nM or 10 nM PA, respectively, for 8, 16, or 24 h. Immunoblot analyses (Fig. 3C and D) revealed that the levels of cyclin B1 and Cdc2 were decreased, while those of p21, Aurora B and survivin increased in a time-dependent manner. These results were consistent with the observed increase in G2/M phase arrest, suggesting that PA might block the proliferation of NSCLC cells by triggering microtubule disorganization and subsequent cell cycle arrest.
Figure 2.

PA inhibits the polymerization of intracellular microtubules. (A) Immunoblot analyses of α-tubulin in the insoluble and soluble fractions of A549 and NCI-H1299 cells treated with PA (20 or 10 nM, respectively) for 24 or 48 h. Con, the mock-treated control. (B) Immunocytochemical staining for α-tubulin in A549 and NCI-H1299 cells treated with PA (20 or 10 nM, respectively) for 24 h. Nuclei are stained blue, while α-tubulin is shown in green.
Figure 3.

PA induces cell cycle arrest. (A and B) Cell cycle analysis of A549 and NCI-H1299 cells treated with PA for 8 and 16 h. Con, the mock-treated control, and 5, 10 and 20 nM the groups treated with 5, 10 and 20 nM PA, respectively. (C and D) Immunoblot analyses of the G2/M arrest-related proteins, cyclin B1, Cdc2, p21, Aurora B, survivin, and β-actin. Con, the mock-treated control, and 8, 16 or 24 h, the times at which cells were harvested after treatment with PA (20 and 10 nM for A549 and NCI-H1299 cells, respectively).
Table I.
Quantitative analysis of cell cycle arrest in A549 and NCI-H1299 cells treated with PA for 8 and 16 h.
| Con | 5 nM | 10 nM | 20 nM | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| A549 | 8 h | 16 h | 8 h | 16 h | 8 h | 16 h | 8 h | 16 h |
| Sub-G1 | 2.1 | 1.4 | 2.8 | 1.4 | 3.2 | 3.5 | 3.1 | 8.1 |
| G1 | 53.1 | 51.5 | 52.1 | 51.6 | 42.1 | 52.2 | 28.2 | 5.9 |
| S | 12.8 | 11.4 | 11.6 | 12.4 | 13.9 | 6.5 | 17.1 | 4.1 |
| G2/M | 22.0 | 19.7 | 23.2 | 20.4 | 30.6 | 18.7 | 39.3 | 58.3 |
|
| ||||||||
| Con | 5 nM | 10 nM | 20 nM | |||||
|
|
|
|
|
|||||
| NCI-H1299 | 8 h | 16 h | 8 h | 16 h | 8 h | 16 h | 8 h | 16 h |
|
| ||||||||
| Sub-G1 | 2.9 | 1.6 | 5.8 | 4.6 | 9.6 | 20.0 | 15.1 | 31.2 |
| G1 | 45.0 | 44.4 | 34.9 | 40.2 | 12.8 | 12.3 | 8.2 | 2.2 |
| S | 7.3 | 15.8 | 14.7 | 16.4 | 19.0 | 6.7 | 17.6 | 4.1 |
| G2/M | 27.5 | 24.3 | 33.9 | 24.3 | 52.1 | 43.2 | 49.6 | 46.0 |
Con, mock-treated control; 5,10 and 20 nM groups treated with 5, 10 and 20 nM PA, respectively.
PA induces DNA damage and cell death via G2/M phase arrest
Next, we performed PI/Annexin V staining to detect cell death in A549 cells treated with 20 nM PA for 24 or 48 h, and in NCI-H1299 cells treated with 10 nM PA for 24 or 48 h. Our results revealed that the cell death of A549 and NCI-H1299 cells increased time-dependently, with maximal cell death seen at 48 h (Fig. 4A and B). Immunoblot analyses against caspase-3, -8 and 9 were performed on A549 and NCI-H1299 cells treated with PA (20 and 10 nM, respectively) for 8, 16, 24 or 48 h (Fig. 4D). Increases in the levels of cleaved caspase-3, -8 and -9 were detected beginning at 16 h of PA treatment. Thus, we concluded that the PA-induced cell cycle arrest triggered cell death. We also observed expression of γ-H2AX under the same treatment conditions in both cell lines, as a means to examine the DNA damage effects of PA. Our results revealed that the expression of γ-H2AX increased beginning at 16 h of PA treatment (Fig. 4C). Taken together, these results suggest that PA induces the cell death of NSCLS cells via microtubule disorganization accompanied by cell cycle G2/M arrest and DNA damage.
Figure 4.


PA induces apoptotic cell death via DNA damage in vitro. (A and B) PI-Annexin V assays were used to detect the cell death of A549 and NCI-H1299 cells treated with or without PA. Con, the mock-treated control, and PA the groups treated with 20 or 10 nM PA for A549 and NCI-H1299 cells, respectively. Cells were harvested at 24 or 48 h post-treatment. (C and D) Immunoblot analyses were used to detect DNA damage-related γ-H2AX and apoptosis-related (caspase-3, -8 and -9) proteins at 8, 16, 24 or 48 h after treatment with 20 and 10 nM PA for A549 and NCI-H1299 cells, respectively.
PA might induce cell death via ER stress and autophagic signaling
To examine whether other cell death machineries (in addition to apoptosis) are involved in the PA-induced elimination of NSCLC cells, we performed various immunoblot analyses. To examine the potential involvement of endoplasmic reticulum (ER) stress, we examined the expression levels of the ER stress-related proteins, binding immunoglobulin protein (BiP), CCAAT-enhancer-binding protein homologous protein (CHOP), protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme-1 (IRE1)-α and phospho-JNK were detected in A549 and NCI-H1299 cells treated with PA (20 and 10 nM, respectively) for 8, 16, 24 or 48 h. Our results revealed that the levels of CHOP, phospho-PERK, IRE1-α and phospho-JNK increased time-dependently in both cell lines following PA treatment, whereas BiP time-dependently increased in A549 cells but not in NCI-H1299 cells (Fig. 5). These results are consistent with a previous report that pro-apoptotic ER stress was involved in the cell death induced by another podophyllotoxin derivative. We also used immunoblot analyses to test the possible involvement of autophagic cell death in our experimental system. Indeed, we detected time-dependent PA-induced increases in the expression levels of autophagy-related gene (Atg)-3, -5 and -7 and beclin-1, and in the cleavage of microtubule-associated protein 1A/1B-light chain 3 (LC3) (Fig. 6).
Figure 5.

PA induces NSCLC cell death via ER stress. (A and B) Immunoblot detection of ER stress signaling molecules in A549 and NCI-H1299 cells receiving mock treatment (con) or treated with 20 and 10 nM PA (for A549 and NCI-H1299 cells, respectively). Samples were harvested at 8, 16, 24 or 48 h post-treatment.
Figure 6.

PA-induced NSCLC cell death is related to the activation of autophagic signaling. (A and B) Immunoblot analysis of the expression levels of autophagic signaling molecules in A549 and NCI-H1299 cells receiving mock treatment (con) or treated with 20 and 10 nM PA (for A549 and NCI-H1299 cells, respectively). Samples were harvested at 8, 16, 24 or 48 h post-treatment.
Discussion
Non-small cell lung cancer (NSCLC), which is a main type of lung cancer, has a particularly low 5-year survival rate (7). NSCLC therapy typically involves surgery and/or chemo- or radiotherapy. Such therapies are not fully effective, however, and chemo- or radiotherapy is often associated with side effects and/or therapeutic resistance (8). Efforts to develop new drugs that have increased treatment efficiency and minimal side effects are a key to the field of cancer treatment. Numerous candidate anticancer reagents have been developed from natural plant products and their secondary metabolites, which are proven sources for many pharmaceutical reagents (1).
In this study, we tested whether PA has anticancer effects against two NSCLC cell lines, and sought to reveal the underlying molecular mechanisms. As shown in Fig. 1B, PA inhibited NSCLC proliferation more effectively than etoposide, which is a representative anticancer reagent and semi-synthetic derivative of podophyllotoxin. The IC50 values of PA against NCI-H1299 and A549 cells were 7.53 and 16.08 nM, respectively, whereas those reported for etoposide were 0.448 and 21.3 μM, respectively (9,10). This indicates that the cell growth inhibition mediated by PA is much stronger (~70–1,000-fold) than that of etoposide. Our results suggested that the inhibitory effect of PA was independent of the p53 or PTEN status, as A549 cells contain wild-type p53 and PTEN, whereas NCI-H1299 cells are null for both p53 and PTEN (6,11,12).
We also found that PA could damage the soluble fraction of intracellular tubulin and destroy tubulin structures (Fig. 2), suggesting that PA might target the polymerization of tubulin, which is closely related to cell cycle progression and cellular proliferation, and is a main target of several anti-cancer reagents (13). On tubulin, the paclitaxel site, the vinca domain and the colchicine domain are the main binding sites for representative microtubule-targeting drugs. Among them, the colchicine domain has attracted recent research interest from groups seeking to develop vessel-targeting reagents for cancer therapy (13). The PA-mediated destruction of tubulins is consistent with the effects of other podophyllotoxin derivatives, including etoposide (14).
Many tubulin-targeting drugs also reportedly inhibit the cell cycle, due to their ability to disturb the rapid microtubule dynamics of mitotic spindles (13). As shown in Fig. 3 and Table I, PA-treated cells showed G2/M arrest at 8 or 16 h post-treatment, as confirmed at the protein level by decreases in cyclin B1 and Cdc2, with corresponding increases in p21, Aurora B and survivin. Moreover, PA treatment induced DNA damage, as shown by time-dependent increases in the levels of the DNA damage marker, γ-H2AX. Similar DNA-damaging effects have been reported for other podophyllotoxin analogs, particularly those known to act as DNA topoisomerase inhibitors (14,15).
PA was found to enhance cell death via apoptosis, as shown by an increase in the sub-G1 population (Fig. 3A and Table I) and by our PI-Annexin V staining analysis (Fig. 4A). Apoptotic signaling involves intrinsic and extrinsic pathways. The former is activated by external stress and triggers mitochondrial permeability changes and caspase-9 activation, while the latter begins with death receptor/ligand binding and proceeds through caspase-8 activation. Both apoptotic pathways feed to the activation of caspase-3 as a common ‘executioner’. Consistent with previous reports that these pathways are triggered by derivatives of podophyllotoxin, we found that PA time-dependently increased the activities of caspase-3, -8 and -9 (16).
In terms of other cell death pathways, Bicknell et al (17) reported that the inhibition of nuclear migration and cell division could induce ER stress, and a report showed that the anticancer effects of podophyllotoxin derivatives involved activation of the ER stress pathway (18). Therefore, we tested whether PA could induce ER stress and its related molecular changes. Indeed, our results showed that PA treatment increased various markers of ER stress, including the expression levels of BiP, CHOP, and IRE1-α and the phosphorylation levels of PERK and JNK (Fig. 5). The IRE1-α serine/threonine kinase regulates the unfolded protein response (UPR) that is part of the ER stress response, while PERK (an eIF2α kinase) is an ER transmembrane protein that, upon activation by ER stress, phosphorylates eIF2α to inhibit protein translation. Phosphorylation of PERK at Thr980 is used as a marker of ER stress. Elevated expression of CHOP, which mediates apoptosis, is also used as an ER stress marker (19). Therefore, our results suggest that the relationship between apoptosis and ER stress pathways might be a main target of PA. The elevated levels of ER stress markers by PA treatment did not coincidence with patterns in our cell lines, however. We estimated these differences of protein expression due to cell line specific differences for ER stress pathway.
As another cell death-inducing pathway, we examined the possible involvement of PA in autophagic cell death. A number of anticancer regents reportedly induce cell death via the autophagic pathway (20,21), which is generally activated by conditions of nutrient deprivation, but has also been associated with pathologies, including cancer (22,23). The main function of the autophagic pathway is to provide the building blocks (e.g., amino acids) for new cellular components via the autophagosome-mediated degradation of abnormal or excess cytoplasmic contents. Several Atg proteins are known to control autophagosome formation, including the Atg12-Atg5 and LC3-II (Atg8-II) complexes. The Atg4 protease cleaves LC3/Atg8 at its C-terminus to yield the cytosolic LC3-I, which is then conjugated to phosphatidylethanolamine (PE) via the actions of Atg7 and Atg3. The other lipidated form of LC3, known as LC3-II, is also attached to sequestosome 1 (SQSTM1, p62) on autophagosome, and it is a ubiquitin binding protein. SQSTM1-containing protein aggregates to the autophagosome and then promotes autophagy. In this study, we observed increases in the expression levels of Atg-3, -5 and -7 and beclin-1, and the cleavage of LC3 (24). These changes suggest that the autophagic cell death pathway is also involved in the effects of PA on the tested NSCLC cell lines. Some investigators have suggested that the functional status of caspase-3 might control the switch between apoptosis and autophagy in MCF-7 (a breast cancer cell line) cells exposed to chemo-therapeutics (25,26). This may suggest the existence of crossroads connecting the apoptotic and autophagic pathways.
This study did not examine whether the low dose of PA that is capable of eliminating NSCLC cells might spare normal tissues. However, our present and previous results collectively show that PA enhances apoptotic death of NSCLC cell lines (e.g., NCI-H460, A549 and NCI-H1299) via multiple cell death routes. This suggests the novel possibility that PA could be a candidate anticancer drug and/or could prove useful for other clinical applications. Our results also suggest a model in which PA induces the following sequence: damage of microtubule polymerization; induction of cell cycle arrest/DNA damage; ER stress/autophagy; and finally apoptotic cell death (Fig. 7). Our findings may facilitate the development of podophyllotoxin derivatives as anticancer reagents. Finally, the various molecules involved in the cytotoxic effects of PA could be useful targets for the development of novel therapeutic agents against cancer.
Figure 7.
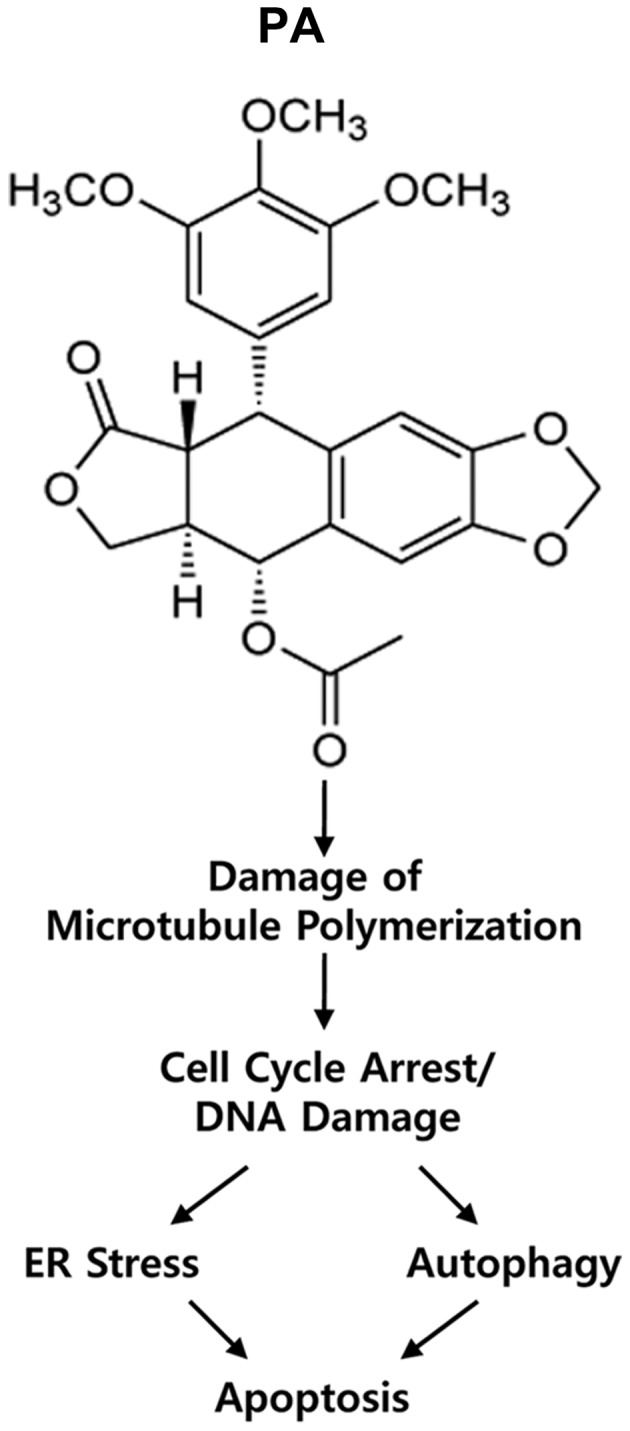
Scheme of the possible cell death routes induced by PA treatment of NSCLC cell lines.
Acknowledgements
This study was supported by the Nuclear Research and Development Program of the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (2012M2A2A7010422) and, in part, by the Basic Science Research Program through the NRF (NRF-2014R1A1A2054985).
References
- 1.Cragg GM, Grothaus PG, Newman DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 2.Gordaliza M, García PA, del Corral JM, Castro MA, Gómez-Zurita MA. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon. 2004;44:441–459. doi: 10.1016/j.toxicon.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Guerram M, Jiang Z-Z, Zhang L-Y. Podophyllotoxin, a medicinal agent of plant origin: Past, present and future. Chin J Nat Med. 2012;10:161–169. doi: 10.3724/SP.J.1009.2012.00161. [DOI] [Google Scholar]
- 4.Choi JY, Cho HJ, Hwang SG, Kim WJ, Kim JI, Um HD, Park JK. Podophyllotoxin acetate enhances γ-ionizing radiation-induced apoptotic cell death by stimulating the ROS/p38/caspase pathway. Biomed Pharmacother. 2015;70:111–118. doi: 10.1016/j.biopha.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 5.Kuo CC, Hsieh HP, Pan WY, Chen CP, Liou JP, Lee SJ, Chang YL, Chen LT, Chen CT, Chang JY. BPR0L075, a novel synthetic indole compound with antimitotic activity in human cancer cells, exerts effective antitumoral activity in vivo. Cancer Res. 2004;64:4621–4628. doi: 10.1158/0008-5472.CAN-03-3474. [DOI] [PubMed] [Google Scholar]
- 6.Park JK, Jung HY, Park SH, Kang SY, Yi MR, Um HD, Hong SH. Combination of PTEN and gamma-ionizing radiation enhances cell death and G(2)/M arrest through regulation of AKT activity and p21 induction in non-small-cell lung cancer cells. Int J Radiat Oncol Biol Phys. 2008;70:1552–1560. doi: 10.1016/j.ijrobp.2007.11.069. [DOI] [PubMed] [Google Scholar]
- 7.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 8.Begg AC, Stewart FA, Vens C. Strategies to improve radio-therapy with targeted drugs. Nat Rev Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 9.Tsai CM, Chang KT, Perng RP, Mitsudomi T, Chen MH, Kadoyama C, Gazdar AF. Correlation of intrinsic chemoresistance of non-small-cell lung cancer cell lines with HER-2/neu gene expression but not with ras gene mutations. J Natl Cancer Inst. 1993;85:897–901. doi: 10.1093/jnci/85.11.897. [DOI] [PubMed] [Google Scholar]
- 10.Xin Y, Yin F, Qi S, Shen L, Xu Y, Luo L, Lan L, Yin Z. Parthenolide reverses doxorubicin resistance in human lung carcinoma A549 cells by attenuating NF-κB activation and HSP70 up-regulation. Toxicol Lett. 2013;221:73–82. doi: 10.1016/j.toxlet.2013.06.215. [DOI] [PubMed] [Google Scholar]
- 11.Breen L, Heenan M, Amberger-Murphy V, Clynes M. Investigation of the role of p53 in chemotherapy resistance of lung cancer cell lines. Anticancer Res. 2007;27A:1361–1364. [PubMed] [Google Scholar]
- 12.Jung IL, Kang HJ, Kim KC, Kim IG. PTEN/pAkt/p53 signaling pathway correlates with the radioresponse of non-small cell lung cancer. Int J Mol Med. 2010;25:517–523. doi: 10.3892/ijmm_00000372. [DOI] [PubMed] [Google Scholar]
- 13.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 14.Montecucco A, Biamonti G. Cellular response to etoposide treatment. Cancer Lett. 2007;252:9–18. doi: 10.1016/j.canlet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Kaufmann SH. Cell death induced by topoisomerase-targeted drugs: More questions than answers. Biochim Biophys Acta. 1998;1400:195–211. doi: 10.1016/S0167-4781(98)00136-5. [DOI] [PubMed] [Google Scholar]
- 16.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bicknell AA, Babour A, Federovitch CM, Niwa M. A novel role in cytokinesis reveals a housekeeping function for the unfolded protein response. J Cell Biol. 2007;177:1017–1027. doi: 10.1083/jcb.200702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JY, Tang YA, Li WS, Chiou YC, Shieh JM, Wang YC. A synthetic podophyllotoxin derivative exerts anti-cancer effects by inducing mitotic arrest and pro-apoptotic ER stress in lung cancer preclinical models. PLoS One. 2013;8:e62082. doi: 10.1371/journal.pone.0062082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 20.Gills JJ, Lopiccolo J, Dennis PA. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy. 2008;4:107–109. doi: 10.4161/auto.5224. [DOI] [PubMed] [Google Scholar]
- 21.Terés S, Lladó V, Higuera M, Barceló-Coblijn G, Martin ML, Noguera-Salvà MA, Marcilla-Etxenike A, García-Verdugo JM, Soriano-Navarro M, Saus C, et al. 2-Hydroxyoleate, a nontoxic membrane binding anticancer drug, induces glioma cell differentiation and autophagy. Proc Natl Acad Sci USA. 2012;109:8489–8494. doi: 10.1073/pnas.1118349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: Therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan CH, Yang YP, Qin ZH, Gu ZL, Reid P, Liang ZQ. Autophagy is involved in cytotoxic effects of crotoxin in human breast cancer cell line MCF-7 cells. Acta Pharmacol Sin. 2007;28:540–548. doi: 10.1111/j.1745-7254.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- 26.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–510. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]