

Abstract
Multikinase inhibitors (MKIs) targeting VEGF receptors and other receptor tyrosine kinases have shown considerable activity in clinical trials of thyroid cancer. Thyroid cancer frequently exhibits activation of the RAS/RAF/MEK/ERK pathway. In other types of cancer, paradoxical ERK activation has emerged as a potential resistance mechanism to RAF-inhibiting drugs including MKIs such as sorafenib and pazopanib. We therefore queried whether the MEK inhibitor trametinib, could augment the activity of pazopanib in thyroid cancer cell lines. Trametinib potently inhibited growth in vitro (GI50 1.1–4.8 nM), whereas pazopanib had more limited in vitro activity, as anticipated (GI50 1.4–7.1 µM). We observed progressive upregulation of ERK activity with pazopanib treatment, an effect abrogated by trametinib. For xenografts (bearing either KRASG12R or BRAFV600E mutations), the combination of trametinib and pazopanib led to sustained shrinkage in tumor volume by 50% or more, compared to pre-treatment baseline. Trametinib also was highly effective as a single agent, compared to pazopanib alone. These preclinical findings support the evaluation of trametinib, alone or in combination with pazopanib or other kinase inhibitors, in thyroid cancer clinical trials. We highlight the importance of pharmacodynamic assessment of the ERK pathway for patients enrolled in trials involving MKIs.
Keywords: differentiated thyroid cancer, MEK, BRAF, ERK, angiogenesis
Introduction
Thyroid cancer accounts for 96% of endocrine malignancies and causes ~1,900 deaths annually in the USA (1). Except for medullary thyroid cancer, thyroid cancers are derived from thyroid follicular cells. In the known mutational spectrum of these follicular cell-derived thyroid cancers, non-overlapping mutations activating the ERK pathway predominate, including BRAF, RAS and RET-PTC gene re-arrangements (2). Additional genetic alterations specifically affect the PI3K pathway, including PIK3CA, PTEN and AKT1 (2,3).
Systemic treatment of patients with advanced, metastatic, radioiodine-refractory thyroid cancer has recently evolved to include multikinase inhibitors (MKIs). These drugs target VEGF receptors (principally VEGFR2), plus overlapping sets of other receptor tyrosine kinases (RTKs). In addition to these main targets of action, several of the kinase inhibitors used in thyroid cancer, including sorafenib and pazopanib, have been shown to more weakly target RAF kinases including BRAF (4,5).
Notably, RAF inhibition, particularly at sub-maximal levels, has been shown to result in paradoxical ERK pathway activation in certain settings. In one well-studied scenario, an upstream RAS mutation was able to overdrive signaling through RAF protein heterodimers, even when one RAF molecule is bound to a BRAF inhibitor (6–9). This concern over paradoxical ERK activation has led to the restriction of potent RAF inhibitor drugs such as vemurafenib to BRAF mutant tumor settings, avoiding RAS mutant tumors in particular. Weaker RAF inhibitors such as sorafenib also have the potential to induce BRAF-CRAF heterodimers and cause paradoxical ERK activation (10). In thyroid cancer cell lines treated with vemurafenib, paradoxical ERK activation also occurs in BRAF mutant settings, due to feedback activation of ERBB3 (11). There are currently insufficient data in thyroid cancer to assess whether paradoxical RAF activation occurs in patients treated with MKIs.
Pazopanib is a small molecule MKI that selectively inhibits VEGFR1-3, PDGFR-α, PDGFR-β, KIT and FGFR1-3 in the low nanomolar range (12). Similar to sorafenib, pazopanib recently has been shown to be a RAF inhibitor as well (5). In a cell-based assay, the IC50 value was 59 nM for wild-type BRAF and 148 nM for BRAFV600E. Pazopanib has been approved for use in advanced renal cell carcinoma and advanced refractory soft tissue sarcoma (13,14), and has been studied in phase II trials for a variety of types of cancer, including breast, thyroid and cervical cancer. Phase II data in advanced, progressive differentiated thyroid cancer (DTC) from Bible et al, indicate a 49% response rate, one of the highest response rates reported for any agent in thyroid cancer (15). In this high-risk patient population screened to have disease progression within 6 months, Bible et al reported a median progression-free survival (PFS) of 11.7 months on pazopanib. Interestingly this study suggested a trend toward a higher response rate in follicular than in papillary thyroid cancer.
Trametinib (GSK1120212) is a potent, highly specific allosteric inhibitor of MEK1 and MEK2 (IC50 value <5 nM). In patients with BRAF-mutant malignant melanoma in a phase III clinical trial, single agent trametinib was associated with enhanced progression-free survival (PFS) and overall survival (OS) compared to chemotherapy (16). Trametinib was approved by the FDA for unresectable or metastatic BRAF-mutant melanoma in May 2013, and in combination with dabrafenib for BRAF-mutant melanoma in January 2014.
Our goal in the present preclinical study was to determine whether adding the potent MEK inhibitor trametanib to the clinically useful MKI, pazopanib, could enhance the activity of pazopanib in preclinical models of thyroid cancer. Based on the reported ability of pazopanib to inhibit RAF in vitro (5), we considered it likely that paradoxical activation of ERK could be occurring in pazopanib-treated tumor cells. Due to the high frequency of mutations activating the ERK pathway in thyroid and the potential for paradoxical ERK activation in some tumors, we hypothesized that addition of trametinib could be a rational complement to pazopanib in treating thyroid cancer.
Materials and methods
Cell lines
BCPAP, 8505C and CAL62 cells were obtained from the German Collection of Microorganisms and Cell Culture (DSMZ, Braunschweig, Germany). Cell lines were cultured in RPMI-1640 with 10% FBS. All media were supplemented with penicillin-streptomycin.
Inhibitor treatments
Trametinib and pazopanib were obtained from GlaxoSmithKline (King of Prussia, PA, USA). Stock solutions (10 mM) were prepared in dimethyl sulfoxide (DMSO). For analysis of ERK pathway inhibition, cultured cells were treated with indicated doses of inhibitors for 4 h or for 5 days with medium change and fresh drug at day 3. For growth inhibition assays, cells were treated for 5 days with medium change and fresh drug at day 3.
Immunoblotting
Cells were treated for 4 h or 5 days as described above, then washed with PBS and harvested by scraping with 1X sodium dodecyl sulfate lysis buffer [2% sodium dodecyl sulfate and 62.5 mM Tris (pH 6.8)]. Lysates were electrophoresed on 4–20% gradient polyacrylamide gels and transferred onto PVDF membranes. Blots were probed at 4°C overnight with a primary antibody to pERK (CST, Beverly, MA, USA; no. 9101) diluted 1:1,000 in 5% milk, total ERK (CST; no. 9102) and GAPDH (Trevigen). Anti-rabbit secondary antibodies (Santa Cruz) were diluted 1:2,000. Blots were visualized using SuperSignal Pico Chemiluminescence (Pierce Chemical Co., Rockford, IL, USA).
Growth analyses
Growth assays were performed in triplicate using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazo-lium bromide (MTT) assay (M2128; Sigma-Aldrich) following the manufacturer's instructions. Cells were seeded in 24-well plates using phenol red-free media. MTT absorbance was determined 5 days after exposure to drugs or DMSO alone. Data are represented as the mean absorbance ± SEM, based on 3–6 independent experiments, normalized to control cells. GI50 was determined as the x-intercept of Log10(fa/fu) plotted vs. Log10(concentration), determined by linear regression (17). GI50 values were reported as the means ± standard deviation of 3–5 independent experiments.
Animal studies
Animal studies were approved by the Johns Hopkins Animal Care and Use Committee and performed in accordance with NIH guidelines. CAL62 or 8505C cells suspended in Matrigel (5×106 cells/200 µl) were inoculated s.c. into the right flank of 4- to 6-week-old female athymic nude (nu/nu) mice (Harlan Laboratories, Indianapolis, IN, USA). Once palpable, tumor volumes were calculated with calipers using the formula: Length × width × height × 0.5236. After tumors reached 0.1–0.2 cm3 in size, animals were sorted into groups of 11 and randomized for drug assignment, to achieve equal distribution of tumor size in all treatment groups. Animals were treated with DMSO vehicle alone, trametinib (1 mg/kg daily), pazopanib (100 mg/kg daily) or a combination of both drugs, by oral gavage. Animals were euthanized by CO2 asphyxiation and cervical dislocation when tumors reached ~1.5 cm3. Kaplan-Meier analysis for tumor progression was performed with GraphPad Prism version 5 software, using a 3-fold increase in tumor volume from onset of treatment as a threshold for tumor progression.
Immunohistochemistry
Four hours following treatment with drugs or DMSO vehicle, mice were sacrificed, and tumors were harvested in 10% paraformaldehyde overnight, followed by incubation in 70% ethanol. Paraffin section slides were incubated with anti-pERK (CST; no. 4376; 1:100), pAKT (CST; no. 3787; 1:50) and developed using a Vectastain ABC kit (Vector Labs, Burlingame, CA, USA) with DAB as chromogen, and a hematoxylin counterstain. Anti-CD31 staining (Abcam, Cambridge, MA, USA; ab28364; 1:50) followed a pressure cooker antigen retrieval method according to the manufacturer's protocol. Microvessel density was estimated as the mean of CD31-reactive foci in ten 200 power fields in the tumor periphery, using a two-tailed t-test with equal variance for significance. Light microscopy was performed with an Olympus Vanox and Nikon DMX1200 CCD camera, using Nikon ACT-1 image capture software.
Results
Growth inhibition of thyroid cancer cell lines by trametinib and pazopanib
In preliminary dose-response studies, we found that trametinib efficiently inhibited ERK activation and growth in thyroid cancer cell lines at low nanomolar doses. Pazopanib treatment caused in vitro growth inhibition at low micromolar dose ranges. We therefore tested dose ranges from 0.1 to 10 nM of trametinib and from 0.5 to 20 µM of pazopanib to determine GI50 values for these drugs. [Trough plasma pazopanib concentrations achieved in clinical trials may exceed 30 µM at the typical 800 mg daily dose (18)]. Table I shows calculated GI50 values for single agent trametinib or pazopanib against the thyroid cancer cell lines CAL62, BCPAP and 8505C. All three cell lines were highly sensitive to trametinib with a GI50 ranging from 1.1 to 4.8 nM. Pazopanib GI50 values ranged from 1.4 to 7.1 µM. Clearly, trametinib alone was highly active in vitro for thyroid cancer cell lines bearing either a RAS- or BRAF-mutant genotype; pazopanib was relatively less active as a single agent in a cell culture setting, as anticipated.
Table I.
Inhibition of thyroid cancer growth in vitro by trametinib and pazopanib.
| Cell line | Genotype | GI50
|
|
|---|---|---|---|
| Trametinib (nM) | Pazopanib (µM) | ||
| CAL62 | KRASG12R/G12R | 4.8±0.3 | 1.4±0.4 |
| BCPAP | BRAFV600E/wt | 4.0±1.7 | 3.2±0.5 |
| 8505C | BRAFV600E/V600E | 1.1±0.6 | 7.1±1.5 |
Trametinib abrogates ERK activation caused by pazopanib
We hypothesized that pazopanib treatment could potentially cause paradoxical ERK pathway activation in thyroid cancer, based on studies performed in breast cancer and other types of tumor (5). To explore the ERK pathway signaling effects of pazopanib and trametinib in vitro, we tested both acute and 5 day responses in CAL62 cells (KRASG12R) and BCPAP cells (BRAFV600E). Initial data indicated an IC50 value for trametinib of 1–2 nM for pERK inhibition in CAL62 cells and complete loss of pERK signal at doses >4 nM (data not shown), confirming earlier data derived from other types of tumor (19). Fig. 1 illustrates that pazopanib alone was associated with upregulation of pERKT202/Y204 at 4 h and 5 days in the RAS mutant line, CAL62. No corresponding changes were observed in total ERK abundance. The pERK upregulation was fully abrogated by combination with trametinib 10 nM at 4 h, and was significantly reduced at 5 days. Pazopanib also was associated with ERK activation in the BRAF-mutant cell line BCPAP at 5 days but not at 4 h. Combination treatment with trametinib again reduced this activation. Since pazopanib has multiple effects in an in vivo tumor setting relevant to ERK, beyond the in vitro effects modeled here, we returned to this phenomenon in tumor xenografts (see below, Fig. 3).
Figure 1.
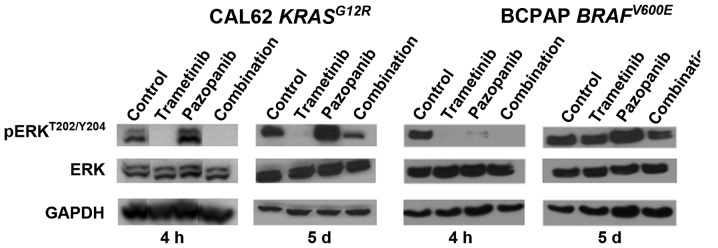
Trametinib reduces basal and pazopanib-associated ERK activation in cultured DTC. CAL62 or BCPAP cells were treated 4 h or 5 days (as described in Materials and methods) with vehicle, trametinib 10 nM, pazopanib 800 nM, or the combination. Lysates were analyzed for pERK by immunoblotting. Control levels of total ERK and GAPDH are also shown. pERK was acutely and chronically upregulated by pazopanib in CAL62 cells in vitro, mitigated by combination with trametinib. DTC, differentiated thyroid cancer.
Figure 3.

Trametinib with pazopanib reduces ERK and AKT activation in vivo. Established 8505C and CAL62 xenograft tumors treated with the indicated drugs for 2–3 weeks were harvested 4 h after dosing. Immunohistochemistry for pERKT202/Y204 and pAKTS473 was performed on formalin-fixed, paraffin-embedded sections. ERK activation was significantly inhibited by trametinib or trametinib plus pazopanib but not by single agent pazopanib. (Scale bar, 20 µm; DAB used as chromogen).
Tumor size reduction in thyroid cancer xenografts treated with trametinib and pazopanib
In limited published studies to date, pazopanib has been shown to inhibit the growth of xenografts derived from anaplastic thyroid cancer cells (20). In order to test the in vivo activity of pazopanib plus trametinib in thyroid cancer, athymic nu/nu mice bearing palpable 8505C or CAL62 tumors were randomized to receive daily trametinib, pazopanib, a combination of both drugs or vehicle alone, by oral gavage. Dose levels were selected optimizing for tolerability and to achieve plasma levels comparable to achievable levels in human clinical trials. For 8505C tumor xenografts bearing a BRAFV600E mutation, the drug combination caused a rapid and sustained decrease in tumor size, reaching a mean tumor volume reduction of 50% at 7 days and a peak reduction of >65% at 25 days, compared to the starting tumor volume (Fig. 2A). Trametinib alone also was highly effective with sustained tumor size reductions. Kaplan-Meier analysis showed highly significant retardation of progression for trametinib or the combination (p<0.0001 vs. vehicle), but not for single agent pazopanib (Fig. 2C).
Figure 2.

Trametinib with pazopanib causes regression of established DTC xenograft tumors. (A) 8505C or (B) CAL62 cells were implanted in the flanks of athymic nu/nu mice and allowed to grow to 0.1 to 0.2 cm3. Groups of 11 mice were randomized to receive trametinib 1 mg/kg, pazopanib 100 mg/kg, the combination or vehicle alone, by daily oral gavage. Dashed lines in A and B indicate 50% regression compared to the starting volume. Kaplan-Meier analyses for tumor progression are shown for (C) 8505C and (D) CAL62 tumors, *p=0.03 vs. vehicle, **p<0.0001 vs. vehicle. DTC, differentiated thyroid cancer.
The combination of trametinib and pazopanib also was highly effective in CAL62 tumors bearing a KRASG12R mutation, reaching a peak reduction in tumor volume by >50% at 25 days compared to the starting tumor volume (Fig. 2B). Trametinib also was effective as a single agent in reducing the volume of CAL62 tumors. Trametinib, pazopanib and the two-drug combination all were associated with significant delays in tumor progression by Kaplan-Meier analysis (p=0.03 vs. vehicle). The drugs were well tolerated and no significant weight loss was noted with the drug combination or with either drug separately.
Pharmacodynamic analysis of treated xenograft tumors
We performed IHC for pERKT202/Y204 and pAKTS473 in order to qualitatively assess the ability of trametinib and pazopanib to inhibit key signaling targets in vivo. As shown in Fig. 3, 8505C or CAL62 xenograft tumors from the vehicle-treated mice had dense, predominantly nuclear reactivity for pERK. Four hours after trametinib treatment, we observed a significant reduction in pERK reactivity. The combination of the two drugs strongly reduced pERK reactivity whereas pazopanib alone had little effect on nuclear pERK compared to the vehicle-treated control. AKT phosphorylation at serine 473 depends partially on PI3K-PDK1 activation, and also on TORC2 activation, which may be independent of PI3K. We observed downregulation of pAKTS473 by trametinib and by the trametinib-pazopanib combination in both the CAL62 and 8505C xenograft models, whereas pazopanib alone had a more limited effect.
Based on the potential of pazopanib to inhibit VEGFR2 signaling, we examined microvessel density (MVD), using immunohistochemistry for CD31 (Fig. 4). In CAL62 and 8505C xenografts, pazopanib resulted in a significant decline in MVD. Trametinib plus pazopanib together or trametinib alone also caused significant reductions in MVD.
Figure 4.
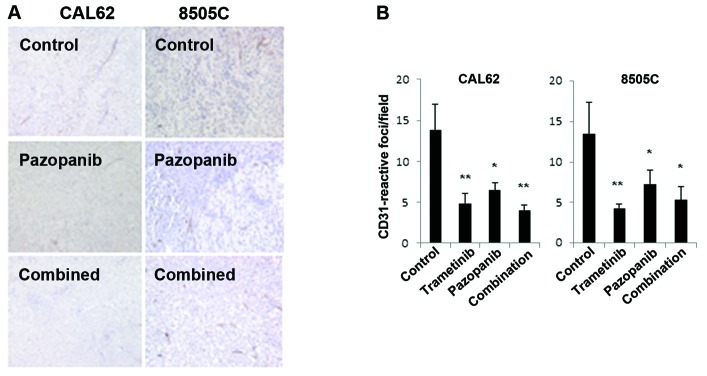
Trametinib and pazopanib reduce microvessel density. Established CAL62 and 8505C xenograft tumors treated with the indicated drugs for 2–3 weeks were harvested 4 h after dosing. (A) CD31 immunohistochemistry was performed on formalin-fixed, paraffin-embedded sections (DAB as chromogen). (B) Quantification of CD31-reactive foci (see Materials and methods). Both trametinib and pazopanib resulted in significant reduction in CD31-reactive foci (*p<0.05 vs. control; **p<0.005 vs. control).
Based on these in vitro and xenograft data, the MEK inhibitor trametinib appears highly effective in BRAF- and RAS-mutant thyroid cancer preclinical models, both singly and in combination with pazopanib. The combination of trametinib and pazopanib appeared more effective than pazopanib alone in downregulating pERK immunoreactivity; effects on microvessel density were comparable.
Discussion
Highly selective MEK inhibitors have shown significant clinical benefit in cancers with defined genotypes, including both BRAF and RAS mutant malignant melanoma (21,22). In published studies, RAF or MEK inhibitors alone or in combination have shown significant activity against DTC cell lines and xenografts (11,23,24). Recently, the MEK inhibitor selumetinib (AZD6244) has shown utility in a pilot study to increase radioiodine uptake in some radioiodine-refractory DTC tumor lesions (25). In this preclinical study, we explored the activity of trametinib, and whether trametinib could augment the antitumor activity of pazopanib, an agent shown to have promising activity in a phase II study of DTC (15). We found that trametinib induced durable shrinkage of thyroid cancer xenografts, either singly or in combination with pazopanib. Pazopanib alone was less effective in the tested xenograft models. We found that pazopanib treatment led to paradoxical ERK activation in vitro. While we did not observe ERK activation by pazopanib in the xenograft models, trametinib potently inhibited pERK immunoreactivity, whereas pazopanib did not.
It is currently unknown whether ERK activation occurs adaptively in the tumors of DTC patients treated with MKIs such as sorafenib, and pazopanib, and whether this phenomenon could contribute to drug resistance. Our preclinical data suggest that ERK activation by pazopanib may occur in patients as well. Given these preclinical data, as well as the high frequency of mutations affecting the ERK pathway and the recent FDA approval of sorafenib in DTC, the question of ERK activation by MKI's seems critical for correlative studies in thyroid cancer clinical trials.
Most impressively in our present study, trametinib showed marked single agent activity against both RAS- and BRAF-mutant thyroid cancer models in vitro and in vivo. This activity was retained or further augmented when trametinib was combined with pazopanib. Notably, trametinib was effective as a single agent against the RAS mutant CAL62 xenograft model. We did not observe significant AKT activation with trametinib in this model. Clinically, the MEK inhibitor MEK162 (Novartis) has shown significant single agent activity in RAS-mutant melanoma patients, a subset not appropriate for BRAF inhibitor treatment (26).
In summary, this preclinical study suggests that single agent trametinib, and trametinib plus pazopanib in combination, are highly active in thyroid cancer models, and may be promising for clinical efficacy in thyroid cancer. Based on early data from this study, we have initiated a phase I clinical trial combining trametinib and pazopanib, including a DTC expansion cohort (NCT01438554).
Acknowledgments
The authors thank Dr Tona Gilmer (GlaxoSmithKline, King of Prussia, PA, USA) for the generous gift of trametinib and pazopanib. The present research was supported by NIH Specialized Programs of Research Excellence (SPORE) in Head and Neck Cancer grant P50 DE19032.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Ricarte-Filho JC, Ryder M, Chitale DA, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–4893. doi: 10.1158/0008-5472.CAN-09-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010;20:697–706. doi: 10.1089/thy.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 5.Gril B, Palmieri D, Qian Y, et al. Pazopanib reveals a role for tumor cell B-Raf in the prevention of HER2+ breast cancer brain metastasis. Clin Cancer Res. 2010;17:142–153. doi: 10.1158/1078-0432.CCR-10-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 8.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lito P, Pratilas CA, Joseph EW, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnault JP, Mateus C, Escudier B, et al. Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin Cancer Res. 2012;18:263–272. doi: 10.1158/1078-0432.CCR-11-1344. [DOI] [PubMed] [Google Scholar]
- 11.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520–533. doi: 10.1158/2159-8290.CD-12-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar R, Knick VB, Rudolph SK, et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther. 2007;6:2012–2021. doi: 10.1158/1535-7163.MCT-07-0193. [DOI] [PubMed] [Google Scholar]
- 13.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 14.van der Graaf WT, Blay JY, Chawla SP, et al. EORTC Soft Tissue and Bone Sarcoma Group. PALETTE study group Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 15.Bible KC, Suman VJ, Molina JR, et al. Endocrine Malignancies Disease Oriented Group. Mayo Clinic Cancer Center. Mayo Phase 2 Consortium Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11:962–972. doi: 10.1016/S1470-2045(10)70203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flaherty KT, Robert C, Hersey P, et al. METRIC Study Group Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 17.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 18.Hurwitz HI, Dowlati A, Saini S, et al. Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res. 2009;15:4220–4227. doi: 10.1158/1078-0432.CCR-08-2740. [DOI] [PubMed] [Google Scholar]
- 19.Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2012;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 20.Bible KC1, Suman VJ, Menefee ME, et al. Mayo Phase 2 Consortium. Mayo Clinic Endocrine Malignances Disease Oriented Group A multiinstitutional phase 2 trial of pazopanib monotherapy in advanced anaplastic thyroid cancer. J Clin Endocrinol Metab. 2012;97:3179–3184. doi: 10.1210/jc.2012-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–489. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ball DW, Jin N, Rosen DM, Dackiw A, Sidransky D, Xing M, Nelkin BD. Selective growth inhibition in BRAF mutant thyroid cancer by the mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244. J Clin Endocrinol Metab. 2007;92:4712–4718. doi: 10.1210/jc.2007-1184. [DOI] [PubMed] [Google Scholar]
- 24.Leboeuf R, Baumgartner JE, Benezra M, et al. BRAFV600E mutation is associated with preferential sensitivity to mitogen-activated protein kinase kinase inhibition in thyroid cancer cell lines. J Clin Endocrinol Metab. 2008;93:2194–2201. doi: 10.1210/jc.2007-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho AL, Grewal RK, Leboeuf R, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368:623–632. doi: 10.1056/NEJMoa1209288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]