

Abstract
Spinal serotonin type 7 (5-HT7) receptors elicit complex effects on motor activity. Whereas 5-HT7 receptor activation gives rise to long-lasting phrenic motor facilitation (pMF), it also constrains 5-HT2 receptor-induced pMF via “cross-talk inhibition.” We hypothesized that divergent cAMP-dependent signaling pathways give rise to these distinct 5-HT7 receptor actions. Specifically, we hypothesized that protein kinase A (PKA) mediates cross-talk inhibition of 5-HT2 receptor-induced pMF whereas 5-HT7 receptor-induced pMF results from exchange protein activated by cAMP (EPAC) signaling. Anesthetized, paralyzed, and ventilated rats receiving intrathecal (C4) 5-HT7 receptor agonist (AS-19) injections expressed pMF for >90 min, an effect abolished by pretreatment with a selective EPAC inhibitor (ESI-05) but not a selective PKA inhibitor (KT-5720). Furthermore, intrathecal injections of a selective EPAC activator (8-pCPT-2′-Me-cAMP) were sufficient to elicit pMF. Finally, spinal mammalian target of rapamycin complex-1 (mTORC1) inhibition via intrathecal rapamycin abolished 5-HT7 receptor- and EPAC-induced pMF, demonstrating that spinal 5-HT7 receptors elicit pMF by an EPAC-mTORC1 signaling pathway. Thus 5-HT7 receptors elicit and constrain spinal phrenic motor plasticity via distinct signaling mechanisms that diverge at cAMP (EPAC vs. PKA). Selective manipulation of these molecules may enable refined regulation of serotonin-dependent spinal motor plasticity for therapeutic advantage.
Keywords: motor neuron, phrenic nerve, spinal cord, respiratory plasticity, neuroplasticity, 5-HT7, receptor, exchange protein activated by cAMP, protein kinase A, rapamycin, mTOR
serotonin plays a key role in important forms of sensory-motor plasticity, including sensitization of the gill withdrawal reflex in Aplysia (reviewed in Kandel 2012). For example, episodic serotonin presentations enhance sensory motor synaptic transmission, giving rise to the gill withdrawal reflex (Brunelli et al. 1976). This well-studied form of plasticity in an invertebrate model system relies on multiple serotonin receptor subtypes, each activating unique kinase signaling cascades (Barbas et al. 2003).
In ways similar to sensory motor facilitation in Aplysia, episodic serotonin receptor activation is necessary and sufficient for important forms of spinal respiratory motor plasticity, such as long-lasting (>90 min) phrenic motor facilitation (pMF) following acute intermittent hypoxia (AIH; reviewed by Mahamed and Mitchell 2007; Dale-Nagle et al. 2010; Devinney et al. 2013) or direct injections of serotonin or serotonin receptor agonists into the cervical spinal cord of rats (Hoffman and Mitchell 2011; MacFarlane et al. 2009, 2011). Indeed, multiple serotonin receptor subtypes elicit pMF via mechanistically distinct signaling cascades named for the G proteins most often coupled with the initiating metabotropic serotonin receptor (Dale-Nagle et al. 2010). Specifically, Gs protein-coupled serotonin type 7 (5-HT7) receptors give rise to pMF through the “S pathway” (Dale-Nagle et al. 2010; Hoffman and Mitchell 2011), whereas Gq protein-coupled 5-HT2 receptors induce the “Q pathway” to pMF (Dale-Nagle et al. 2010; MacFarlane et al. 2011).
While the S and Q pathways elicit pMF through distinct signaling cascades, they interact via mutual “cross-talk inhibition” (Dale-Nagle et al. 2010; Devinney et al. 2013; Hoffman and Mitchell 2013). One manifestation of cross-talk inhibition is the bell-shaped dose-response curve of pMF in response to intermittent intrathecal serotonin injections (MacFarlane and Mitchell 2009). Although low serotonin doses elicit pMF by activating spinal 5-HT2 receptors (Fuller et al. 2001; MacFarlane et al. 2011; MacFarlane and Mitchell 2009), high doses do not unless spinal 5-HT7 receptors are inhibited. Thus, when activated alone, 5-HT2 (MacFarlane et al. 2011) and 5-HT7 (Hoffman and Mitchell 2011) receptors each give rise to distinct forms of pMF. When activated concurrently, they effectively cancel each other because of balanced cross-talk inhibition (Devinney et al. 2013; Hoffman and Mitchell 2013; MacFarlane and Mitchell 2009). Cross-talk inhibition from 5-HT7 receptors to Q pathway-induced pMF is mediated by protein kinase A (PKA) activity (Hoffman and Mitchell 2013); however, it is not known how 5-HT7 receptor activation initiates pMF when acting alone (Hoffman and Mitchell 2011).
An alternative, but only recently appreciated, cAMP-dependent signaling cascade operating through exchange protein activated by cAMP (EPAC; de Rooij et al. 1998; Kawasaki et al. 1998) contributes to at least some forms of cAMP-dependent synaptic plasticity (Fernandes et al. 2015; Ster et al. 2007; Woolfrey et al. 2009). A key downstream molecule differentiating the PKA vs. EPAC signaling pathway is protein kinase B or Akt, a molecule necessary for 5-HT7-induced pMF (Hoffman and Mitchell 2011). Whereas PKA inhibits neuronal Akt activity, EPAC stabilizes Akt's association with scaffolding proteins to enhance its kinase function (Nijholt et al. 2008). Thus we hypothesized that EPAC (not PKA) is both necessary and sufficient for 5-HT7 receptor-induced pMF, whereas PKA mediates cross-talk inhibition of the Q pathway to pMF. Furthermore, Akt may elicit plasticity by increasing activity of mammalian target of rapamycin complex-1 (mTORC1), a major regulator of new protein synthesis (Ma and Blenis 2009). Thus we tested the hypothesis that spinal 5-HT7 receptors give rise to pMF through an EPAC-mTORC1 signaling pathway.
We found that spinal EPAC activation is necessary and sufficient for 5-HT7 receptor-induced pMF and that these effects require mTORC1 signaling. We confirm that PKA is not necessary for 5-HT7 receptor-induced pMF and conclude that divergent cAMP signaling pathways underlie the distinct functions of 5-HT7 receptors as they elicit (EPAC) and constrain (PKA) spinal serotonin-dependent respiratory motor plasticity.
METHODS
Experiments were performed on anesthetized 300- to 400-g male Lewis rats (Colony 202c, Harlan, Indianapolis, IN). The Animal Care and Use Committee at the University of Wisconsin, Madison approved all procedures.
Immunohistochemistry.
Although PKA constrains 5-HT2 receptor-dependent pMF through cross-talk inhibition (Hoffman and Mitchell 2013), several lines of evidence suggest that EPAC (not PKA) mediates 5-HT7 receptor-induced pMF. However, no study has attempted to localize these proteins in spinal respiratory neurons in general, or phrenic motor neurons (PMNs) in specific. Thus we investigated 5-HT7 receptor, PKA, and EPAC protein expression within PMNs.
To identify PMNs, four untreated rats received intrapleural injections of the retrograde tracer cholera toxin B fragment (CtB; List Biological Laboratories, Campbell, CA; Mantilla et al. 2009). Briefly, each rat was anesthetized with isoflurane (1–1.5% in 100% O2), and a 50-μl Hamilton syringe with a sterile, slightly beveled 27-gauge needle was used to inject 12.5 μl of 0.2% CtB in sterile saline. Bilateral injections were made at the fifth intercostal space (25 μl of 0.2% CtB total). After injections rats were carefully monitored for signs of respiratory distress associated with potential pneumothorax as isoflurane was discontinued (15 min). Seven days after injection rats were again anesthetized with isoflurane, euthanized, and transcardially perfused with 0.1 M phosphate-buffered saline (PBS; pH 7.4) followed by 4.0% paraformaldehyde (PFA; freshly made in 0.1 M phosphate buffer, pH 7.4). The cervical spinal cord was removed and placed in 4.0% PFA overnight at 4°C and then transferred to 20% and 30% sucrose in PBS buffer until sinking. Serial coronal sections (40 μm) of cervical C3–C5 were prepared with a microtome (Leica SM2000R) before storage in a cryoprotectant solution (30% ethylene glycol, 30% glycerol in 0.1 M PBS).
Free-floating sections were washed with 0.1 M PBS and incubated in a blocking solution (2% normal donkey serum, 0.1% Triton in PBS) for 60 min. Afterwards, tissues were costained for CtB:EPAC, CtB:PKA, or CtB:5-HT7 with the following antibodies: goat anti-CtB (1:5,000; Calbiochem), rabbit anti-EPAC (1:100; Santa Cruz), rabbit anti-PKA (1:100; Abcam), and rabbit anti 5-HT7 (1:100; Abcam). Unbound primary antibodies were removed with several PBS (0.1 M, pH 7.4) washes prior to incubation with secondary antibodies (Alexa 594 anti-goat 1:1,000 for CtB, Alexa 488 anti-rabbit 1:500 for EPAC and 5-HT7, Alexa 488 anti-rabbit 1:1,000 for PKA; Invitrogen) for 2 h at room temperature. Afterwards, sections were washed in PBS and mounted on charged slides with an antifade solution (Prolong Gold antifade reagent; Invitrogen). Control staining with each primary antibody (without secondary antibodies) and all secondary antibodies (without primary antibodies) ruled out nonspecific, off-target staining (see Fig. 1).
Fig. 1.

Identified phrenic motor neurons (PMNs) express exchange protein activated by cAMP (EPAC), protein kinase A (PKA), and serotonin type 7 (5-HT7) receptors: immunofluorescence images (×100) from 3 rats (left to right) for EPAC (A–C), PKA (D–F), and 5-HT7 receptor (G–I) protein (red) with retrograde cholera toxin B fragment (CtB; green). A–C: EPAC + CtB staining of PMNs reveals perinuclear EPAC distribution (*). D–F: PKA + CtB staining of PMNs exhibits diffuse cytoplasmic PKA distribution (*). G–I: 5-HT7 receptor + CtB staining shows abundant neuropil (*) but limited cytoplasmic/cell body 5-HT7 receptor distribution. CtB staining was consistent in all groups, with prominent cytoplasmic staining and limited immunofluorescence in the nucleus or neuropil.
Slides were examined with a confocal microscope (Eclipse TE 2000-U, Nikon) using EZ-C1 software (Nikon). Z stacks were taken (26 μm thick, 2-μm steps) with a ×100 magnification objective. Images were rendered and finished with EZ-C1 software.
Experimental preparation.
Rat anesthesia was induced with isoflurane (3.5%) before placement on a heated stainless steel surgical table. The inspired oxygen concentration was continuously monitored (TED 60T; Teledyne Analytical Instruments), with adjustments made as necessary by changing nitrogen and/or oxygen flow rates. A tail vein catheter (24 gauge; Surflo, Elkton, MD) was inserted, and an infusion pump (Cole-Palmer, Vernon Hills, IL) was used to deliver intravenous fluids throughout the experiment; the solution used was 9:1 lactated Ringer solution (Baxter)-sodium bicarbonate solution (8.4%; Hospira; 1.5–2 ml/h). Rats were tracheotomized and bilaterally vagotomized through a ventral midline incision. A polyethylene catheter (PE50, ID/OD 0.58 mm/0.965 mm; Intramedic) was inserted into the femoral artery to monitor blood pressure (Gould Pressure Transducer, P23) and allow for arterial blood sampling (ABL 500; Radiometer, Copenhagen, Denmark). Dorsal laminectomy and durotomy (C1/C2) were performed to enable intrathecal drug delivery. Muscles overlying the cervical spinal cord were separated to expose the C1-C2 cervical vertebrae; after C2 laminectomy, a silicone catheter (OD 0.6 mm; Access Technologies; primed with drug/vehicle) was inserted through a small hole in the dura and advanced caudally (3 mm), resting just over spinal regions C3-C4. This catheter was not placed in position until the end of surgical preparations to minimize unintended drug diffusion from the catheter. The left phrenic and hypoglossal (XII) nerves were dissected via a dorsal approach, desheathed, and covered with saline-soaked cotton to prevent desiccation. The rat was then slowly converted to urethane anesthesia (1.8 g/kg) while simultaneously being weaned from isoflurane (over 15–20 min). During anesthetic conversion, a rectal thermometer (Fisher Scientific) was inserted and body temperature was maintained at 37.5 ± 1.0°C throughout the experiment by adjusting the temperature of the surgical table. After conversion to urethane anesthesia, the rat was allowed at least 1 h for stabilization before experimental protocols started.
During the stabilization period the intrathecal catheter was inserted while both nerves were placed on bipolar silver recording electrodes and submerged in mineral oil. Once adequacy of anesthesia was confirmed (absent blood pressure spike in response to toe pinch) and respiratory nerve activity was detected, the rat was paralyzed with pancuronium bromide (3 mg/kg; Sicor Pharmaceuticals); 20 min was allowed for further stabilization. Nerve activity was amplified (gain, 10,000; A-M Systems, Everett, WA), band-pass filtered (100 Hz to 10 kHz), and integrated (CWE 821 filter; Paynter, Ardmore, PA; time constant 50 ms). The signal was digitized and recorded with a WINDAQ data acquisition system (DATAQ Instruments, Akron, OH). The resulting signal was analyzed with custom-designed software on a LabVIEW platform.
Throughout surgery and experimental protocols rats were ventilated with 60% inspired oxygen. End-tidal CO2 was monitored with a flow-through capnoguard with sufficient response time to detect end-tidal CO2 levels in a rat (Novametrix, Wallingford, CT). Rats were ventilated at a frequency of 70 breaths/min and a tidal volume of 2.5 ml or less (rodent ventilator model 683; Harvard Apparatus, South Natick, MA). This ventilation level caused hypocapnia (reduced end-tidal CO2); thus inspired CO2 was increased until the desired end-tidal value was attained. The CO2 apneic threshold was determined by progressively lowering inspired CO2 until respiratory nerve activity ceased; the recruitment threshold was subsequently determined by progressively raising inspired CO2 until nerve activity resumed. To establish baseline levels, end-tidal CO2 was set 2–3 mmHg above the CO2 recruitment threshold as done previously (Bach and Mitchell 1996). Arterial partial pressure of CO2 (PaCO2) was maintained within 1 mmHg of baseline values throughout the experiment by manipulating inspired CO2 and monitoring arterial blood gases. Base excess values more negative than −3.0 meq/l were corrected with intravenous sodium bicarbonate (8.4%; Hospira) prior to baseline measurements. Blood was sampled (0.3 ml using a heparinized capillary) once baseline nerve recordings had stabilized and then again at 30, 60 and 90 min after injection of intrathecal drugs (see below). Measurements of nerve activity (burst amplitude and frequency) were monitored continuously and evaluated immediately prior to blood samples in 1-min bins. At the end of experiments rats were euthanized by urethane overdose.
Physiological variables for respective groups are summarized in Table 1. While several groups showed a significant increase in firing frequency, this was mild relative to phrenic nerve amplitude changes. Because of the inconsistency and faintness of this difference we did not pursue this further.
Table 1.
Physiological variables at baseline and 30, 60, and 90 min after final injection
| Time, min | Control | 5-HT7 Agonist | EPACi + 5-HT7 Agonist | Rapamycin + 5-HT7 Agonist | PKAi + 5-HT7 Agonist | EPACa | EPACi + EPACa | Rapamycin + EPACa | |
|---|---|---|---|---|---|---|---|---|---|
| PaCO2, mmHg | Baseline | 49.8 ± 1.5 | 47.2 ± 1.5 | 52.3 ± 2.0 | 52.6 ± 0.9 | 50.5 ± 1.7 | 52.7 ± 0.9 | 55.5 ± 0.6 | 52.6 ± 0.9 |
| 30 | 48.7 ± 2.4 | 48.2 ± 1.7 | 52.5 ± 1.9 | 52.2 ± 1.2 | 50.6 ± 1.7 | 53.4 ± 1.1 | 55.5 ± 0.9 | 52.2 ± 1.2 | |
| 60 | 50.7 ± 1.5 | 47.5 ± 1.6 | 54.0 ± 2.1 | 52.9 ± 1.5 | 50.5 ± 1.5 | 53.0 ± 0.8 | 55.4 ± 0.7 | 52.9 ± 1.5 | |
| 90 | 50.6 ± 1.9 | 47.0 ± 1.5 | 51.9 ± 1.9 | 52.5 ± 1.4 | 50.7 ± 1.8 | 52.8 ± 1.0 | 55.7 ± 0.8 | 52.5 ± 1.4 | |
| PaO2, mmHg | Baseline | 310 ± 27 | 301 ± 11 | 306 ± 17 | 341 ± 25 | 310 ± 8 | 322 ± 12 | 369 ± 18 | 309 ± 23 |
| 30 | 291 ± 29 | 303 ± 18 | 297 ± 22 | 337 ± 26 | 296 ± 6.0 | 302 ± 11 | 347 ± 22 | 299 ± 16 | |
| 60 | 289 ± 26 | 289 ± 9.9 | 295 ± 25 | 359 ± 26 | 291 ± 7.5 | 304 ± 14 | 344 ± 25 | 302 ± 15 | |
| 90 | 291 ± 29 | 283 ± 11 | 289 ± 30 | 364 ± 27 | 305 ± 15 | 288 ± 7.7 | 344 ± 27 | 305 ± 15 | |
| MAP, mmHg | Baseline | 50.7 ± 3.0 | 49.8 ± 4.6 | 73.4 ± 14 | 58.5 ± 3.6 | 54.1 ± 4.8 | 44.5 ± 2.6 | 49.3 ± 3.3 | 39.8 ± 3.5 |
| 30 | 50.0 ± 3.4 | 48.5 ± 3.8 | 69.5 ± 12 | 47.9 ± 3.9 | 50.5 ± 4.8 | 40.4 ± 2.3 | 41.1 ± 2.9 | 31.7 ± 3.4 | |
| 60 | 42.5 ± 3.4 | 44.6 ± 3.6 | 67.4 ± 11 | 45.9 ± 7.3 | 45.3 ± 3.0 | 37.5 ± 2.5 | 41.1 ± 2.5 | 28.5 ± 3.0 | |
| 90 | 41.9 ± 2.5 | 44.9 ± 2.4 | 66.0 ± 12 | 43.7 ± 6.5 | 42.32.8 | 31.1 ± 3.4 | 40.9 ± 3.8 | 24.5 ± 3.4 | |
| Phrenic burst frequency | Baseline | 55.8 ± 8.4 | 44.8 ± 1.39 | 49.3 ± 0.5 | 47.9 ± 1.1 | 50.0 ± 0.8 | 44.4 ± 0.8 | 41.0 ± 0.9 | 45.1 ± 1.1 |
| 30 | 54.9 ± 8.5 | 47.4 ± 1.3 | 48.3 ± 1.8 | 48.3 ± 1.1 | 49.4 ± 0.9 | 47.1 ± 1.0 | 41.9 ± 1.2 | 44.5 ± 1.5 | |
| 60 | 55.6 ± 9.5 | 48.6 ± 1.4* | 48.8 ± 1.4 | 49.3 ± 2.1 | 51.0 ± 1.1 | 47.5 ± 1.1* | 43.3 ± 0.8 | 43.4 ± 1.5 | |
| 90 | 56.3 ± 10.2 | 48.8 ± 1.5* | 47.1 ± 0.5 | 50.0 ± 1.8 | 49.6 ± 1.9 | 46.3 ± 1.2 | 44.6 ± 0.8* | 43.0 ± 1.5 |
Values are expressed as means ± SE. EPAC, exchange protein activated by cAMP; PKA, protein kinase A; EPACi, EPAC-selective inhibitor; PKAi, PKA-selective inhibitor; EPACa, EPAC-selective activator; PaCO2, arterial partial pressure of CO2; PaO2, arterial partial pressure of O2; MAP, mean arterial pressure.
Significantly different from baseline within same group (P ≤ 0.05).
Drugs and vehicles.
The following drugs were obtained from Santa Cruz (Dallas, TX): AS-19 (5-HT7 receptor agonist), 8-pCPT-2′-Me-cAMP [8-pCPT; EPAC-selective activator (EPACa)], and KT-5720 [PKA-selective inhibitor (PKAi)]. Rapamycin (mTORC1 inhibitor) was obtained from Thermo-Fisher (Waltham, MA), while ESI-05 [EPAC-selective inhibitor (EPACi)] was obtained from BioLog Life Science Institute (Germany). All drugs were initially dissolved in dimethyl sulfoxide (DMSO) before dilution with vehicle (20% DMSO in sterile saline) before use. Aliquots of stock solutions remained viable for up to 1 wk if stored frozen (−20°C) in 100% DMSO; after this time, unused drug solutions were discarded. Prior studies confirm that EPACa is a selective EPAC activator (Christensen et al. 2003; Poppe et al. 2008); conversely, EPACi, PKAi, and rapamycin are regarded as selective inhibitors of EPAC (Rehmann 2013; Tsalkova et al. 2012), PKA (Davies et al. 2000), and mTORC1 (Davies et al. 2000), respectively.
Experimental protocols.
After stabilization of nerve signals a baseline blood sample was drawn, followed by a control intrathecal injection of vehicle (12 μl), a 15-min gap, and three consecutive intrathecal injections (C4) of 5-HT7 receptor agonist (3 × 5 μl, 100 μM; 5-min intervals) or a single injection of EPACa (10 μl, 100 μM). The 5-HT7 receptor agonist dose was determined from a previous study using the same experimental protocol (Hoffman and Mitchell 2011), while a limited dose-response curve was completed for EPACa (data not shown).
Whereas the 5-HT7 receptor agonist gave rise to pMF when injected intermittently (not as a single bolus), intrathecal EPACa gave rise to pMF when given as a single bolus (not intermittently). This requirement for intermittent 5-HT7 receptor activation is consistent with previous studies demonstrating pattern sensitivity of both serotonin-induced and serotonin-dependent pMF (Baker and Mitchell 2000; MacFarlane and Mitchell 2009). In contrast, single (vs. intermittent) injection requirements for 8-pCPT (EPACa) are consistent with previous studies of EPAC-induced plasticity (Ster et al. 2007).
To identify molecules necessary for 5-HT7 receptor- and EPAC-induced pMF, additional groups received intrathecal injections of selective inhibitors prior to the 5-HT7 receptor agonist or EPAC activator. All inhibitors were given intrathecally via a second catheter (over a period of 2 min) 15 min prior to 5-HT7 receptor agonist or EPACa injections. To determine whether PKA is necessary for 5-HT7 receptor-induced pMF KT-5720, a PKAi (n = 6; 12 μl, 100 μM), was given prior to 5-HT7 receptor agonist injections. We previously demonstrated that KT-5720 at this dose prevents PKA-mediated constraint of 5-HT2A receptor-dependent, AIH-induced pMF (Hoffman and Mitchell 2013). In addition, this dose prevents PKA- but not EPAC-dependent signaling within cell cultures (Davies et al. 2000). Using an EPACi (12 μl, 2 mM), ESI-05, we tested whether EPAC is necessary for 5-HT7 receptor (n = 7)- and EPACa (n = 6)-induced pMF. Finally, by pretreating with the highly selective inhibitor rapamycin (12 μl, 100 μM) we determined whether mTORC1 was necessary for 5-HT7 receptor (n = 6)- or EPACa (n = 6)-induced pMF.
Additional control groups were completed for vehicle (n = 6), PKAi (n = 5), EPACi (n = 5), and rapamycin (n = 4) in which the drug was given followed by 3 × 5-μl injections of vehicle 15 min later. None of the control groups affected phrenic nerve activity, and they were not significantly different from each other; thus these groups were assembled into a single control group (n = 20; P ≥ 0.26).
Statistical analyses.
Peak integrated amplitude and frequency of phrenic and XII nerves were averaged in 1-min bins at baseline (preinjection) and then 30, 60, and 90 min after intrathecal injections. Amplitude values are expressed as percent change from baseline, while frequency is expressed as change from baseline in bursts per minute. Statistical comparisons were made for control and experimental groups with a two-way repeated-measures ANOVA; a Tukey post hoc test was used to identify pairwise differences. Significance was accepted as P < 0.05. All values are expressed as means ± SE.
RESULTS
Phrenic motor neuron expression of EPAC, PKA, and 5-HT7 receptors.
PMNs were identified via intrapleural CtB injections. Antibodies targeting EPAC2, PKA, and 5-HT7 receptors all revealed immunofluorescence within/near CtB-identified PMNs. EPAC2 immunoreactivity was cytoplasmic, with the densest staining in the perinuclear region (Fig. 1, A–C). PKA immunoreactivity was observed within the cytoplasm of CtB-colabeled PMNs (Fig. 1, D–F). Immunofluorescence for 5-HT7 receptors was localized to the neuropil, with relatively limited cytoplasmic or nuclear staining (Fig. 1, G–I). Thus PMNs can directly respond to 5-HT7 receptor agonists via EPAC and/or PKA signaling, although we cannot rule out additional contributions to pMF from surrounding cells: glia, presynaptic neurons, or interneurons.
EPAC, but not PKA, is necessary for 5-HT7-induced pMF.
Intermittent intrathecal injections of 5-HT7 receptor agonist (3 × 5 μl, 100 μM) AS-19 elicited long-lasting pMF expressed as a progressive enhancement in phrenic nerve amplitude (>90 min: 88.8 ± 7.2%; Fig. 2, A and Bii). The dose/selectivity of this same 5-HT7 receptor agonist was determined in a previous study using the same experimental protocol (Hoffman and Mitchell 2011).
Fig. 2.
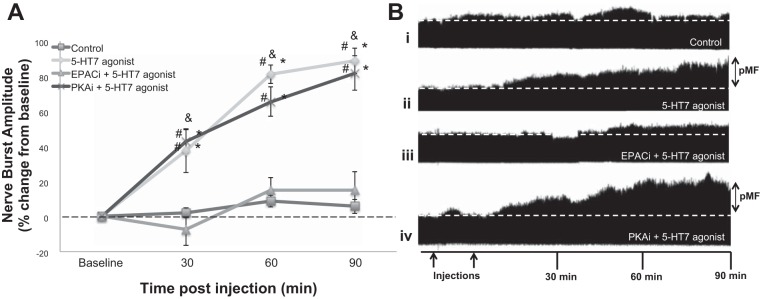
5-HT7 receptor-induced phrenic motor facilitation (pMF) requires EPAC but not PKA activity. A: average change in phrenic burst amplitude from baseline to 90 min after injections. Lines are from rats with intrathecal injections of the 5-HT7 receptor agonist (AS-19; 3 × 5 μl, 100 μM) with saline (◇, n = 7; 10 μl), EPAC-selective inhibitor (EPACi, △, n = 7; 10 μl, 2 mM), or PKA-selective inhibitor (PKAi, ×, n = 5; 10 μl, 100 μM) pretreatment or control rats that did not receive the 5-HT7 receptor agonist (□, n = 20; 10 μl). Data represent mean ± SE values. Significant differences from #baseline, *control, or &PKAi + 5-HT7 agonist: all P ≤ 0.05. B: representative traces of phrenic neurograms are shown before, during, and after intrathecal injections of vehicle + 5-HT7 receptor agonist, EPACi + 5-HT7 receptor agonist, or PKAi + 5-HT7 receptor agonist. First arrow (below trace iv) represents the pretreatment injection; 2nd arrow represents the first of 3 5-HT7 receptor agonist injections. Data from control and 5-HT7 agonist groups are repeated within multiple figures.
Pretreatment with an intrathecal EPACi (10 μl, 2 mM), ESI-05, abolished 5-HT7 receptor-induced pMF (90 min: 15.1 ± 10.5%, P = 0.0002; Fig. 2, A and Biii) demonstrating EPAC as necessary for 5-HT7 receptor-induced pMF. In contrast, pretreating with a PKAi (10 μl, 100 μM), KT-5720, did not significantly reduce pMF relative to the vehicle-pretreated 5-HT7 receptor agonist group (81.8 ± 10.0%, P = 0.61; Fig. 2, A and Biv). The KT-5720 dose is based on previous work using the same in vivo protocol (Hoffman and Mitchell 2013) and PKA cell culture activity assays (Davies et al. 2000). Cervical spinal injections of the 5-HT7 receptor agonist did not affect hypoglossal nerve (XII) activity (n = 4; 90 min: −0.1 ± 13.1%, P = 0.95), suggesting that 5-HT7 receptor agonist-induced pMF requires activation of spinal (vs. brain stem) receptors (Baker-Herman and Mitchell 2002; MacFarlane and Mitchell 2009).
Spinal EPAC activation elicits pMF.
Intrathecal injection of an EPACa (10 μl, 100 μM), 8-pCPT, elicited robust pMF lasting at least 90 min (104.4 ± 7.8%, P = 0.0000040; Fig. 3, A and Bii). Pretreatment with an intrathecal EPACi (10 μl, 2 mM), ESI-05, significantly attenuated EPACa-induced pMF (90 min: 24.5 ± 7.8%, P = 0.00064; Fig. 3, A and Biii), confirming the selectivity of EPACa. Although cervical spinal 5-HT7 receptor agonist injections did not cause XII facilitation, similar EPACa injections did elicit significant XII burst amplitude facilitation (data not shown) at 10 μM (n = 2; 90 min: 59.5 ± 8.2%) and 100 μM (n = 4; 90 min: 77.2 ± 23.1%); this effect was blocked by intrathecal EPACi pretreatment (n = 3; 90 min: 7.9 ± 11.6%, P = 0.00064). These differential 5-HT7 vs. EPAC effects on XII activity are difficult to explain, and we did not pursue this issue further in the present study.
Fig. 3.
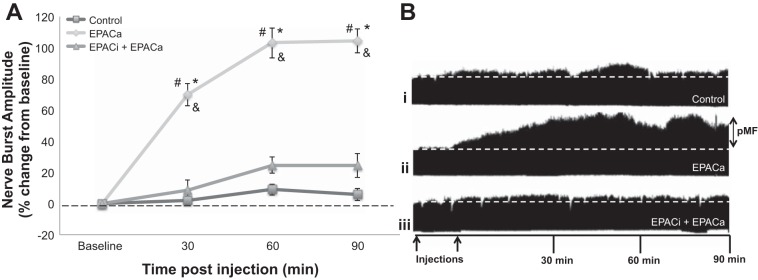
Spinal EPAC activation is sufficient to elicit pMF. A: average change in phrenic burst amplitude from baseline. Individual curves represent EPACa-treated rats (10 μl, 100 μM) with saline (◇, n = 7; 10 μl) or with EPACi pretreatment (△, n = 6; 10 μl, 2 mM) or control injections alone (□; n = 20; 10 μl). Data represent mean ± SE values. Significant differences from #baseline, *control, or &EPACi + 5-HT7 receptor agonist: all P ≤ 0.05. B: representative phrenic neurograms: time control (i), vehicle + EPACa (ii), or EPACi + EPACa (iii). First arrow represents pretreatment injection; 2nd arrow represents single EPACa injection. Data from control and EPACa groups are shown within multiple figures.
mTORC1 is necessary for 5-HT7- and EPAC-induced pMF.
Pretreatment with the mTORC1 inhibitor rapamycin (10 μl; 100 μM) blocked both 5-HT7 receptor (11.2 ± 9.2%, P = 0.00007)- and EPACa (9.9 ± 10.0%, P = 0.000039)-induced pMF (Fig. 4, A and B, respectively), demonstrating that mTORC1 is necessary for both processes.
Fig. 4.
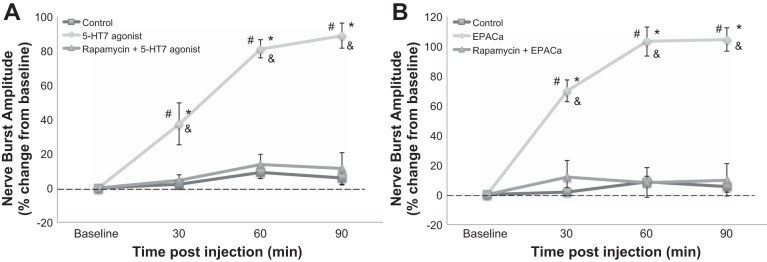
Mammalian target of rapamycin complex-1 (mTORC1) activity is necessary for 5-HT7 receptor- and EPAC-induced pMF. A: average change in phrenic burst amplitude from baseline for 5-HT7 receptor agonist (10 μl, 100 μM) with saline (◇, n = 7; 10 μl) or rapamycin (△, n = 6; 10 μl, 100 μM) pretreatment or control injection alone (□, n = 20; 10 μl). Data represent mean ± SE values. Significant differences from #baseline, *controls, and &groups with rapamycin pretreatment: all P ≤ 0.05. Data from control, 5-HT7 agonist, and EPACa groups are shown in multiple figures.
DISCUSSION
Although cervical spinal 5-HT7 receptor activation elicits pMF, it also constrains 5-HT2 receptor-induced pMF via cross-talk inhibition (Hoffman and Mitchell 2013; MacFarlane and Mitchell 2009). Here we show that these diverse actions result from divergent cAMP signaling. Specifically, 5-HT7 receptors constrain 5-HT2 receptor-induced pMF through cAMP/PKA signaling (Hoffman and Mitchell 2013) but elicit pMF via a mechanistically distinct cAMP/EPAC pathway. Since 5-HT7 receptor- and EPAC-induced pMF are rapamycin sensitive, this pathway requires mTORC1 signaling. We conclude that serotonin elicits spinal plasticity via diverse mechanisms. First, serotonin elicits phrenic motor plasticity via unique mechanisms associated with distinct serotonin receptor subtypes (e.g., 5-HT7 vs. 5-HT2 receptors). Second, 5-HT7 receptors elicit heterogeneous effects (presumably) within the same cell via divergent cAMP signaling (PKA and EPAC). Understanding distinct serotonin and cAMP functions may be critical as we attempt to harness serotonin-dependent plasticity as a treatment for severe neuro-motor disorders that compromise breathing, such as spinal injury and amyotrophic lateral sclerosis (Dale et al. 2014; Mitchell 2007).
While EPAC activation is necessary and sufficient for some forms of cAMP-dependent plasticity, selective PKA activation (EPAC independent) also gives rise to plasticity in some model systems (Castellucci et al. 1980). Thus EPAC and PKA may give rise to alternate, parallel signaling cascades for cAMP-dependent plasticity. Although PKA-dependent plasticity has been well studied, considerably less is known concerning how EPAC activation elicits plasticity. Here we demonstrate that EPAC-dependent, 5-HT7 receptor-induced pMF is mechanistically distinct from the serotonin-induced PKA-ERK-brain-derived neurotrophic factor (BDNF) signaling pathway described in invertebrate model systems (for review see Kandel 2012). As shown by the available evidence concerning 5-HT7 receptor-induced pMF, 5-HT7 receptors elicit pMF via EPAC-Akt-mTORC1 signaling, resulting in new synthesis of an immature TrkB isoform (Golder et al. 2008; Hoffman and Mitchell 2011). This mechanism is independent of PKA, ERK signaling, or new BDNF synthesis (Hoffman and Mitchell 2011).
We are not aware of any previous studies connecting EPAC or mTORC1 signaling with serotonin-induced plasticity. By showing that the canonical pathway of PKA-ERK-BDNF does not play a role in 5-HT7 receptor-induced spinal respiratory motor plasticity we have uncovered an interesting question: what purpose do parallel plasticity pathways operating downstream from a single receptor serve? We suggest that mechanistic heterogeneity in spinal motor plasticity signaling is important to maintain appropriate network activity throughout maturation, during changing environmental conditions, and during severe disease states (Dale et al. 2014). Although PKA signaling does not contribute to 5-HT7-induced pMF, it may serve as a “reserve” pathway when EPAC-dependent signaling is impaired. For example, while wild-type mice express EPAC-dependent (PKA-independent) mossy fiber plasticity, EPAC2-knockout mice retain certain forms of mossy fiber plasticity by shifting to PKA-dependent signaling (Fernandes et al. 2015). Thus in some conditions PKA activity is sufficient to induce plasticity during compromised EPAC signaling.
Finally, since PKA does not normally contribute to 5-HT7 receptor-induced pMF our findings suggest that reduced PKA signaling may prevent cross-talk inhibition without compromising 5-HT7 receptor-induced pMF. By minimizing cross-talk inhibition with PKA-selective inhibitors it may be possible for both 5-HT2 and 5-HT7 receptors to independently contribute to an enhanced form of pMF (i.e., metaplasticity; Fields and Mitchell 2015). In agreement, whereas PKA mediates cross-talk inhibition (Hoffman and Mitchell 2013), EPAC enables concurrent activation of signaling pathways operating downstream from 5-HT2 and 5-HT7 receptors (Johnson-Farley et al. 2005). The respective contributions of reduced PKA and/or enhanced EPAC activity to enhanced AIH-induced pMF following intermittent hypoxia preconditioning (Fields and Mitchell 2015) remain to be explored.
Although we are only beginning to understand interpathway interactions in serotonin receptor-induced spinal motor plasticity, the ability of nonessential accessory signaling pathways to regulate plasticity has considerable significance, from both a biological and a therapeutic perspective.
GRANTS
This work was funded by National Institutes of Health (NIH) Grants HL-80209 and HL-69064. D. P. Fields was supported by an Advanced Opportunity Fellowship at the University of Wisconsin and the Medical Scientist Training Program (NIH T32 GM-008692).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.P.F. and G.S.M. conception and design of research; D.P.F. and G.S.M. interpreted results of experiments; D.P.F. and G.S.M. edited and revised manuscript; D.P.F. and G.S.M. approved final version of manuscript; S.R.S. and G.S.M. performed experiments; S.R.S. and G.S.M. prepared figures; G.S.M. analyzed data; G.S.M. drafted manuscript.
REFERENCES
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol 104: 251–260, 1996. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol 529: 215–219, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci 7: 48–55, 2004. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci 22: 6239–6246, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbas D, DesGroseillers L, Castellucci VF, Carew TJ, Marinesco S. Multiple serotonergic mechanisms contributing to sensitization in Aplysia: evidence of diverse serotonin receptor subtypes. Learn Mem 10: 373–386, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelli M, Castellucci V, Kandel ER. Synaptic facilitation and behavioral sensitization in Aplysia: possible role of serotonin and cyclic AMP. Science 194: 1178–1181, 1976. [DOI] [PubMed] [Google Scholar]
- Castellucci VF, Kandel ER, Schwartz JH, Wilson FD, Nairn AC, Greengard P. Intracellular injection of the catalytic subunit of cyclic AMP-dependent protein kinase simulates facilitation of transmitter release underlying behavioral sensitization in Aplysia. Proc Natl Acad Sci USA 77: 7492–7496, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Døskeland SO. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem 278: 35394–35402, 2003. [DOI] [PubMed] [Google Scholar]
- Dale EA, Ben Mabrouk F, Mitchell GS. Unexpected benefits of intermittent hypoxia: enhanced respiratory and nonrespiratory motor function. J Physiol 29: 39–48, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol 669: 225–230, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396: 474–477, 1998. [DOI] [PubMed] [Google Scholar]
- Devinney MJ, Huxtable AG, Nichols NL, Mitchell GS. Hypoxia-induced phrenic long-term facilitation: emergent properties. Ann NY Acad Sci 1279: 143–153, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci 26: 239–266, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes S, Marshall JJ, Kukreja L, Vassar R, Contractor A. Epac2 mediates cAMP-dependent potentiation of neruotransmission in the hippocampus. J Neurosci 35: 6544–6553, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields DP, Mitchell GS. Spinal metaplasticity in respiratory motor control. Front Neural Circuits 9: 2, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol 90: 2001–2006, 2001. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci 28: 2033–2042, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33: 67–75, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J Physiol 589: 1397–1407, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptors and protein kinase A constrain intermittent hypoxia-induced phrenic long-term facilitation. Neuroscience 250: 632–643, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Nichols NL, Macfarlane PM, Mitchell GS. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation but not TrkB synthesis. J Appl Physiol 113: 1184–1193, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Farley NN, Kertesy SB, Dubyak GR, Cowen DS. Enhanced activation of Akt and extracellular-regulated kinase pathways by simultaneous occupancy of Gq-coupled 5-HT2A receptors and Gs-coupled 5-HT7A receptors in PC12 cells. J Neurochem 92: 72–82, 2005. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5: 14, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science 282: 2275–2279, 1998. [DOI] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10: 307–318, 2009. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol 587: 5469–5481, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience 178: 45–55, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol 92: 27–37, 2007. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods 182: 244–249, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Skorova EY. Stimulation of bursting in pre-Bötzinger neurons by Epac through calcium release and modulation of TRPM4 and K-ATP channels. J Neurochem 117: 295–308, 2011. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Skorova EY, Kügler S. Epac-mediated cAMP-signalling in the mouse model of Rett Syndrome. Neuropharmacology 60: 869–877, 2011. [DOI] [PubMed] [Google Scholar]
- Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Genetic Basis for Respiratory Control Disorders, edited by Gaultier C. New York: Springer, 2007. [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB Jr. Intermittent hypoxia and respiratory plasticity. J Appl Physiol 90: 2466–2475, 2001. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Shewan DA. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol Cell Neurosci 38: 578–588, 2008. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Tucker SJ, Shewan DA. cAMP-dependent axon guidance is distinctly regulated by Epac and protein kinase A. J Neurosci 29: 15434–15444, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrette-Opaza A, Mitchell GS. Therapeutic potential of intermittent hypoxia: a matter of dose. Am J Physiol Regul Integr Comp Physiol 307: R1181–R1197, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Dale EA, Mitchell GS. Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J Appl Physiol 112: 1678–1688, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijholt IM, Dolga AM, Ostroveanu A, Luiten PG, Schmidt M, Eisel UL. Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell Signal 20: 1715–1724, 2008. [DOI] [PubMed] [Google Scholar]
- Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods 5: 277–278, 2008. [DOI] [PubMed] [Google Scholar]
- Rehmann H. Epac-inhibitors: facts and artefacts. Sci Rep 3: 3032, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava DP, Jones KA, Woolfrey KM, Burgdorf J, Russell TA, Kalmbach A, Lee H, Yang C, Bradberry MM, Wokosin D, Moskal JR, Casanova MF, Waters J, Penzes P. Social, communication, and cortical structural impairments in Epac2-deficient mice. J Neurosci 32: 11864–11878, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ster J, De Bock F, Guérineau NC, Janossy A, Barrère-Lemaire S, Bos JL, Bockaert J, Fagni L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc Natl Acad Sci USA 104: 2519–2524, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL Jr, Cheng X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci USA 109: 18613–18618, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, Xie Z, Jones KA, Quilliam LA, Prakriya M, Penzes P. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci 12: 1275–1284, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shu X, Liu D, Shang Y, Wu Y, Pei L, Xu X, Tian Q, Zhang J, Qian K, Wang YX, Petralia RS, Tu W, Zhu LQ, Wang JZ, Lu Y. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron 73: 774–788, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]