

Abstract
Cisplatin is a commonly used drug for cancer treatment by crosslinking DNA, leading to apoptosis of cancer cells, resistance to cisplatin treatment often occurs, leading to relapse. Therefore, there is a need for the development of more effective treatment strategies that can overcome chemoresistance. Myricetin is a flavonoid from fruits and vegetables, showing anticancer activity in various cancer cells. In this study, we found myricetin exhibited greater cytotoxicity than cisplatin in two cisplatin-resistant ovarian cancer cell lines, OVCAR-3 and A2780/CP70, and it was less cytotoxic to the normal ovarian cell line IOSE-364. Myricetin selectively induced apoptosis in both cisplatin-resistant cancer cell lines, but did not induce apoptosis in the normal ovarian cell line. It induced both Bcl-2 family-dependent intrinsic and DR5 dependent extrinsic apoptosis in OVCAR-3 cells. P53, a multifunctional tumor suppressor, regulated apoptosis in OVCAR-3 cells through a Bcl-2 family protein-dependent pathway. Myricetin did not induce cell cycle arrest in either ovarian cancer cell line. Because of its potency and selectivity against cisplatin-resistant cancer cells, myricetin could potentially be used to overcome cancer chemoresistance against platinum-based therapy.
Keywords: ovarian cancer, apoptosis, p53, Bcl-xl, Bax
Introduction
The human health and life are under constant serious threat of cancer. Surgery, radiation, and chemotherapy have been the most commonly utilized methods for therapy of ovarian cancer. Recently, chemotherapy has become the predominant mode of treatment. Platinum drugs, such as cisplatin and its analogues, are the most frequently used agents for the treatment of human cancer. Initially, cisplatin responsiveness is high, however, a large number of cancer patients will relapse with cisplatin-resistant disease due to many mechanisms including increased detoxification of the drug, changes in cellular efflux and uptake of the drug, increased DNA repair and inhibition of apoptosis (1). Moreover, it causes unacceptable rates of normal cell toxicity and a number of side-effects, such as nephrotoxicity, neurotoxicity, ototoxicity electrolyte disturbance, myelotoxicity, hemolytic anemia, nausea and vomiting (2–5). Therefore, novel therapies and more selective drugs are needed for the treatment of human cancer (6).
The cell cycle is the series of events that take place in a cell leading to its division and duplication, resulting in two daughter cells. It can be divided into three periods: interphase (G1, S and G2 phases), the mitotic phase (M phase) and cytokinesis. A dysregulation of cell cycle components may lead to tumor formation (7), and investigational anticancer drugs have recently focused on the molecular targets involved in cell cycle control mechanisms (8,9).
Apoptosis is a process of programmed cell death (PCD) that occurs in multicellular organisms. Excessive apoptosis causes atrophy, whereas defective apoptotic processes result in uncontrolled cell proliferation, implicated in a wide variety of diseases such as cancer. It has been reported that a decreased susceptibility of cancer cells to apoptosis was tightly associated with drug resistance. Therefore, inducing apoptosis in cancer cells may be a promising strategy to overcome resistance (6). Many mechanisms are connected to the failure to develop apoptosis in cancer cells, including the expression of P-glycoprotein and p53 mutations. Two major pathways leading to apoptosis, the intrinsic (mitochondrial) and extrinsic (receptor-mediated) pathways, have been delineated. The Bcl-2 protein family regulates the intrinsic apoptotic pathway by controlling the permeability of the mitochondrial membrane and the release of the pro-apoptotic factors (10). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), including Apo2L/TRAIL, regulates the extrinsic apoptotic pathway by engaging its receptor.
Flavonoids are natural polyphenols present in a variety of foods, especially fruits and vegetables. The anticancer activity of flavonoids isolated from plants have been widely studied. It has been reported that flavonoids display anticancer characteristics and might be able to prevent oxidation and inflammation, diminish angiogenesis and cell proliferation, and induce apoptosis (11). Myricetin (Fig. 1A) is a member of the flavonoid class of phenolic compounds with antioxidant properties (12). It can be found in fruits, vegetables, nuts, berries, tea and red wine (13,14). Previous studies have reported myricetin induces apoptosis in various cancer cells, such as in hepatoma, pancreatic cancer, esophageal cancer and colon carcinoma cells (15–18).
Figure 1.

Myricetin inhibits cell viability in ovarian cancer cells. (A) Structure of myricetin. (B) Cells (1×104/well) were seeded in 96-well plates, incubated overnight, and then treated with myricetin for 24 h. Cell viability was determined by an MTT-based method and expressed as percentages of control. (C) Myricetin suppressed OVCAR-3 cells growth in vivo. Chicken chorioallantoic membrane (CAM) assay shows tumor without myricetin treatment, and treated with 20 μM myricetin. After 5 days of treatment, the tumors were excised. Weight of xenografted tumor implanted on CAM treated with and without myricetin (0 and 20 μM). *p<0.05 as compared to control.
In this study, we examined the effects of myricetin apoptosis induction and cell cycle arrest in two platinum-resistant ovarian cancer cell lines: A2780/CP70 and OVCAR-3. The mechanisms involved in the effects of myricetin were also investigated.
Materials and methods
Cell culture and reagents
Human ovarian cancer cell lines, OVCAR-3 and A2780/CP70, were kindly provided by Dr B. Jiang, Department of Microbiology, Immunology, and Cell Biology, West Virginia University, Morgantown, WV, USA. IOSE-364, normal ovarian surface epithelial cells from healthy women, but immortalized with SV40 T/t, were a gift from Dr N. Auersperg at the University of British Columbia, Canada (19). All cell lines were maintained in RPMI-1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10% US-qualified fetal bovine serum (Invitrogen, Grand Island, NY, USA). All cells were maintained in a humidified incubator with 5% CO2 at 37°C. Myricetin was purchased from J&K Chemical Technology (Beijing, China). It was dissolved in dimethyl sulfoxide (DMSO) to make stock solutions of 100 mM, and equal amounts of DMSO were included in controls for every experiment. The primary antibodies against caspase-3, -7, -8, and -9, Bax, Bcl-2, DR5, Puma, FADD, and p21 were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). The primary antibodies against p53, cmyc, Bcl-xl and GAPDH were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
Cell viability
Cell growth inhibition was determined by measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye absorbance. The cells (1×104) were seeded into 96-well, incubated at 37°C. Cells were incubated overnight for attaching to the bottom and then treated with different concentrations of myricetin (5–80 μM) or DMSO (as vehicle). After 24 h, the medium was removed, and 100 μl MTT (1 mg/ml) were added to each well and then incubated at 37°C for 4 h in the dark. After removing the supernatant, the formed formazan crystals were dissolved in 200 μl DMSO, and absorbance was measured at 570 nm.
Chicken chorioallantoic membrane assay
Specific pathogen-free fertile chicken eggs (Charles River Laboratories, North Franklin, CT, USA) were slowly turned by an automatic egg turner (G.Q.F. Manufacturing Co., Savannah, GA, USA) and incubated at 37.5°C. The cells (1.2×106) were suspended in 20 μl FBS-free medium, mixed with 80 μl of Matrigel (BD Bioscience) and 0- or 20-μM myricetin, incubated on an autoclaved silicone mat for 30 min for gelation, and implanted into the chorioallantoic membrane (CAM) of a 9-day-old chicken embryo. After incubating another 5 days, the tumor implants were measured for tumor weight.
Flow cytometry analysis of cell apoptosis
The apoptosis effects of myricetin on ovarian cells were determined by Alexa Fluor 488 Annexin V/Dead Cell Apoptosis kit from Invitrogen. A2780/CP70, OVCAR-3 and IOSE-364 cells (106/dish) treated with myricetin or DMSO for 24 h were washed with cold PBS twice and re-suspended in binding buffer. An aliquot of 100 μl of the cell solution (1×105 cells) was transferred to a 5-ml tissue culture tube. Subsequently, 5 μl of FITC Annexin V and 1 μl propidium iodide (PI) were added to the cells. The cells were gently vortexed and incubated for 15 min at room temperature in the dark. The next step involved the addition of 400 μl of 1× binding buffer to each tube. The samples were analyzed by flow cytometry (FACSCalibur system, BD Biosciences, San Jose, CA, USA).
Apoptosis assessment by Hoechst 33342 staining
OVCAR-3 cells (106/dish) treated with various concentrations of myricetin and pifithrin-α (PFT-α) (Sigma) for 24 h were washed with cold PBS and stained with 10 μg/ml Hoechst 33342 (Sigma) in PBS for 10 min in the dark at 37°C. Cell apoptosis was examined under a fluorescence microscope (Zeiss).
Flow cytometry analysis of the cell cycle
Cells treated with various concentrations of myricetin for 24 h were digested by trypsin, collected by 1,000 rpm centrifugation for 10 min, and then washed with cold PBS. The cell pellets were suspended with 70% ethanol, stored at −20°C. After centrifugation at 1,000 rpm for 6 min, the cell pellets were re-suspended in PBS, collected by centrifugation, and incubated with 180 μg/ml RNase A at 37°C for 15 min. Flow cytometry (FACSCalibur system, BD Biosciences) was used for detection after 50 μg/ml propidium iodide (final concentration) was added to cell pellets for 15-min staining. Data were plotted and analyzed by using FCS software (De Novo Software, Los Angeles, CA, USA).
Caspase-3/7 and caspase-9 assay
A2780/CP70 and OVCAR-3 cells were seeded into 96-well plates (1×104/well) and incubated overnight at 37°C. Cells were treated with different concentrations of myricetin (5–20 μM) or DMSO for 4 h. After treatment, Caspase-Glo 3/7 or Caspase-Glo 9 Assay kit (Promega) was used to detect the caspase-3/7 or caspase-9 enzymatic activities in both ovarian cancer cell lines. Enzymatic activities were normalized by total protein levels and were expressed as a percentage of the untreated control.
Western blotting
Ovarian cancer cells were seeded in 60-mm dishes at the concentration of 106/dish, incubated overnight, and then treated with various concentrations of myricetin for 24 h. The cells were lysed in 100 μl mammalian protein extraction reagent including 1 μl Halt protease, 1 μl phosphatase inhibitor, and 2 μl EDTA (M-PER, Pierce, Rockford, IL, USA). Total protein levels were determined by a BCA Protein Assay kit (Pierce). The cell lysates were separated by SDS-PAGE and blotted onto a nitrocellulose membrane with a Mini-Protean 3 system (Bio-Rad, Hercules, CA, USA). The membranes were blocked in 5% non-fat milk (dissolved in Tris-buffer saline containing 0.1% Tween-20) for 1 h at room temperature. The membranes were incubated with the primary antibodies and secondary antibody dilutions, washed with TBST. The antigen-antibody complex was visualized with the SuperSignal West Dura Extended Duration Substrate (Pierce).
Transfection with small interfering RNA (siRNA)
OVCAR-3 cells were seeded in 60-mm dishes at 5×105/dish and incubated overnight before transfection with p53 siRNA, p21 siRNA or control siRNA (Santa Cruz) using jetPRIME™ DNA and siRNA transfection reagent (VWR International, Radnor, PA, USA) according to the manufacturer's protocol. After 24 h, cells were treated with myricetin or DMSO. Cell lysates were collected for western blotting to test Bad, Bax, Bcl-xl, cmyc, p53 and p21 proteins.
Statistical analysis
The experiments were performed at least three times. Results were expressed as mean ± standard error of mean (SEM) using Microsoft Excel (2007). SPSS (Version 18.0 for Windows) was used to perform statistical assessment. The results were analyzed using post hoc test (2-sided Dunnett's test) and one-way analysis of variance (ANOVA) to test differences between each treatment and control. A p-value of <0.05 was considered statistically significant.
Results
Effect of myricetin on ovarian cancer cell proliferation
MTT assay was performed after treatment of ovarian cancer and normal ovarian cells with myricetin to investigate the effect of myricetin on the viability of platinum-resistant ovarian cancer cells. We observed that compared with controls (myricetin 0 μM), myricetin significantly reduced the viability of A2780/CP70 and OVCAR-3. Cell viability with myricetin treatment (5–80 μM) for 24 h ranged from 93.8 to 17.4% (p<0.05) for A2780/CP70 cells (Fig. 1B). Similarly, myricetin also reduced viability of OVCAR-3 cells at concentrations of 10 μM myricetin and above. Cell viability was significantly decreased from 71.5% at a concentration of 10 μM myricetin to 12.5% at a concentration of 80 μM myricetin (p<0.05) (Fig. 1B). We also examined the growth-inhibitory activity of myricetin on IOSE-364 normal ovarian cells (Fig. 1B). Myricetin had higher level of cytotoxicity in ovarian cancer cells A2780/CP70 and OVCAR-3 than on normal ovarian cells IOSE-364, indicating that cisplatin-resistant ovarian cancer cells were more sensitive to myricetin than normal ovarian cells. In CAM models, myricetin reduced typical tumor growth of OVCAR-3 cells. The tumors treated with 20 μM myricetin were smaller and lighter than corresponding controls (Fig. 1C).
Myricetin induces apoptosis in A2780/CP70 and OVCAR-3 cells
For the purpose of investigating whether myricetin inhibited cell growth by inducing apoptosis, the apoptosis rates of ovarian cancer cell lines A2780/CP70, OVCAR-3, and normal ovarian cells IOSE-364 were treated with myricetin (5–30 μM) for 24 h, and analyzed by flow cytometry after Annexin V and propidium iodide (PI) staining. As shown in Fig. 2A, myricetin significantly induced apoptosis in the two ovarian cancer cells, especially in OVCAR-3, but it did not induce apoptosis in the normal ovarian cells IOSE-364. For A2780/CP70 and OVCAR-3, the apoptosis rates were 3.0 and 3.8% when not treated with myricetin, which was increased to the maximum apoptosis rates of 42.1 and 84.2% at the concentration of 30 μM myricetin. However, the treatment of myricetin did not increase the IOSE-364 cell apoptosis rate (Fig. 2A). Caspase-Glo 3/7 Assay kit was used to confirm that myricetin induces apoptosis in ovarian cancer cells. As shown in Fig. 2B, compared to controls, the caspase-3/7 enzymatic activities were maximally increased to 1.68- and 2.20-fold in A2780/CP70 and OVCAR-3 when treated with 20 μM myricetin for 4 h. These results indicate that myricetin inhibited cell growth of ovarian cancer cells at least partly through the induction of apoptosis.
Figure 2.
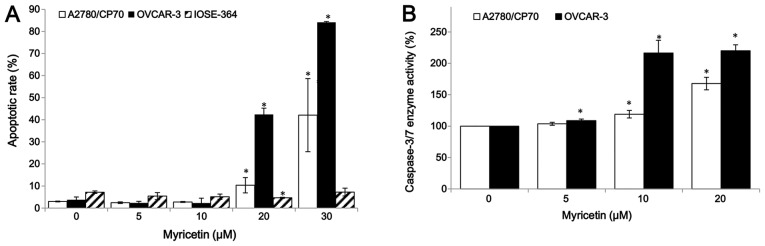
Myricetin induced apoptosis in ovarian cancer cells A2780/CP70 and OVCAR-3 but not the normal ovarian cells IOSE-364. (A) Cells treated with myricetin for 24 h, were collected, stained with PI and Annexin V, analyzed by flow cytometry to measure the apoptotic rate. (B) Cells were treated with myricetin for 4 h, and caspase-3/7 enzymatic activity was determined using Caspase-Glo 3/7 assay kit. *p<0.05 as compared to control.
Myricetin does not induce cell cycle arrest in A2780/CP70 and OVCAR-3 cells
To investigate whether the growth-inhibitory effect of myricetin on ovarian cancer cells was caused by cell cycle arrest, cell cycle phase distribution of cells treated with myricetin (5–20 μM) for 24 h was analyzed by flow cytometry after PI staining. As shown in Fig. 3, myricetin had little effect on inducing cell cycle arrest in A2780/CP70 and OVCAR-3 cells. These results indicate that myricetin inhibited ovarian cancer cell growth through a mechanism separate from cell cycle arrest.
Figure 3.
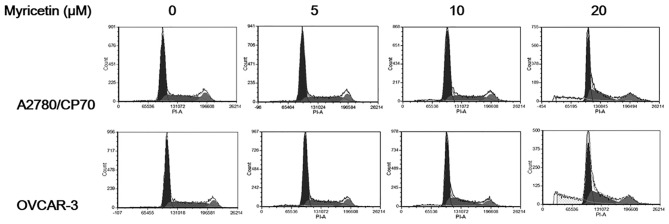
Myricetin did not induce cell cycle arrest in ovarian cancer cells A2780/CP70 and OVCAR-3. Cells treated with myricetin for 24 h, were collected, stained with PI, analyzed by flow cytometry to measure cell phase division.
Effect of myricetin on the intrinsic and extrinsic apoptotic pathway
The intrinsic apoptotic pathway and extrinsic apoptotic pathway are the two main pathways which induce apoptosis in cancer cells. An investigation was conducted to determine whether myricetin induces apoptosis through the intrinsic pathway in ovarian cancer cell lines A2780/CP70 and OVCAR-3. The expression of Puma, Bcl-2, Bcl-xl, Bax, Bad and caspase-9 proteins were detected by western blotting. As shown in Fig. 4A, the expression of Bax and Bad proteins was increased, while the Bcl-2 and Bcl-xl protein levels were decreased when treated with myricetin. However, the procaspase-9 and Puma protein levels were not changed by treatment with myricetin. The cleaved caspase-9 protein was not detected, suggesting that it may be degraded after activation from procaspase-9. Caspase-Glo 9 Assay kit was used to detect changes in caspase-9 enzymatic activities when treated with myricetin. The test was found to be consistent with the western blotting result, which shows that myricetin had no effect on the caspase-9 enzymatic activities of A2780/CP70 and OVCAR-3 cells (Fig. 4B). These results indicate that myricetin might induce the intrinsic pathway in A2780/CP70 and OVCAR-3 cells through Bcl-2 family proteins.
Figure 4.

Myricetin activated the intrinsic and extrinsic apoptotic pathway. (A) Myricetin decreased the levels of procaspase-9, Bcl-2 and Bcl-xl and increased levels of Bad and Bax in A2780/CP70 and OVCAR-3 ovarian cancer cells. However, myricetin had no effect on the expression of Puma protein. (B) Cells were treated with myricetin 4 h, and determined caspase-9 enzymatic activities using Caspase-Glo 9 Assay kit. *p<0.05 as compared to control. (C) Myricetin decreased the levels of procaspase-8 and increased levels of DR5 (p40) in OVCAR-3 but not A2780/CP70 cells. However, myricetin had no effect on the expression of FADD in either of the ovarian cancer cells.
Next, the role of the extrinsic pathway in myricetin-induced apoptosis was determined. Myricetin increased the expression of matured DR5 and decreased the levels of procaspase-8 in OVCAR-3 cells but not A2780/CP70 cells (Fig. 4C). FADD protein levels were not changed in both ovarian cancer cell lines treated with myricetin. Myricetin might induce the extrinsic pathway in OVCAR-3 cells through a DR5 and caspase-8 dependent pathway. However, the induction of apoptosis in A2780/CP70 cells might be through an extrinsic-independent pathway.
Role of p53 in the myricetin-induced apoptosis in OVCAR-3 cells
P53, a tumor suppressor protein, is involved in several cellular outcomes such as apoptosis and angiogenesis (20). Therefore, we examined some proteins which are associated with p53 to clarify whether myricetin inhibits viability in A2780/CP70 and OVCAR-3 cells via the p53 protein (Fig. 5A). We found that myricetin upregulated expression of the p53 and p21 proteins and downregulated expression of the oncogene cmyc protein.
Figure 5.

Myricetin induces apoptosis on ovarian cancer cells in a p53-dependent pathway. (A) Myricetin decreased the expression of protein cmyc, and increased levels of protein p21 and p53. (B) Cells were treated with myricetin and p53 inhibitor pifithrin-α (PFT-α) for 24 h, stained with Hoechst 33342, apoptotic rate measured by fluorescence microscope. *p<0.05 as compared to myricetin-treated control. (C) Cells were treated with myricetin and PFT-α for 4 h, and caspase-3/7 enzymatic activity was determined using Caspase-Glo 3/7 assay kit. *p<0.05 as compared to myricetin-treated control. (D) Knockdown p53 resulted in abrogation of myricetin-increased levels of Bad, Bax, p21 and p53 and decreased levels of cmyc, Bcl-2 and Bcl-xl in OVCAR-3 cells. Cells were seeded in 60-mm dishes, incubated overnight, and then transfected with p53 siRNA or control siRNA. After 24 h, cells were treated with myricetin or DMSO. Cell lysates were collected for western blotting.
The inhibitor of pifithrin-α (PFT-α) was used to investigate the role of p53 on myricetin-induced apoptosis in OVCAR-3 cells. Treatment with 20 μM pifithrin-α significantly reduced the apoptotic rates and caspase-3/7 enzymatic activities in OVCAR-3 cells induced by myricetin (Fig. 5B and C). Fig. 5D shows knockdown of p53 by specific siRNA (50 nM) resulted in abrogation of myricetin-increased levels of Bax, Bad, and p21 proteins and myricetin-decreased levels of cmyc and Bcl-xl proteins. These results indicate that p53-associated intrinsic pathways were, at least partly, involved in myricetin-induced apoptosis in ovarian cancer cells.
Role of p21 in the myricetin-induced apoptosis in OVCAR-3 cells
The p21 siRNA was used to investigate the role of p21 on myricetin-induced apoptosis in OVCAR-3 cells. Knockdown of p21 by siRNA (50 nM) did not abrogate the apoptosis and caspase-3/7 enzymatic activation in OVCAR-3 cells which was induced by treatment of myricetin (Fig. 6). These results indicate that myricetin induced apoptosis in OVCAR-3 cells through a p21-independent pathway.
Figure 6.
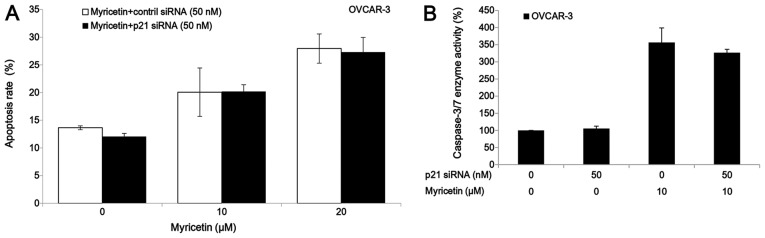
Myricetin induces apoptosis on ovarian cancer cells in a p21-independent pathway. (A) Knockdown p21 had no effect on the myricetin-induced apoptosis in OVCAR -3 cells. Cells were seeded in 60-mm dishes, incubated overnight, and then transfected with p21 siRNA or control siRNA. After 24 h, cells were treated with myricetin or DMSO for another 24 h, stained with Hoechst 33342, apoptotic rate was measured by fluorescence microscope. *p<0.05 as compared to myricetin-treated control. (B) Cells were transfected with p21 siRNA or control siRNA for 24 h, treated with myricetin or DMSO for another 24 h, caspase-3/7 enzymatic activity was determined using Caspase-Glo 3/7 assay kit. *p<0.05 as compared to myricetin-treated control.
Discussion
A pressing problem in cancer treatment is that platinum-based chemotherapy is seriously hampered by high rates of adverse effects and chemoresistance (21). There exists a dire need for more selective drugs in the treatment of cancer. Previous studies have reported that myricetin induces apoptosis in various cancer cells, such as hepatoma, pancreatic cancer, esophageal cancer, and colon carcinoma cells (15–18). However, its effects on ovarian cancer are currently unknown. In this study, it was found that myricetin was more cytotoxic to two cisplatin-resistant ovarian cancer cell lines A2780/CP70 and OVCAR-3 than normal ovarian cells IOSE-364, and that myricetin had stronger anticancer activity than cisplatin in the two ovarian cancer cell lines when compared to our previous study (19).
The mechanism through which cancer cells develop resistance to chemotherapy is associated with increased resistance to apoptosis. In pre-clinical disease models, agents that induce apoptosis have been reported to sensitize tumor cells to chemotherapy and radiotherapy (22). There are two major pathways leading to apoptosis: the intrinsic or mitochondrial and extrinsic or receptor-mediated pathways. The permeability of the mitochondria and release of cytochrome c into the cytoplasm will be increased when the intrinsic apoptotic pathway is activated. After that, cytochrome c forms a multi-protein complex, known as the apoptosome, initiating activation of caspase-9. Bcl-2 protein family plays an important role in the regulation of the intrinsic apoptotic pathway through controlling the permeability of the mitochondrial membrane and the release of pro-apoptotic factors (10). Whether or not cells will undergo apoptosis is dependent on the balance between the pro- (such as Bax and Bad) and anti-apoptotic (such as Bcl-xl and Bcl-2) proteins of the family members. In the extrinsic pathway, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), including the Apo2L/TRAIL, regulates the extrinsic apoptotic pathway by engaging its receptor, such as DR5. The receptor homotypically binds to FAS-associated death domain protein (FADD) to form death inducing signaling complex (DISC), activating caspase-8 and -10. Activation of either the intrinsic pathway of apoptosis or the extrinsic pathway results in activation of caspase-3 and -7 culminating in apoptosis. In this study, myricetin was found to induce intrinsic apoptosis through the Bcl-2 protein but not the caspase-9 enzyme in A2780/CP70 and OVCAR-3 cells. A previous study indicated that myricetin induces apoptosis in colon cancer cells through increasing the ratio of Bax/Bcl-2 proteins (18), which agrees with the results obtained here. The effect of myricetin on the extrinsic apoptotic pathway was also examined by testing the expression of DR5, FADD, and caspase-8 proteins. It was found that myricetin increased the levels of DR5 protein and decreased the levels of procaspase-8 in the OVCAR-3 but not the A2780/CP70 cells, suggesting that myricetin induces apoptosis in OVCAR-3 cells though a DR5-associated extrinsic pathway. The alterations in the balance of Bcl-2/Bax proteins was associated with the differential induction of apoptosis in cancer versus normal cells (23). Evidence from clinical trials has indicated that normal cells are resistant to the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and targeting DR5 selectively eliminates tumor cells while sparing normal cells (24). This study has shown that myricetin induced apoptosis in ovarian cancer cells A2780/CP70 and OVCAR-3 but not normal ovarian cells IOSE-364, which might be due to its effects on the expression of the Bcl-2 family and DR5 protein.
The cell cycle represents a series of events that allow the cell to replicate into two daughter cells. Many cancer cells have defective G1 checkpoint mechanisms and are more dependent on the G2 checkpoint during replication than normal cells. Cancer represents a dysregulation of the cell cycle, such as an overexpression of cyclins or insufficient expression of CDKIs, which result in cell growth and tumor formation (7). Therefore, the innovative strategy of cell cycle arrest, which activates the apoptotic cascade and leads to cell death, was developed. Novel anticancer drugs have been focused on as the target of cell cycle control mechanisms (8,9). Previous studies indicated that myricetin induces G2/M blockage in human squamous cell carcinoma cell lines SCC-25 and human colon cancer cell lines HCT116 (25,26). However, in the present study, the cell cycle in human ovarian cancer cells was not affected by treatment of myricetin, which means myricetin inhibited ovarian cancer cell growth through a mechanism separate from inducing cell cycle arrest.
p53, as a multifunctional tumor suppressor, regulates cell cycle arrest, transcription, DNA repair, genomic instability, senescence, differentiation, angiogenesis, apoptosis, and glucose metabolism (20). If the p53 gene is damaged, tumor suppression will be under serious threat. As shown in previous studies, p53 alterations, such as loss of function dominant-negative activity and gain of oncogene function, has been connected with the failure of chemotherapy and radiotherapy in a number of cancers (27). More than 50% of human tumors contain p53 gene that is mutated or deleted (28), and people who have only one functional copy of the p53 gene will probably suffer from tumors in early adulthood. Therefore, increasing the amount of p53 might be a new strategy for treatment of tumors. The intrinsic pathways of apoptosis such as Bax, Puma, and Noxa are regulated by p53 (29). It has been reported that the Bcl-2 family genes are regulated by p53, and cancer cells with loss of p53 function are expected to contain relatively low levels of Bax, Bad and high levels of Bcl-2 and Bcl-xl (30,31). In ovarian cancer cells, p53 also has an effect on cell apoptosis (32). In the present study, myricetin was observed to increase p53 protein expression in the ovarian cancer cells. In OVCAR-3 cells, the myricetin-induced balance alterations of Bcl-2 family proteins were associated with increased p53 protein, indicating that p53 played an important role in the myricetin-activated intrinsic pathway of apoptosis. The transcription factor cmyc protein is encoded by a regulator. It is a nuclear protein which plays a role in the cell cycle, apoptosis, progression, and cellular transformation. Previous studies indicated that p53-dependent repression of cmyc was involved in cell cycle arrest and apoptosis (33,34). In this study, myricetin decreased the cmyc protein expression in A2780/CP70 and OVCAR-3 cells, and knockdown of protein p53 neutralized the repressive effect myricetin had on cmyc expression in OVCAR-3 cells. Therefore, it is reasonable to think that p53 induced apoptosis through a cmyc-dependent manner. Of course, more studies are needed to clarify the role of cmyc in myricetin-induced apoptosis in ovarian cancer cells. Additionally, it was seen that myricetin increased the expression of p21 protein in both ovarian cancer cell lines, and it was mediated by p53 in OVCAR-3 cells. Therefore, it was inferred that p21 might be involved in myricetin-induced apoptosis and the inhibition of angiogenesis in OVCAR-3 cells.
P21, known as CDK-interacting protein 1 or cyclin-dependent kinase inhibitor 1, regulates cell cycle progression, tightly regulated by p53 protein (35). It has been reported that myricetin increased levels of p21 protein in HepG2 cells which resulted in cell cycle arrest at the G2/M phase (36). However, in the present study, the increased levels of p21 protein had little effect on the cell cycle in A2780/CP70 and OVCAR-3 cells. Some previous studies indicated that p21 was a positive regulator of apoptosis in either p53-dependent or independent pathways, while other studies showed p21 inhibited p53-dependent apoptosis (37). In our study, although myricetin increased levels of p21 protein in OVCAR-3 cells, it was not associated with apoptosis.
In conclusion, the present study suggests that myricetin, which exhibited higher cytotoxicity to two cisplatin-resistant ovarian cancer cell lines than in normal ovarian cells, might be a potential candidate for the chemoprevention of ovarian cancer. It induces apoptosis in both ovarian cancer cells through a Bcl-2 family protein-associated intrinsic pathway. In OVCAR-3, myricetin also induces DR-5 associated extrinsic apoptosis. P53 plays an important role in Bcl-2 family protein-dependent apoptosis, induced by myricetin, in OVCAR-3 cells. Ostensibly, further studies in animal models and human trials are needed to determine the efficacy of this compound for the treatment of ovarian cancer.
Acknowledgements
We thank Dr Kathy Brundage from the Flow Cytometry Core at West Virgina University for providing technical help on apoptosis and cell cycle analysis, and Yu Fu for giving a critical review of the manuscript. This study was supported by a West Virginia Experimental Program to Stimulate Competitive Research grant and NIH grants (P20RR016477 and P20GM103434) from the National Institutes of Health awarded to the West Virginia IDeA Network of Biomedical Research Excellence. This study was also supported by the Chinese National Key Technologies R&D Program of 12th Five-year Plan (2012BAD31B06).
Abbreviations
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- PFT-α
pifithrin-α
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- siRNA
small interfering RNA
- DR5
death receptor 5
References
- 1.Stordal B, Davey M. Understanding cisplatin resistance using cellular models. IUBMB Life. 2007;59:696–699. doi: 10.1080/15216540701636287. [DOI] [PubMed] [Google Scholar]
- 2.Loehrer PJ, Einhorn LH. Drugs five years later. Cisplatin Ann Intern Med. 1984;100:704–713. doi: 10.7326/0003-4819-100-5-704. [DOI] [PubMed] [Google Scholar]
- 3.Milosavljevic N, Duranton C, Djerbi N, Puech PH, Gounon P, Lagadic-Gossmann D, Dimanche-Boitrel MT, Rauch C, Tauc M, Counillon L, et al. Nongenomic effects of cisplatin: Acute inhibition of mechanosensitive transporters and channels without actin remodeling. Cancer Res. 2010;70:7514–7522. doi: 10.1158/0008-5472.CAN-10-1253. [DOI] [PubMed] [Google Scholar]
- 4.Windsor RE, Strauss SJ, Kallis C, Wood NE, Whelan JS. Germline genetic polymorphisms may influence chemotherapy response and disease outcome in osteosarcoma: A pilot study. Cancer. 2012;118:1856–1867. doi: 10.1002/cncr.26472. [DOI] [PubMed] [Google Scholar]
- 5.Levi JA, Aroney RS, Dalley DN. Haemolytic anaemia after cisplatin treatment. Br Med J (Clin Res Ed) 1981;282:2003–2004. doi: 10.1136/bmj.282.6281.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu Rev Pharmacol Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 7.Champeris Tsaniras S, Kanellakis N, Symeonidou IE, Nikolopoulou P, Lygerou Z, Taraviras S. Licensing of DNA replication, cancer, pluripotency and differentiation: An interlinked world? Semin Cell Dev Biol. 2014;30:174–180. doi: 10.1016/j.semcdb.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 8.Buolamwini JK. Cell cycle molecular targets in novel anticancer drug discovery. Curr Pharm Des. 2000;6:379–392. doi: 10.2174/1381612003400948. [DOI] [PubMed] [Google Scholar]
- 9.Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138:255–271. doi: 10.1016/j.pharmthera.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gates MA, Vitonis AF, Tworoger SS, Rosner B, Titus-Ernstoff L, Hankinson SE, Cramer DW. Flavonoid intake and ovarian cancer risk in a population-based case-control study. Int J Cancer. 2009;124:1918–1925. doi: 10.1002/ijc.24151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong KC, Khoo HE. Biological effects of myricetin. Gen Pharmacol. 1997;29:121–126. doi: 10.1016/S0306-3623(96)00421-1. [DOI] [PubMed] [Google Scholar]
- 13.Ross JA, Kasum CM. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu Rev Nutr. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]
- 14.Basli A, Soulet S, Chaher N, Mérillon JM, Chibane M, Monti JP, Richard T. Wine polyphenols: Potential agents in neuro-protection. Oxid Med Cell Longev. 2012;2012:805762. doi: 10.1155/2012/805762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang XH, Chen SY, Tang L, Shen YZ, Luo L, Xu CW, Liu Q, Li D. Myricetin induces apoptosis in HepG2 cells through Akt/p70S6K/bad signaling and mitochondrial apoptotic pathway. Anticancer Agents Med Chem. 2013;13:1575–1581. doi: 10.2174/1871520613666131125123059. [DOI] [PubMed] [Google Scholar]
- 16.Phillips PA, Sangwan V, Borja-Cacho D, Dudeja V, Vickers SM, Saluja AK. Myricetin induces pancreatic cancer cell death via the induction of apoptosis and inhibition of the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Cancer Lett. 2011;308:181–188. doi: 10.1016/j.canlet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zang W, Wang T, Wang Y, Li M, Xuan X, Ma Y, Du Y, Liu K, Dong Z, Zhao G. Myricetin exerts anti-proliferative, anti-invasive, and pro-apoptotic effects on esophageal carcinoma EC9706 and KYSE30 cells via RSK2. Tumour Biol. 2014;35:12583–12592. doi: 10.1007/s13277-014-2579-4. [DOI] [PubMed] [Google Scholar]
- 18.Kim ME, Ha TK, Yoon JH, Lee JS. Myricetin induces cell death of human colon cancer cells via BAX/BCL2-dependent pathway. Anticancer Res. 2014;34:701–706. [PubMed] [Google Scholar]
- 19.Li B, Gao Y, Rankin GO, Rojanasakul Y, Cutler SJ, Tu Y, Chen YC. Chaetoglobosin K induces apoptosis and G2 cell cycle arrest through p53-dependent pathway in cisplatin-resistant ovarian cancer cells. Cancer Lett. 2015;356:418–433. doi: 10.1016/j.canlet.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darcy KM, Brady WE, McBroom JW, Bell JG, Young RC, McGuire WP, Linnoila RI, Hendricks D, Bonome T, Farley JH Gynecologic Oncology Group. Associations between p53 overexpression and multiple measures of clinical outcome in high-risk, early stage or suboptimally-resected, advanced stage epithelial ovarian cancers A Gynecologic Oncology Group study. Gynecol Oncol. 2008;111:487–495. doi: 10.1016/j.ygyno.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang YI, Kim JH, Lee KT, Choi JH. Costunolide induces apoptosis in platinum-resistant human ovarian cancer cells by generating reactive oxygen species. Gynecol Oncol. 2011;123:588–596. doi: 10.1016/j.ygyno.2011.08.031. [DOI] [PubMed] [Google Scholar]
- 22.Wilson TR, Johnston PG, Longley DB. Anti-apoptotic mechanisms of drug resistance in cancer. Curr Cancer Drug Targets. 2009;9:307–319. doi: 10.2174/156800909788166547. [DOI] [PubMed] [Google Scholar]
- 23.Liu T, Hannafon B, Gill L, Kelly W, Benbrook D. Flex-Hets differentially induce apoptosis in cancer over normal cells by directly targeting mitochondria. Mol Cancer Ther. 2007;6:1814–1822. doi: 10.1158/1535-7163.MCT-06-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozören N, El-Deiry WS. Cell surface death receptor signaling in normal and cancer cells. Semin Cancer Biol. 2003;13:135–147. doi: 10.1016/S1044-579X(02)00131-1. [DOI] [PubMed] [Google Scholar]
- 25.Maggioni D, Nicolini G, Rigolio R, Biffi L, Pignataro L, Gaini R, Garavello W. Myricetin and naringenin inhibit human squamous cell carcinoma proliferation and migration in vitro. Nutr Cancer. 2014;66:1257–1267. doi: 10.1080/01635581.2014.951732. [DOI] [PubMed] [Google Scholar]
- 26.Shiomi K, Kuriyama I, Yoshida H, Mizushina Y. Inhibitory effects of myricetin on mammalian DNA polymerase, topoisomerase and human cancer cell proliferation. Food Chem. 2013;139:910–918. doi: 10.1016/j.foodchem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 27.Kong D, Ma S, Liang B, Yi H, Zhao Y, Xin R, Cui L, Jia L, Liu X, Liu X. The different regulatory effects of p53 status on multidrug resistance are determined by autophagy in ovarian cancer cells. Biomed Pharmacother. 2012;66:271–278. doi: 10.1016/j.biopha.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 29.Kuo YC, Kuo PL, Hsu YL, Cho CY, Lin CC. Ellipticine induces apoptosis through p53-dependent pathway in human hepatocellular carcinoma HepG2 cells. Life Sci. 2006;78:2550–2557. doi: 10.1016/j.lfs.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 30.Basu A, Haldar S. The relationship between BcI2, Bax and p53: Consequences for cell cycle progression and cell death. Mol Hum Reprod. 1998;4:1099–1109. doi: 10.1093/molehr/4.12.1099. [DOI] [PubMed] [Google Scholar]
- 31.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/S1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 32.Guan YQ, Li Z, Yang A, Huang Z, Zheng Z, Zhang L, Li L, Liu JM. Cell cycle arrest and apoptosis of OVCAR-3 and MCF-7 cells induced by co-immobilized TNF-α plus IFN-γ on polystyrene and the role of p53 activation. Biomaterials. 2012;33:6162–6171. doi: 10.1016/j.biomaterials.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 33.Ho JS, Ma W, Mao DY, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol. 2005;25:7423–7431. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 35.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- 36.Zhang XH, Zou ZQ, Xu CW, Shen YZ, Li D. Myricetin induces G2/M phase arrest in HepG2 cells by inhibiting the activity of the cyclin B/Cdc2 complex. Mol Med Rep. 2011;4:273–277. doi: 10.3892/mmr.2011.417. [DOI] [PubMed] [Google Scholar]
- 37.Piccolo MT, Crispi S. The dual role played by p21 may influence the apoptotic or anti-apoptotic fate in cancer. J Cancer Res Updates. 2012;1:189–202. [Google Scholar]